

Abstract
A fundamental aspect of the adaptive immune system is the generation and maintenance of a diverse and self-tolerant T cell repertoire. Through its regulation of T cell development, homeostasis, tolerance, and differentiation, the highly evolutionarily conserved cytokine transforming growth factor-β (TGF-β) critically supports a functional T cell pool. The pleiotropic nature of this regulation is likely due to the elaborate control of TGF-β production and activation in the immune system, and the intricacy of TGF-β signaling pathways. This review discusses our current understanding of TGF-β regulation of T cells.
Introduction
The highly evolutionarily conserved cytokine transforming growth factor-β has three known mammalian family members (TGF-β1, -β2, and -β3) that regulate multiple physiological processes. TGF-β is synthesized in a latent form that must be activated to allow for engagement of a tetrameric receptor complex composed of TGF-β receptors I and II (TGF-βRI, -βRII). The production and activation of TGF-β can be mediated by distinct cellular sources, providing additional complexity to the regulation of this pleiotropic cytokine. Binding of active TGF-β to its receptor complex triggers receptor serine/threonine kinase activity, allowing for the phosphorylation of downstream signaling targets. TGF-β signaling is primarily mediated through the Smad family of transcription factors, but is also known to engage Smad-independent pathways.
TGF-β1 is the primary isoform expressed in the immune system, and its widespread regulatory activity affects multiple types of immune cells (1). T cells were established as critical targets of TGF-β in its control of immune tolerance by the finding that mice with T cell-specific deletion of TGF-βRII phenocopied the lethal inflammatory disorder that develops in Tgfb1 knockout mice (2-5). Nevertheless, TGF-β is more than an immunosuppressive cytokine. For instance, early studies revealed that TGF-β induces stimulatory or inhibitory effects in human T cells, which is dependent on the T cell differentiation status and the stimulation conditions (6). This context-dependent function of TGF-β allows for its distinct roles in T cell development, homeostasis, tolerance, and differentiation (Figure 1). This review discusses our current understanding of TGF-β regulation of T cells with a focus on recent discoveries.
Figure 1. TGF-β Regulation of T Cells.
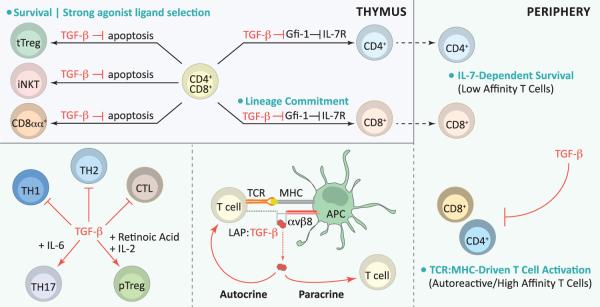
During thymic development, TGF-β-supported survival of tTreg, iNKT, and CD8αα+ T cell precursors fosters agonist ligand-induced T cell development. TGF-β also critically regulates thymocyte IL-7R expression by inhibiting the transcriptional repressor Gfi-1, and promotes conventional CD8+ T cell lineage commitment. In the periphery, TGF-β regulates T cell homeostasis by promoting IL-7-dependent survival of low affinity T cells (resulting from thymic conditioning of IL-7R expression) and by inhibiting TCR-driven activation of autoreactive/high affinity T cells. The T cell and antigen presenting cell (APC) pairing is essential for in vivo TGF-β function. T cells are a critical source of TGF-β, and APCs engaging T cells by TCR:peptide-MHC interactions are key providers of the αvβ8 integrins required for TGF-β activation. Activated TGF-β regulates T cells by autocrine and paracrine signaling. TGF-β inhibits TH1, TH2, and CTL differentiation, but in concert with other factors promotes TH17 or pTreg cell differentiation.
T Cell Development
During thymic development, T cell precursors undergo an orchestrated series of changes resulting in the differentiation of distinct mature T cell subsets. TGF-β has been shown to play important roles in the development of conventional, regulatory, and innate-like T cells.
CD8+ T Cells
In addition to T cell receptor (TCR) engagement, signaling via the common γ-chain family cytokine IL-7 is critical for the thymic development of CD8+ T cells (7). Whether TGF-β plays a role in CD8+ T cell lineage commitment was unclear given contradictory reports of reduced and normal thymic CD8+ T cell populations in mice with T cell-specific deletion of Tgfbr2 during the CD4+CD8+ thymocyte stage (4, 5). Although TGF-βRII-deficient mice were analyzed before the onset of overt autoimmunity, the confounding effects of systemic inflammation were a concern in both studies. The generation of mice with T cell-specific Tgfbr2 deletion and an HY transgenic TCR restricted repertoire allowed for the study of TGF-β regulation of CD8+ T cell development in the absence of autoimmune inflammation. The HY TCR recognizes a male mouse-specific antigen, but is also positively selected by low-affinity self antigens in female mice. TGF-βRII-deficient female HY transgenic mice exhibited impaired CD8+ T cell development relative to their wild-type counterparts (8). Intriguingly, TGF-β promoted the specification of CD8+ T cell fate largely through its control of thymocyte IL-7Rα expression (and by extension IL-7 signaling) by suppressing the transcriptional repressor Gfi-1, a known Il7ra inhibitor in CD8+ T cells (9). These findings reveal a mechanism of CD8+ T cell lineage commitment via the crosstalk of TGF-β and IL-7 cytokine signaling pathways.
Strong Agonist Ligand-Induced Selection: Regulatory and Innate-like T Cells
A combination of stringent TCR interactions, costimulation, and cytokine signals controls the development of thymus-derived CD4+CD25+Foxp3+ regulatory T (tTreg) cells, which are essential for immune tolerance (10). TGF-β signaling was thought to be dispensable for tTreg cell development based on reports that mice with T cell-specific Tgfbr2 deletion possessed normal thymocyte Foxp3+ populations (4, 5). However, the finding that Foxp3+ thymocyte frequency was dramatically reduced in 3 to 5 day old mice with T cell-specific Tgfbr1 deletion indicated that TGF-β does contribute to early tTreg cell development (11). IL-2-driven expansion of existing Foxp3+ thymocytes resulted in increased tTreg cell frequencies in older TGF-βRI-deficient mice (similar to observations in TGF-βRII-deficient mice). The discovery of a conserved Smad3 binding sequence in the Foxp3 gene prompted the hypothesis that TGF-β signaling induces Foxp3 expression in tTreg cells (12). However, deletion of the enhancer region containing the Smad3 binding site (conserved non-coding sequence 1, CNS1), or specific ablation of the Smad binding sequence revealed defects in only peripheral Treg (pTreg) cells, demonstrating that TGF-β is not required for Foxp3 induction in tTreg cells (13, 14). Rather, TGF-β has been shown to support tTreg cell development by antagonizing thymic negative selection. TGF-βRII-deficient thymocytes were found to express high levels of pro-apoptotic molecules, undergo enhanced negative selection, and contain a reduced frequency of Foxp3+ cells (15). Deletion of the pro-apoptotic molecule Bim prevented enhanced apoptosis of TGF-βRII-deficient thymocytes and rescued tTreg cell development.
Invariant Natural Killer T (iNKT) cells, which possess innate and adaptive-like properties, recognize lipids presented by the MHC Class I-like molecule CD1d. iNKT cells arise from CD4+CD8+ thymocytes, and akin to tTreg cells, are thought be induced by strong agonist ligand interactions (16). TGF-β signaling was implicated in iNKT cell development based on the observations that thymic and peripheral iNKT cells were lost in mice with T cell-specific deletion of Tgfbr2 (4, 5, 17). TGF-β was found to promote iNKT cell development in part by inhibiting the apoptosis of immature cells via a TIF-1γ-dependent, but Smad4-independent, pathway (17). Indeed, constitutive TGF-β signaling led to an increase in iNKT cell frequency; however, the cells were arrested at an immature stage of development, suggesting that while TGF-β signaling promotes this innate-like T cell lineage, it must also be abrogated for proper iNKT cell maturation.
CD8αα+TCRαβ+ intraepithelial lymphocytes (IELs), which are important contributors to intestinal homeostasis, are thought to be generated by both thymic and extrathymic differentiation pathways (18). A recent study reported that enhancing autoreactive thymocyte survival by inhibiting clonal deletion led to increased Runx3-dependent CD8αα+TCRαβ+ IEL development (19), indicating that CD8αα+TCRαβ+ IEL selection is also driven by strong TCR interactions. A role for TGF-β in the development of this innate-like T cell lineage was suggested by TGF-β signaling gain and loss of function studies showing increased and decreased frequencies of CD8αα+TCRαβ+ IELs respectively (20). The loss of CD8αα+TCRαβ+ IELs in the absence of TGF-β signaling appeared to result in part from a reduction in thymic precursor cells, which were found to express increased amounts of the pro-apoptotic molecule Bim relative to their wild-type counterparts. Given the role of high affinity TCR interactions in tTreg, iNKT, and CD8αα+ T cell ontogeny, TGF-β-supported survival may be a unifying mechanism that promotes agonist ligand-induced T cell development (21). TGF-β has also been shown to induce CD8α expression on mature CD4+ T cells by regulating the transcription factors ThPOK and Runx3 (20, 22), but whether this pathway promotes Runx3 expression during thymic CD8αα+TCRαβ+ IEL development is unknown.
Naïve T Cell Homeostasis
An effective immune system must establish and maintain a diverse naïve T cell pool within the confines of a fairly constant number of peripheral T cells. Critical contributions of TGF-β in the maintenance of T cell homeostasis and repertoire diversity have recently been described.
Low Affinity CD4+ T Cells
The severe inflammatory disease that develops in polyclonal mice with T cell-specific TGF-βRII deficiency precludes the precise study of TGF-β control of naïve T cell homeostasis (4, 5). By contrast, TGF-βRII-deficient mice with a restricted T cell repertoire in which CD4+ T cells (ovalbumin-specific OT-II) have high affinity for a non-self peptide do not develop autoimmunity, allowing for the study of CD4+ T cell homeostasis without the complications of systemic inflammation. The absence of TGF-β signaling did not alter OT-II thymic selection or the naïve phenotype of peripheral T cells; however, peripheral T cell frequency was dramatically reduced (4, 8). Impaired homeostasis of TGF-βRII-deficient OTII T cells was attributed to loss of IL-7Rα expression and the consequent abrogation of IL-7 signaling. Thus, in addition to its role in CD8+ T cell lineage commitment, TGF-β mediated conditioning of thymocyte IL-7Rα expression critically promotes IL-7-dependent homeostasis of peripheral CD4+ T cells (8). Interestingly, TGF-β was found to be particularly important for the maintenance of low affinity CD4+ T cells. In the absence of TGF-β, IL-7Rα expression positively correlated with TCR affinity, as TGF-βRII-deficient T cells bearing higher affinity TCRs expressed increased amounts of IL-7Rα (and thus exhibited better homeostatic survival) than their lower affinity counterparts. Accordingly, a report that TCR diversity is altered only in the periphery, and not the thymus, of Tgfb1−/− mice likely reflects repertoire changes resulting from preferential loss of low affinity T cells (23). TGF-β maintenance of low affinity T cells may also be important for a novel regulatory population of “deletor” CD4+ T cells that restricts antigen-specific T cells by competing for endogenous self-peptides, including low affinity ligands that may mediate positive selection (24).
CD8+ T Cells
TGF-β critically regulates thymocyte IL-7Rα expression, and while loss of this conditioning causes impaired CD8-lineage commitment, it likely also has consequences for peripheral CD8+ T cells. Indeed, TGF-βRII-deficient HY CD8+ T cells exhibit dramatic defects in peripheral homeostasis (Ouyang W and Li MO, unpublished observations). Recent studies have also reported altered homeostasis and aberrant activation of TGF-β signaling defective OT-I T cells (25, 26). Consistent with the finding that TGF-β promotes IL-7Rα expression and IL-7 sensitivity (8), the increased homeostatic proliferation of TGF-β signaling defective OT-I T cells did not result from enhanced responsiveness to IL-7 (or IL-15). However, although homeostatic proliferation of OT-I T cells expressing the dominant negative TGF-βRII (DNRII) was freed from classical cytokine requirements, it remained dependent on TCR and MHC Class I interactions (25). This requirement for TCR engagement may explain the discrepancies in CD8+ T cell phenotypes from DNRII mice expressing distinct transgenic TCRs. In contrast to the phenotype of OT-I mice, TGF-β signaling-deficient mice with HY or 2C TCR restricted repertoires did not exhibit an increased frequency of effector or memory-like CD8+ T cells (27, 28). This difference likely results from the increased homeostatic proliferation capacity of OT-I T cells relative to 2C or HY T cells (OT-I>2C>HY), which is positively correlated with TCR affinity (29). Collectively, these studies suggest that TGF-β plays a dual role in T cell homeostasis by promoting IL-7 driven, but inhibiting TCR driven, expansion. By limiting the outgrowth of specific TCRs, TGF-β preserves the naïve state and repertoire diversity of the CD8+ T cell pool.
T Cell Tolerance
The immune system has multiple mechanisms in place to prevent autoimmunity. Central tolerance, whereby overtly self-reactive T cells are deleted in the thymus, is an essential, but incomplete part of this process (30). Thus peripheral tolerance mechanisms have evolved to keep autoreactive T cells in check (31). The lethal inflammatory disorders that develop in mice with global Tgfb1 deficiency or T cell-specific deletion of Tgfbr2 demonstrate the essential role of TGF-β in immune tolerance (2-5). Loss of tolerance in the absence of TGF-β is not solely attributable to defective Treg cell activity, as the provision of wild-type Treg cells (either by adoptive transfer or the generation of mixed bone marrow chimeras) failed to completely rescue the inflammatory disorder in TGF-βRII-deficient mice (4, 5). Indeed, TGF-β has been shown to control peripheral tolerance by both direct and indirect regulation of autoreactive T cells.
Direct Regulation of T Cells
During thymic development, highly self-reactive T cells undergo negative selection or are diverted to a regulatory lineage. However, these processes are not foolproof, and autoreactive T cells that have evaded central tolerance mechanisms exist in the peripheral T cell pool. Recent studies demonstrate that TGF-β regulation of these “escaped” autoreactive T cells is essential for the maintenance of immune tolerance. In mice with an OT-II T cell restricted repertoire that also express membrane-bound ovalbumin under the control of the rat insulin promoter (RIP-mOva), T cell-specific loss of TGF-β signaling results in enhanced antigen-induced negative selection of OT-II thymocytes (15). Nevertheless, TGF-βRII-deficient OT-II RIP-mOva mice possess peripheral OT-II T cells that eventually induce autoimmune diabetes. By contrast, wild-type OT-II RIP-mOva mice (which have a higher frequency of peripheral OT-II T cells than their TGF-βRII-deficient counterparts) do not develop diabetes, demonstrating that TGF-β critically regulates diabetogenic OT-II T cells. TGF-β appears to prevent disease by controlling effector T cell differentiation (Oh SA and Li MO, unpublished observations), which is consistent with recent findings in another transgenic diabetes model (BDC2.5 TCR mice). In BDC2.5 mice, Foxp3-Cre mediated deletion of Tgbr2 in Treg cells did not induce diabetes, but Ox40-Cre mediated deletion, which targets both Treg and activated CD4+ T cells, triggered disease, indicating that direct regulation of diabetogenic T cells by TGF-β is the dominant tolerance mechanism in this model (32). Additionally, in a transfer model of disease, TGF-βRII-deficient Treg cells could prevent diabetes, whereas TGF-βRII-deficient effector CD4+ T cells were refractory to suppression (32).
Despite the evidence that TGF-β directly regulates T cells, the exact mechanisms of action remain unclear. One mechanism of TGF-β-mediated tolerance that may be specific to mucosal sites was recently elucidated. In the intestine, TGF-β (with retinoic acid) has been shown to limit inflammation by inducing pathological CD4+ T cells to express CD8α in a manner that requires upregulation of Runx3 and downregulation of ThPOK (22). This redirection of CD4+ T cells to a non-pathogenic phenotype fails to occur in TGF-βRII-deficient T cells.
Although TGF-β is essential for T cell tolerance, it has recently been proposed that loss of TGF-β signaling alone is insufficient to induce autoimmunity. In striking contrast to mice in which Tgfbr2 deletion occurs during the double positive thymocyte stage, mice in which Tgfbr2 undergoes slow deletion (by the distal Lck-Cre, dLck-Cre) in peripheral T cells do not develop autoimmune disease (26). However, adoptively transferred dLck-Cre Tgfbr2fl/fl T cells could induce disease in Rag-deficient recipients, suggesting that an additional insult, such as lymphopenia, is required for the development of autoimmunity in the absence of TGF-β signaling. A noteworthy aspect of the dLck-Cre model is that tTreg and iNKT cells, which undergo agonist-ligand induced selection, are reported to express normal amounts of TGF-βRII. This raises the question of whether the dLck-Cre favors deletion in T cells bearing lower affinity TCRs, such that autoreactive CD4+ T cells possessing high affinity TCRs remain under the control of TGF-β signaling. It would be of interest to compare the expression profiles of TGF-βRII and Nur77GFP, which reflects TCR signal strength (33), to determine the pattern of dLck-Cre activity in T cells.
pTreg Cell Controlled Tolerance
Peripheral Treg cell induction is driven by a combination of suboptimal TCR signaling and other environmental stimuli, including TGF-β (10). TGF-β promotion of pTreg cell development can occur in part through the inhibitory effects of TGF-β signaling on TCR activation (34). The key role of TGF-β in inducing Foxp3 expression in pTreg cells was identified by studies showing that deleting the Foxp3 CNS1 region (which contains the conserved Smad3 binding sequence) or the Smad binding site alone results in a reduction of pTreg cells (13, 14). Interestingly, in addition to promoting pTreg cell development, TGF-β also induces T helper 17 (TH17) differentiation (covered later in this review). The disparate effects of TGF-β on pTreg versus TH17 cell fate reflect the context-dependent function of this pleiotropic cytokine. Indeed, other environmental cues, such as IL-2 and retinoic acid for pTreg cells and IL-6 for TH17 cells, influence the development of a specific lineage. Moreover, certain cell types can also favor pTreg cell differentiation; CD103+ dendritic cells from gut associated lymphoid tissues and mesenteric lymph nodes have been shown to preferentially induce pTreg cells in a TGF-β and retinoic acid dependent manner (35-37). On a molecular level, it has been shown that TGF-β can induce the same CD4+ T cells to express both Foxp3 and RORγt (the key TH17 transcription factor), but Foxp3-mediated inhibition of RORγt activity, which appears to result in part from physical interactions between the two transcription factors, can favor pTreg cell development (38).
In contrast to the insights into pTreg cell differentiation, the unique contributions of this population to immune tolerance have only recently been revealed. The Foxp3 CNS1 element was found to be highly conserved in placental mammals, and CNS1-deficient female mice bred to allogeneic males exhibited increased fetal resorption relative to wild-type females, indicating that pTreg cells are important regulators of maternal-fetal tolerance (39). Peripheral Treg cells were also shown to regulate tolerance at mucosal sites, as mice lacking CNS1 developed T helper 2 (TH2) driven autoimmunity in the gastrointestinal tract and lungs (40).
Effector T Cell Differentiation
TGF-β can broadly impede T cell activation by inhibiting TCR signaling (34), but also inhibits specific T helper subsets by suppressing lineage-defining transcription factors, such as T-bet and GATA-3, which are critical for T helper 1 (TH1) and TH2 CD4+ T cell differentiation respectively (41, 42). Indeed, TGF-β signaling-deficient CD4+ T cells produce both TH1 and TH2 cytokines (4, 5, 43). The molecular mechanisms by which TGF-β controls T cell differentiation remain poorly understood. However, recent work has elucidated the redundant roles of the transcription factors Smad 2 and Smad 3 in TGF-β inhibition of TH1 differentiation (44, 45). The Smad signaling pathway has also been shown to mediate TGF-β inhibition of TH2 differentiation by inducing the transcription factor Sox4, which interferes with GATA-3 activity (46). In contrast to its role in inhibiting effector T cell differentiation, TGF-β has been shown to promote the TH17 lineage. The following sections specifically focus on TGF-β regulation of TH17 cells and cytotoxic T cells.
TH17 Cell Differentiation
A role for TGF-β in the induction of TH17 cells was established through in vitro differentiation assays and in vivo observations that mice with defective TGF-β signaling possessed fewer IL-17-producing T cells, whereas overexpression of Tgfb1 led to an increase in TH17 cells (47-50). Mechanistically, TGF-β was suggested to promote the TH17 lineage by inhibiting the differentiation of other helper subsets, as stimulation with IL-6 alone was sufficient to induce IL-17 production in CD4+ T cells lacking essential TH1 and TH2 molecules (51). In support of this, TGF-β-mediated suppression of Gfi-1 and Eomesodermin (which promote TH2- and TH1-associated cytokines, respectively) was shown to enhance IL-17 production in CD4+ T cells (52, 53). Indeed, although TGF-β stimulation induces RORγt expression in CD4+ T cells, activation of STAT3 signaling promotes much stronger expression of this key TH17 transcription factor (54). Accordingly, in findings that paralleled studies of human TH17 differentiation, TGF-β was shown to be dispensable for murine TH17 differentiation, as stimulation with IL-23, IL-6, and IL-1β was sufficient to induce IL-17 production by CD4+ T cells (55, 56).
Interestingly, a series of studies examining the ability of in vitro differentiated TH17 cells to induce EAE suggests that TGF-β stimulation imprints a regulatory phenotype that renders the cells unable to cause disease. The initial study reported that myelin-reactive T cells isolated from myelin-immunized mice and expanded in the presence of IL-23, but not those cultured with TGF-β and IL-6, could induce EAE in a transfer model of disease (57). In this system, nonpathogenic, TGF-β induced TH17 cells were associated with high levels of IL-10 production. Another group reported similar findings that TH17 cells induced by IL-23, IL-6, and IL-1β, but not those generated by TGF-β and IL-6, could induce EAE in a transfer model of disease (55). In contrast to TGF-β and IL-6 induced TH17 cells, pathogenic TH17 cells were found to produce little IL-10 and express high levels of T-bet. Both studies used TGF-β1, the dominant isoform expressed in the immune system, to induce TH17 differentiation. Interestingly, a recent report suggests that an alternate TGF-β family member, TGF-β3, in conjunction with IL-6, promotes the in vitro differentiation of pathogenic, EAE-inducing TH17 cells by signaling through distinct Smads from TGF-β1 (58). Determining a definitive role for TGF-β3 in TH17 differentiation will require the study of T cell-specific Tgfb3 knockouts; notably, however, the pathogenicity of TGF-β3-derived TH17 cells also required IL-23 signaling, as IL23R-deficient cells did not induce disease. The reports that TGF-β1 inhibits the pathogenic activity of in vitro generated TH17 cells in a transfer model of EAE conflicts with studies showing that T cell-derived Tgfb1 is required for in vivo TH17 differentiation and disease development in a non-transfer model of EAE (59, 60). Whether these discrepancies result from differences between in vitro and in vivo TH17 differentiation, or between transfer and non-transfer models of EAE remains to be determined.
Cytotoxic T Cell Differentiation
Observations of diminished in vitro CD8+ T cell cytolytic activity in the presence of TGF-β provided early evidence for TGF-β-mediated inhibition of CD8+ T cell responses (61). The inhibitory effects of TGF-β on CD8+ T cell function were also indicated in vivo by studies showing that interfering with TGF-β signaling either by T cell-specific DNRII expression, or by administration of a soluble TGF-βRII, led to enhanced immune responses against transplantable tumors (62, 63). That TGF-β directly acts upon CD8+ T cells to control their differentiation was definitively established by the finding that TGF-βRII-deficient CD8+ T cells produce increased amounts of IFN-γ and cytolytic molecules (4, 5). Recent work in the TRAMP spontaneous prostate cancer model has provided additional evidence that TGF-β negatively regulates CD8+ T cell responses. TRAMP mice with DNRII-expressing CD8+ T cells exhibited tumor protection that correlated with decreased expression of the inhibitory marker PD-1 and increased expression of the cytolytic molecule Granzyme B on tumor-infiltrating CD8+ T cells (64). In a model of adoptive cell therapy in tumor-bearing TRAMP mice, TGF-βRII-deficient CD8+ T cells demonstrated increased tumor infiltration and enhanced effector properties relative to their wild-type counterparts (65). However, studies in chronic viral and acute bacterial infections indicate the context-dependent nature of TGF-β control of CD8+ T cell responses, as DNRII-expressing CD8+ T cells showed enhanced effector properties only during a chronic viral infection (66, 67). In both infections, however, effector DNRII-expressing CD8+ T cells underwent decreased apoptosis, suggesting that TGF-β may broadly promote effector CD8+ T cell death. It will be important to understand the factors that dictate disparate effects of TGF-β signaling on CD8+ T cell responses. Indeed, the complexity of TGF-β function has been emphasized by a recent study reporting that memory CD8+ T cells expressing the DNRII, but not those with Tgbr2 deletion, exhibit dysregulated homeostasis and undergo cellular transformation (68). This finding suggests that the dosage of TGF-β signaling may critically influence the resulting T cell phenotype.
TGF-β Production and Activation
TGF-β is synthesized in a latent form that must be activated to engage its receptors and initiate signaling. Distinct cellular sources can mediate the production and activation of TGF-β, providing another layer of complexity in the regulation and function of this pleiotropic cytokine.
T Cell-Derived TGF-β Controls T Cell Tolerance
Although many different cell types produce TGF-β, the discovery that mice with T cell-specific Tgfb1 deficiency develop autoimmunity and exhibit early mortality around six months of age demonstrated the critical contribution of T cell-derived TGF-β1 to immune tolerance (59). The tolerogenic function of T cell-produced TGF-β1 was corroborated by work showing that TRAMP mice with T cell-specific Tgfb1 deletion exhibited a tumor protection phenotype that correlated with increased CD8+ T cell effector function (64). It was subsequently shown that deletion of Tgfb1 in activated CD4+ T and Treg cells, but not in Treg or CD8+ T cells alone, was sufficient for protection from B16-OVA melanoma metastasis, which correlated with increased CD8+ T cell cytolytic activity (69). The identification of activated CD4+ T cells as the essential source of TGF-β in this model indicated that paracrine TGF-β signaling plays an important role in imprinting tolerogenic CD8+ T cell responses. Indeed, deletion of Tgfb1 in activated CD4+ T and Treg cells was also found to be sufficient for tumor protection in the TRAMP spontaneous prostate cancer model. Notably, tumor-produced TGF-β was intact in both B16-OVA melanoma and TRAMP models, underscoring the importance of T cell-derived TGF-β in the regulation of T cell tolerance.
Effector T Cell-Derived TGF-β Controls TH17 Differentiation
In addition to controlling T cell tolerance, T cell-derived TGF-β1 has been found to play an essential role during in vivo TH17 differentiation. This was demonstrated by work showing that mice with T cell-specific deletion of Tgfb1 harbored decreased frequencies of IL-17-producing T cells and exhibited protection from EAE (59). The CD4-Cre used in this system deletes Tgfb1 in conventional CD4+ and CD8+ T cells as well as Treg cells. Although Treg cells could provide sufficient amounts of TGF-β1 to induce in vitro TH17 differentiation (50), Treg cell-derived TGF-β1 was not required for in vivo TH17 differentiation, as TH17 cell frequencies and EAE pathogenesis were comparable in wild-type mice and mice with Foxp3-Cre mediated Tgfb1 deletion (60). By contrast, deletion of Tgfb1 using an Ox40-Cre that targets both Treg and activated CD4+ T cells led to a reduction in IL-17-producing CD4+ T cells and EAE protection, identifying the encephalitogenic T cells themselves as critical TGF-β1 sources. Notably, among the CD4+ T helper cell subsets, TH17 cells were found to express the highest amounts of TGF-β1. These findings suggested that autocrine TGF-β1 signaling is essential for in vivo TH17 differentiation. This was confirmed by mixed bone marrow chimera experiments showing that Tgfb1-deficient T cells failed to produce IL-17 even in the presence of wild-type cells.
Activation of TGF-β
The latent complex of TGF-β consists of mature TGF-β that is noncovalently associated with the latency-associated protein (LAP). This latent form of TGF-β can be secreted or may associate with latent-TGF-β-binding protein (LTBP), which mediates deposition of the complex to the extracellular matrix. More recent work has shown that latent TGF-β can also associate with glycoprotein-A repetitions predominant protein (GARP), resulting in cell surface expression of TGF-β on Treg cells and platelets (70). TGF-β can be activated from the membrane-bound complex of GARP and latent TGF-β, suggesting that GARP may function as an important regulator of TGF-β availability at cell surfaces (71). Release of TGF-β from its association with LAP is an essential step in TGF-β activation and function. Although multiple mechanisms, including thrombospondin and matrix metalloprotease activity, have been implicated in TGF-β activation, the major role of integrins in this process was demonstrated by studies showing that mice expressing a mutant Tgfb1 allele that abolishes integrin binding to LAP phenocopy Tgfb1-deficient mice (72). The particular importance of myeloid cell integrin activity in TGF-β function was demonstrated by the observations that specific loss of αv or β8 integrins in myeloid or dendritic cells was sufficient to induce murine colitis (73, 74). Myeloid cell integrin-mediated activation of TGF-β has also been shown to play a critical role during in vivo TH17 differentiation, as mice with myeloid cell-specific αv or β8 integrin deficiency exhibit a reduction in TH17 cells and are protected from EAE (75, 76). The promotion of TH17 differentiation was specific to myeloid cell integrin activity, as T cell-specific integrin deficiency affected neither TH17 cell frequency nor EAE development. Thus, although effector T cells are critical sources of TGF-β during in vivo TH17 differentiation, a distinct innate cell type is required for TGF-β activation (60, 75). Notably, the same myeloid cell must present antigen and express integrins, suggesting tight spatiotemporal regulation of TGF-β activation during T cell stimulation (75, 76). Determining the precise innate cell types that express the αvβ8 integrins, and how integrin expression is regulated during myeloid cell differentiation and activation will provide important insights into the complex biology of this cytokine.
Conclusions
Through its widespread regulation of T cells (Figure 1), TGF-β ensures a diverse, self-tolerant, and functional T cell repertoire. The complex cellular network of TGF-β production and activation, and the intricacy of TGF-β signaling pathways likely enable the pleotropic functions of this cytokine. Yet, how this complicated regulation results in TGF-β control of T cells is incompletely understood. On a molecular level, although recent work has demonstrated the redundant roles of Smad 2 and Smad 3 in T cell regulation (44, 45), fundamental questions of how the Smad-dependent transcriptional program controls T cells remain unanswered. On a cellular level, the milder autoimmune phenotypes of mice with T cell-specific Tgfb1 deficiency or dendritic cell-specific αv or β8 integrin deficiency versus mice with global defects in TGF-β production or activation indicate that other, unknown cell types are important for both of these processes. Although this review has focused on TGF-β control of T cells, the pervasive regulatory circuit of this cytokine affects other immune cells as well, and its regulation of non-T cell populations might also be critical for the prevention of autoimmunity. Expanding from a T cell-centric view of TGF-β regulation to include other cell types will provide increased insight into the mechanisms that govern TGF-β control of the immune system, and potential therapeutic applications.
Acknowledgments
Studies were supported by grants from the NIAMS (RO1 AR060723 to M.O.L.), the Rita Allen Foundation (M.O.L.), and NIH (T32-CA9149-35 to S.A.O.).
References
- 1.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 2.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 6.de Jong R, van Lier RA, Ruscetti FW, Schmitt C, Debre P, Mossalayi MD. Differential effect of transforming growth factor-beta 1 on the activation of human naive and memory CD4+ T lymphocytes. Int. Immunol. 1994;6:631–638. doi: 10.1093/intimm/6.4.631. [DOI] [PubMed] [Google Scholar]
- 7.Park JH, Adoro S, Guinter T, Erman B, Alag AS, Catalfamo M, Kimura MY, Cui Y, Lucas PJ, Gress RE, Kubo M, Hennighausen L, Feigenbaum L, Singer A. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 2010;11:257–264. doi: 10.1038/ni.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouyang W, Oh SA, Ma Q, Bivona MR, Zhu J, Li MO. TGF-beta Signaling Promotes CD8+ T Cell Development and Low-Affinity CD4+ T Cell Homeostasis by Regulation of Interleukin-7 Receptor Alpha Expression. Immunity. 2013 doi: 10.1016/j.immuni.2013.07.016. http://dx.doi.org/10.1016/j.immuni.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, Singer A. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21:289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 10.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol. 2008;9:632–640. doi: 10.1038/ni.1607. [DOI] [PubMed] [Google Scholar]
- 12.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- 13.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature. 2010;463:808–812. doi: 10.1038/nature08750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlenner SM, Weigmann B, Ruan Q, Chen Y, von Boehmer H. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J. Exp. Med. 2012;209:1529–1535. doi: 10.1084/jem.20112646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouyang W, Beckett O, Ma Q, Li MO. Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity. 2010;32:642–653. doi: 10.1016/j.immuni.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013;13:101–117. doi: 10.1038/nri3369. [DOI] [PubMed] [Google Scholar]
- 17.Doisne JM, Bartholin L, Yan KP, Garcia CN, Duarte N, Le Luduec JB, Vincent D, Cyprian F, Horvat B, Martel S, Rimokh R, Losson R, Benlagha K, Marie JC. iNKT cell development is orchestrated by different branches of TGF-beta signaling. J. Exp. Med. 2009;206:1365–1378. doi: 10.1084/jem.20090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2011;11:445–456. doi: 10.1038/nri3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pobezinsky LA, Angelov GS, Tai X, Jeurling S, Van Laethem F, Feigenbaum L, Park JH, Singer A. Clonal deletion and the fate of autoreactive thymocytes that survive negative selection. Nat. Immunol. 2012;13:569–578. doi: 10.1038/ni.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konkel JE, Maruyama T, Carpenter AC, Xiong Y, Zamarron BF, Hall BE, Kulkarni AB, Zhang P, Bosselut R, Chen W. Control of the development of CD8alphaalpha+ intestinal intraepithelial lymphocytes by TGF-beta. Nat. Immunol. 2011;12:312–319. doi: 10.1038/ni.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu. Rev. Immunol. 2012;30:95–114. doi: 10.1146/annurev-immunol-020711-075035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat. Immunol. 2013;14:271–280. doi: 10.1038/ni.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson RT, Gorham JD. TGF-beta 1 regulates antigen-specific CD4+ T cell responses in the periphery. J Immunol. 2007;179:71–79. doi: 10.4049/jimmunol.179.1.71. [DOI] [PubMed] [Google Scholar]
- 24.Singh NJ, Bando JK, Schwartz RH. Subsets of nonclonal neighboring CD4+ T cells specifically regulate the frequency of individual antigen-reactive T cells. Immunity. 2012;37:735–746. doi: 10.1016/j.immuni.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson LD, Jameson SC. TGF-beta sensitivity restrains CD8+ T cell homeostatic proliferation by enforcing sensitivity to IL-7 and IL-15. PLoS One. 2012;7:e42268. doi: 10.1371/journal.pone.0042268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang N, Bevan MJ. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012;13:667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucas PJ, Kim SJ, Melby SJ, Gress RE. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J. Exp. Med. 2000;191:1187–1196. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas PJ, Kim SJ, Mackall CL, Telford WG, Chu YW, Hakim FT, Gress RE. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-beta dominant-negative receptor transgenic mice. Blood. 2006;108:2789–2795. doi: 10.1182/blood-2006-05-025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J. Immunol. 2004;172:40–44. doi: 10.4049/jimmunol.172.1.40. [DOI] [PubMed] [Google Scholar]
- 30.Hogquist KA, Baldwin TA, Jameson SC. Central tolerance: learning self-control in the thymus. Nat. Rev. Immunol. 2005;5:772–782. doi: 10.1038/nri1707. [DOI] [PubMed] [Google Scholar]
- 31.Mueller DL. Mechanisms maintaining peripheral tolerance. Nat. Immunol. 2010;11:21–27. doi: 10.1038/ni.1817. [DOI] [PubMed] [Google Scholar]
- 32.Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ, Flavell RA. Excessive Th1 responses due to the absence of TGF-beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc. Natl. Acad. Sci. USA. 2013;110:6961–6966. doi: 10.1073/pnas.1304498110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CH, Seguin-Devaux C, Burke NA, Oriss TB, Watkins SC, Clipstone N, Ray A. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J. Exp. Med. 2003;197:1689–1699. doi: 10.1084/jem.20021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paidassi H, Acharya M, Zhang A, Mukhopadhyay S, Kwon M, Chow C, Stuart LM, Savill J, Lacy-Hulbert A. Preferential expression of integrin alphavbeta8 promotes generation of regulatory T cells by mouse CD103+ dendritic cells. Gastroenterology. 2011;141:1813–1820. doi: 10.1053/j.gastro.2011.06.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L, Lopes JE, Chong MM, Ivanov, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. 2012;150:29–38. doi: 10.1016/j.cell.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorelik L, Fields PE, Flavell RA. Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J. Immunol. 2000;165:4773–4777. doi: 10.4049/jimmunol.165.9.4773. [DOI] [PubMed] [Google Scholar]
- 42.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 44.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Saika S, Hara T, Nomura M, Yoshimura A. Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol. 2010;185:842–855. doi: 10.4049/jimmunol.0904100. [DOI] [PubMed] [Google Scholar]
- 45.Gu AD, Wang Y, Lin L, Zhang SS, Wan YY. Requirements of transcription factor Smad-dependent and -independent TGF-beta signaling to control discrete T-cell functions. Proc. Natl. Acad. Sci. USA. 2012;109:905–910. doi: 10.1073/pnas.1108352109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuwahara M, Yamashita M, Shinoda K, Tofukuji S, Onodera A, Shinnakasu R, Motohashi S, Hosokawa H, Tumes D, Iwamura C, Lefebvre V, Nakayama T. The transcription factor Sox4 is a downstream target of signaling by the cytokine TGF-beta and suppresses T(H)2 differentiation. Nat. Immunol. 2012;13:778–786. doi: 10.1038/ni.2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 48.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 49.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 50.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, Van Kaer L, Shi Y, Das G. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J. Exp. Med. 2009;206:2407–2416. doi: 10.1084/jem.20082286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, Guo L, Zhao K, Shevach EM, Paul WE. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009;206:329–341. doi: 10.1084/jem.20081666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, Morita R, Muto G, Shichita T, Takahashi R, Yoshimura A. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-beta is mediated by suppression of eomesodermin. Immunity. 2011;34:741–754. doi: 10.1016/j.immuni.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L, Ivanov, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 55.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romagnani S, Maggi E, Liotta F, Cosmi L, Annunziato F. Properties and origin of human Th17 cells. Mol. Immunol. 2009;47:3–7. doi: 10.1016/j.molimm.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 57.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 58.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 60.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rich S, Seelig M, Lee HM, Lin J. Transforming growth factor beta 1 costimulated growth and regulatory function of staphylococcal enterotoxin B-responsive CD8+ T cells. J. Immunol. 1995;155:609–618. [PubMed] [Google Scholar]
- 62.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 63.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 64.Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, Allison JP, Li MO. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity. 2011;35:123–134. doi: 10.1016/j.immuni.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chou CK, Schietinger A, Liggitt HD, Tan X, Funk S, Freeman GJ, Ratliff TL, Greenberg NM, Greenberg PD. Cell-intrinsic abrogation of TGF-beta signaling delays but does not prevent dysfunction of self/tumor-specific CD8 T cells in a murine model of autochthonous prostate cancer. J. Immunol. 2012;189:3936–3946. doi: 10.4049/jimmunol.1201415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity. 2009;31:131–144. doi: 10.1016/j.immuni.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–157. doi: 10.1016/j.immuni.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishigame H, Mosaheb MM, Sanjabi S, Flavell RA. Truncated form of TGF-betaRII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice. J. Immunol. 2013;190:6340–6350. doi: 10.4049/jimmunol.1300397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donkor MK, Sarkar A, Li MO. Tgf-beta1 produced by activated CD4(+) T Cells Antagonizes T Cell Surveillance of Tumor Development. Oncoimmunology. 2012;1:162–171. doi: 10.4161/onci.1.2.18481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFbeta. Mol. Biol. Cell. 2012;23:1129–1139. doi: 10.1091/mbc.E11-12-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, Munger JS. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J. Cell. Biol. 2007;176:787–793. doi: 10.1083/jcb.200611044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, Bluestone JA, Sheppard D. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, Roes JT, Savill JS, Hynes RO. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc. Natl. Acad. Sci. USA. 2007;104:15823–15828. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acharya M, Mukhopadhyay S, Paidassi H, Jamil T, Chow C, Kissler S, Stuart LM, Hynes RO, Lacy-Hulbert A. alphav Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 2010;120:4445–4452. doi: 10.1172/JCI43796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Melton AC, Bailey-Bucktrout SL, Travis MA, Fife BT, Bluestone JA, Sheppard D. Expression of alphavbeta8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 2010;120:4436–4444. doi: 10.1172/JCI43786. [DOI] [PMC free article] [PubMed] [Google Scholar]