

Abstract
Background
Little is known about the Phasmatodea gut microbial community, including whether phasmids have symbiotic bacteria aiding in their digestion. While symbionts are near ubiquitous in herbivorous insects, the Phasmatodea’s distinctively thin body shape precludes the gut enlargements needed for microbial fermentation. High-throughput sequencing was used to characterize the entire microbiota of the fat bodies, salivary glands, and anterior and posterior midguts of two species of walking stick.
Results
Most bacterial sequences belonged to a strain of Spiroplasma (Tenericutes) found primarily in the posterior midgut of the parthenogenetic species Ramulus artemis (Phasmatidae). Beyond this, no significant differences were found between the R. artemis midgut sections or between that species and Peruphasma schultei (Pseudophasmatidae). Histological analysis further indicated a lack of bacteriocytes.
Conclusions
Phasmids are unlikely to depend on bacteria for digestion, suggesting they produce enzymes endogenously that most other herbivorous insects obtain from symbionts. This conclusion matches predictions based on phasmid anatomy. The role of Spiroplasma in insects warrants further study.
Keywords: Phasmatodea, Microbiota, 16S rDNA, Symbionts, Digestive system
Background
Research on insect endosymbionts has historically focused on insects with limited diets, such as wood-feeders or the nitrogen-limited phloem feeders [1,2]. This body of work has revealed several obligate symbioses, such as the aphid-Buchnera system where the insect cannot survive without its bacterial mutualist [3], as well as the many termite symbioses with gut bacteria and/or flagellates that assist in lignocellulose digestion [4]. Many such insects have specialized cells called bacteriocytes in which the microbes are housed [5], while in others the symbionts persist within the midgut lining [2]. As most microbes, including the majority of endocellular symbionts, cannot be cultured [6], high-throughput sequencing has been used to characterize insect microbiota and identify possible symbionts [7], with successes in groups such as honey bees [8] and moths [9]. Gut microbes and their enzymes have received much recent attention by the biofuel industry’s search for novel cellulases and xylanases, which has biased the gut microbe literature towards xylophages like termites, roaches, and beetles [10,11].
Less understood are the digestive mechanisms and gut microbes of leaf eaters such as Lepidoptera larvae and orthopteroids. While leaves are not as difficult a diet as dry wood, they still contain abundant cellulase as plant cell walls [12], as well as toxic secondary chemicals, waxes, trichomes, and other obstacles to consumption and digestion by insects [13]. Symbiotic microbes would be beneficial to leaf eaters by assisting in cellulose breakdown [14], nitrogen fixation or amino-acid metabolism [15], detoxifying or neutralizing plant defensive compounds [16], and recycling nitrogenous wastes [17].
While some microbial work has been performed on Orthoptera sensu stricto[18-20], and a few papers have looked at specific microbes within phasmids [21,22], no complete microbial inventory of the Phasmatodea gut has ever been performed. As obligate leaf eaters (as opposed to certain periodically cannibalistic orthopteroids), the phasmids would certainly stand to benefit from having microbial symbionts. However, several factors suggest that phasmids would not have digestive symbionts. Their characteristic body shape places restrictions on their gut morphology, which is straight and narrow tubes (Figure 1) with very short gastric caecae and no obvious diverticulae or fermentation chambers [23,24] of the kind that house microbes in other insects [1]. The only likely places for a phasmid symbiont to exist would be the midgut (the main site of phasmid digestion), the salivary glands, and the fat bodies. One paper studying insect fibre digestion suggested phasmids do not rely on microbes for digestion, but it based this on counts of culturable microbes, meaning up to 99% of the microbe diversity in the gut may have been ignored [21]. In our paper, microscopy and next generation sequencing was used to catalog the entire microbiota of the aforementioned phasmid organs from two species, with the goal of finding potential symbionts and possibly elucidating their functions, as well as increasing our knowledge of a poorly described alimentary canal.
Figure 1.
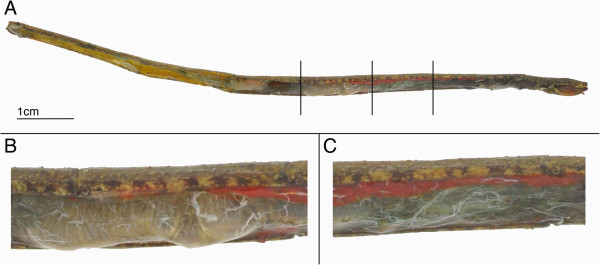
The digestive tract within a partially-dissected adult female Ramulus artemis. (A) Whole gut in body cavity, minus head. Inserts are close-ups of the (B) anterior and (C) posterior midguts.
Methods
The phasmids used were lab-reared specimens of Ramulus artemis (Westwood) (Phasmatidae), fed rose (Rosa) leaves, and Peruphasma schultei (Conle & Hennemann) (Pseudophasmatidae), fed privet (Ligustrum). All insects were maintained and used as per the University of California, Davis’ Institutional Animal Care and Use Committee guidelines.
To check for the presence of bacteriocytes, histological analysis was performed [25]. Whole insects with longitudinal slits across the body wall were fixed in Bouin’s Fluid for three days and stored in 70% ethanol. Tissue samples were dissected out, dehydrated in an ethanol-butanol series, and embedded in paraffin. Sections were slide mounted using Meyer’s albumin and stained in Giemsa or Heidenhaim’s haematoxylin and eosin for light microscopy [26,27].
For molecular analysis, adult females were starved for two days to clear their guts and reduce chloroplast contamination of the samples. The digestive tract and its contents were dissected out, and the midguts divided into two sections reflecting the phasmid’s unique gut anatomy (Figure 1): the heavily pleated anterior midgut (AMG) and the unpleated posterior midgut (PMG) studded with tubules of a currently unknown function [23,28]. The gut sections as well as the fat bodies and salivary glands were preserved in 100% ethanol prior to DNA extraction.
The total DNA from each sample was extracted using the Wizard® Genomic DNA Purification Kit (Promega; Fitchburg, Wisconsin, USA) following the manufacturer’s protocol. To identify gut microbes, PCR was done to amplify the 16S ribosomal DNA using the universal primers 27F (AGAGTTTGATCMTGGCTCAG) and 511R (GCGGCTGCTGGCACRKAGT) with the appropriate 454 Life Sciences adaptor sequence. In addition, the forward primer used for each sample contained a unique 6-bp barcode for multiplexed sequencing. The barcodes used are described in NCBI BioSamples SAMN02318746-SAMN02318757. To minimize biases that might occur in individual PCR reactions, three independent reactions were performed for each sample and the products were pooled before sequencing. Each of the 50 μL of PCR mixture consisted of 1 μL PfuUltra II Fusion HS DNA polymerase (Stratagene; La Jolla, California, USA), 5 μL of supplied 10× buffer, 2.5 μL of 5 mM dNTP mix (MBI Fermentas; Burlington, Ontario, Canada), 0.5 μL of 10 mg/mL BSA (New England Biolabs; Ipswich, Massachusetts, USA), 1 μL of each 10 μM primer, and 50 ng of template DNA. The PCR program included one denaturing step at 95°C for 3 min, 25 cycles of 95°C for 40 sec, 55°C for 40 sec, and 72°C for 40 sec, followed by a final extension at 72°C for 7 min. Gel electrophoresis was used to check the existence of a single band of expected size for each PCR product. For the positive samples, PCR products were purified with the QIAquick PCR Purification Kit (Qiagen; Venlo, Netherlands). To further confirm the successful amplification of bacterial 16S rDNA in our broad range PCR, the purified PCR products were cloned using the CloneJet PCR Cloning Kit (Fermentas Life Science; Burlington, Ontario, Canada) and transformed into HIT-JM 109 competent cells (RBC Bioscience; Zhonghe City, Taipei County, Taiwan). A limited number of clones were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI Prism 3700 Genetic Analyzer (Applied Biosystems; Foster City, California, USA) to verify the presence of expected 16S rDNA fragment, multiplexing barcodes, and the adapters for 454 sequencing.
For high-throughput DNA sequencing, the positive samples were pooled in equal proportions and sequenced on a 454 Jr. sequencer (454 Life Sciences; Branford, Connecticut, USA). The pyrosequencing flowgrams were converted to sequence reads with corresponding quality scores using the standard software provided by 454 Life Sciences. All raw reads were deposited in NCBI Sequence Read Archive (accession number SRR955712). The sequences were quality-trimmed using the default settings of LUCY [29]. Reads that were shorter than 400-bp after the quality trimming were removed from the data set. After the quality trimming, the sample-specific barcode and the primer sequence were identified and trimmed from each sequence; sequences that lacked a recognizable barcode and PCR primer were discarded. To identify the operational taxonomic units (OTUs) in these samples, the partial 16S rDNA sequences were hierarchically clustered at 100%, 99%, and 97% sequence identity using USEARCH version 5.2.32 [30]. The 97% sequence identity threshold was chosen because it is commonly used to define bacterial species [31,32]. For taxonomic assignment, the representative sequence of each OTU was used as the query for the CLASSIFIER [33] program provided by the Ribosomal Database Project [34] with the 16S rRNA training set (version 2.5). The OTUs that were identified as originating from plant chloroplasts or mitochondria were excluded from downstream analyses. Furthermore, OTUs that could not be assigned to a particular genus with at least 70% confidence level were removed because these sequences are likely to represent chimeras or other artifacts introduced during the PCR or pyrosequencing process [35]. For verification of the CLASSIFIER results and taxonomic assignment at species level, BLASTN [36,37] similarity search against the NCBI nt database [38] was performed for the representative sequence of each OTU. We limited the BLASTN search to the subject sequences from Bacteria (‘taxid2’). Additionally, subject sequences from environmental samples or metagenomes were excluded because these sequences often do not contain reliable taxonomic assignments. The e-value cutoff and other parameters of the BLASTN search were set to default; the top one hit was used as the representative for each query.
To compare the microbiota composition among individual samples, we utilized the software package Fast UniFrac [39] to perform Principal Coordinate Analysis (PCoA) and hierarchical clustering. The OTUs were weighted by abundance and the branch lengths were normalized. To generate a reference tree for the Fast UniFrac analyses, the representative sequences from all OTUs were aligned using the RDP ALIGNER [34]. The resulting multiple sequence alignment was examined to ensure that the 5’-end of each sequence was mapping to the expected location of 16S rDNA. The program FastTree [40] was then used to infer a maximum likelihood phylogeny of the OTUs. Additionally, the PVCLUST package [41] for R Statistical Software [42] was used to perform an alternative hierarchical clustering analysis with a phylogeny-independent approach.
Results
The broad range PCR amplification results for bacterial 16S rDNA were negative for the fat body and salivary gland samples examined, so they were excluded after the initial quality check steps. This negative result is consistent with phasmid histological studies, which did not show obvious mycetocytes in fat body or salivary gland tissue, nor any obvious endosymbionts in the midgut tissues (Figure 2). All seven P. schultei PMG samples tested negative for bacterial DNA as well. In total, bacterial 16S rDNA was recovered from four P. schultei AMGs, four R. artemis AMGs, and three R. artemis PMGs, and these samples were included in the 454 sequencing and final analysis. In total, we obtained 26,006 high quality sequence reads for these 11 midgut samples.
Figure 2.

Histological sections from a late instar male Peruphasma schultei, stained in Giemsa solution. Scale bars for all are 1 mm in length. The images shown here are exemplary of all observed sections. (A) Fat body. (B) Salivary Glands. (C) Anterior midgut, with notable pleating. (D) Posterior midgut, unpleated and studded with ampullae-tubules of unknown function (labeled with arrows).
Among the midgut samples, an overwhelming majority of the sequencing reads originated from the 16S rDNA of plant chloroplasts or mitochondria (Table 1). After removing these contaminants, we obtained a total of 162 bacterial OTUs covering 64 genera (Table 2). Rarefaction curves (Figure 3) suggest that the sampling depths achieved in this study may not be sufficient to fully characterize the microbiota of Phasmatodea guts. With the exception of a large Tenericute population in the R. artemis PMGs, most of the microbes isolated were Proteobacteria, followed by Actinobacteria and Firmicutes (Table 1 and Figure 4). Nearly all of the Tenericute reads (4,902 out of 5,412 Tenericute sequences) belonged to a single OTU (OTU_ID: CXTIA; Additional file 1) found mostly in the R. artemis posterior midgut. The BLASTN search against the NCBI nucleotide database showed that this sequence is 99.0% identical to Spiroplasma sp. ‘Gent’ (GenBank AY569829), an uncultured, male-killing entomopathogen identified from the housefly Fannia manicata. The next most common OTU had only 161 reads, and all other OTUs had less than 50 reads (Additional file 1). All but one of the 10 most abundant OTUs appeared to be Spiroplasma, which are known to be associated with a wide range of insects [43,44]. The most abundant non-Spiroplasma OTU was represented by 31 reads and showed high similarity (99.3% sequence identity) to Sphingobium sp. KR5 (GenBank JQ433940).
Table 1.
Summary of reads received from phasmid samples
| P. schultei anterior midgut | R. artemis anterior midgut | R. artemis posterior midgut | Total | |
|---|---|---|---|---|
| # of positive samples |
4 |
4 |
3 |
|
|
All sequences: | ||||
| # reads passed quality control |
9,255 |
9,697 |
7,054 |
26,006 |
| Average # reads per sample |
2,313.75 |
2,424.25 |
2,351.33 |
|
| # 100% id OTUs |
|
|
|
8,090 |
| # 99% id OTUs |
|
|
|
2,702 |
| # 97% id OTUs |
|
|
|
844 |
|
After RDP CLASSIFIER: | ||||
| # chloroplast 16S |
|
|
|
14,541 |
| # mitochondrial 16S and other contaminants |
|
|
|
5,786 |
| # bacterial 16S reads |
112 |
171 |
5,396 |
5,679 |
| Average # reads per sample |
28 |
43 |
1,799 |
|
| # reads (# OTUs) by phylum | ||||
| Actinobacteria |
3(2) |
13(9) |
13(4) |
29(13) |
| Armatimonadetes |
0 |
1(1) |
0 |
1(1) |
| Bacteroidetes |
1(1) |
5(3) |
0 |
6(4) |
| Firmicutes |
3(3) |
16(9) |
5(4) |
24(15) |
| Lentisphaerae |
3(1) |
0 |
0 |
3(1) |
| Proteobacteria |
87(37) |
65(39) |
45(26) |
197(75) |
| Tenericutes |
15(6) |
65(10) |
5,332(47) |
5,412(47) |
| TM7 | 0 | 6(5) | 1(1) | 7(6) |
Phyla Based on RDP CLASSIFIER results.
Table 2.
The number of reads for each microbe genus per gut type (pooled samples)
| Phylum | Genus | Ps AMG | Ra AMG | Ra PMG | Total |
|---|---|---|---|---|---|
| Actinobacteria |
Arthrobacter |
|
3 |
|
3 |
| Actinobacteria |
Brevibacterium |
1 |
1 |
2 |
4 |
| Actinobacteria |
Conexibacter |
|
2 |
|
2 |
| Actinobacteria |
Janibacter |
|
2 |
|
2 |
| Actinobacteria |
Propionibacterium |
2 |
3 |
11 |
16 |
| Actinobacteria |
Sanguibacter |
|
1 |
|
1 |
| Actinobacteria |
Streptomyces |
|
1 |
|
1 |
| Armatimonadetes |
Armatimonadetes_gp5 |
|
1 |
|
1 |
| Bacteroidetes |
Bacteroides |
1 |
|
|
1 |
| Bacteroidetes |
Cloacibacterium |
|
4 |
|
4 |
| Bacteroidetes |
Flavobacterium |
|
1 |
|
1 |
| Firmicutes |
Bacillus |
|
3 |
1 |
4 |
| Firmicutes |
Clostridium sensu stricto |
|
|
1 |
1 |
| Firmicutes |
Clostridium XI |
1 |
|
|
1 |
| Firmicutes |
Exiguobacterium |
|
1 |
|
1 |
| Firmicutes |
Lactococcus |
|
1 |
|
1 |
| Firmicutes |
Pelospora |
|
2 |
|
2 |
| Firmicutes |
Planifilum |
|
|
2 |
2 |
| Firmicutes |
Planococcaceae_incertae_sedis |
|
|
1 |
1 |
| Firmicutes |
Sedimentibacter |
1 |
|
|
1 |
| Firmicutes |
Staphylococcus |
1 |
6 |
|
7 |
| Firmicutes |
Streptococcus |
|
2 |
|
2 |
| Firmicutes |
Weissella |
|
1 |
|
1 |
| Lentisphaerae |
Victivallis |
3 |
|
|
3 |
| Proteobacteria |
Acidovorax |
4 |
4 |
4 |
12 |
| Proteobacteria |
Acinetobacter |
7 |
2 |
4 |
13 |
| Proteobacteria |
Alcanivorax |
|
|
1 |
1 |
| Proteobacteria |
Aquabacterium |
10 |
4 |
6 |
20 |
| Proteobacteria |
Arcobacter |
10 |
4 |
3 |
17 |
| Proteobacteria |
Azospira |
|
|
1 |
1 |
| Proteobacteria |
Belnapia |
|
1 |
|
1 |
| Proteobacteria |
Bordetella |
|
1 |
|
1 |
| Proteobacteria |
Brevundimonas |
2 |
1 |
|
3 |
| Proteobacteria |
Burkholderia |
|
3 |
|
3 |
| Proteobacteria |
Caulobacter |
1 |
3 |
|
4 |
| Proteobacteria |
Comamonas |
1 |
1 |
1 |
3 |
| Proteobacteria |
Cupriavidus |
2 |
|
2 |
4 |
| Proteobacteria |
Enhydrobacter |
2 |
|
|
2 |
| Proteobacteria |
Escherichia/Shigella |
1 |
|
1 |
2 |
| Proteobacteria |
Herbaspirillum |
|
2 |
1 |
3 |
| Proteobacteria |
Hyphomicrobium |
1 |
|
|
1 |
| Proteobacteria |
Legionella |
3 |
|
|
3 |
| Proteobacteria |
Methylobacterium |
2 |
2 |
|
4 |
| Proteobacteria |
Methyloversatilis |
|
2 |
1 |
3 |
| Proteobacteria |
Novosphingobium |
5 |
2 |
|
7 |
| Proteobacteria |
Ochrobactrum |
1 |
2 |
|
3 |
| Proteobacteria |
Pantoea |
|
3 |
|
3 |
| Proteobacteria |
Paracoccus |
|
2 |
|
2 |
| Proteobacteria |
Pelomonas |
1 |
|
|
1 |
| Proteobacteria |
Pseudomonas |
4 |
8 |
4 |
16 |
| Proteobacteria |
Raoultella |
|
|
1 |
1 |
| Proteobacteria |
Rheinheimera |
|
|
1 |
1 |
| Proteobacteria |
Rhizobacter |
|
4 |
|
4 |
| Proteobacteria |
Shewanella |
|
1 |
|
1 |
| Proteobacteria |
Simplicispira |
|
4 |
|
4 |
| Proteobacteria |
Smithella |
1 |
1 |
|
2 |
| Proteobacteria |
Sphingobium |
21 |
6 |
9 |
36 |
| Proteobacteria |
Sphingomonas |
4 |
1 |
2 |
7 |
| Proteobacteria |
Stenotrophomonas |
|
1 |
|
1 |
| Proteobacteria |
Undibacterium |
1 |
|
2 |
3 |
| Proteobacteria |
Zoogloea |
3 |
|
1 |
4 |
| Tenericutes |
Haloplasma |
|
|
1 |
1 |
| Tenericutes |
Spiroplasma |
15 |
65 |
5,331 |
5,411 |
| TM7 |
TM7_genera_incertae_sedis |
|
6 |
1 |
7 |
| Sum | 112 | 171 | 5,396 | 5,679 |
The taxomic assignment at genus level was based on RDP CLASSIFIER. Ps = P. schultei. Ra = R. artemis.
Figure 3.
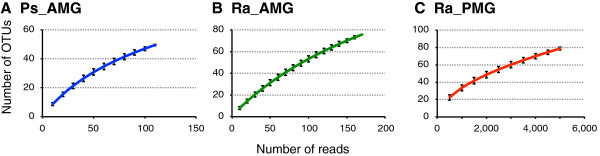
Rarefaction curves of the three sample types. The individual samples of the same type were pooled and resampled 10,000 times to determine the number of OTUs found with different numbers of sequencing reads. The error bars indicate ± one standard deviation. (A)P. schultei anterior midgut (Ps_AMG) samples. (B)R. artemis anterior midgut (Ra_AMG) samples. (C)R. artemis posterior midgut (Ra_PMG) samples.
Figure 4.

Relative abundances of reads of the different phyla. (A) All 5,679 bacterial 16S rDNA reads. (B) Excluding Spiroplasma reads.
Comparison of the microbiota composition based on the PCoA plots (Figure 5) indicated that the relative abundance of Spiroplasma reads is the major determinant that differentiates different sample types. When all 5,679 bacterial reads are considered, the three R. artemis PMG samples form a tight cluster and the PCO1 explains 81.77% of the variance (Figure 5A). Although one P. schultei AMG sample (from individual #5) appears to share a similar microbiota composition with the three R. artemis PMG samples, this P. schultei AMG sample should be considered as an outlier because it contains only one reads assigned to the most abundant Spiroplasma OTU. When the putative Spiroplasma reads were excluded, no clear pattern exists to distinguish among sample types (Figure 5B).
Figure 5.
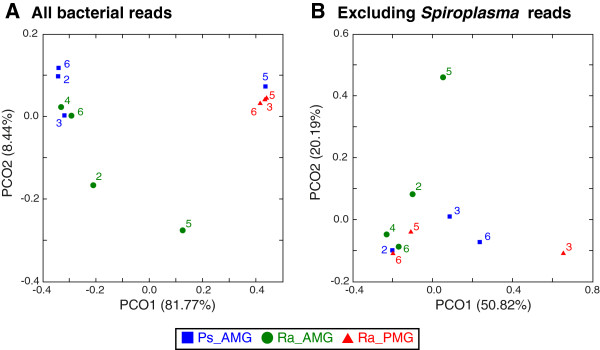
Principal Coordinate Analyses (PCoA) of the microbiota. The samples are color-coded by type: blue squares, P. schultei anterior midgut (Ps_AMG) samples; green circles, R. artemis anterior midgut (Ra_AMG) samples; red triangles, R. artemis posterior midgut (Ra_PMG) samples. The label indicates the individual id within each species. The numbers in parentheses in axis labels indicate the percentage of variance explained. (A) All 5,679 bacterial 16S rDNA reads. (B) Excluding Spiroplasma reads.
The hierarchical clustering analyses based on either phylogeny-dependent approach (Figure 6 panels A and B) or phylogeny-independent approach (Figure 6 panels C and D) produced the same patterns inferred from the PCoA plots. When all bacterial reads are considered (Figure 6 panels A and C), the three R. artemis PMG samples (together with the Ps5_AMG sample that contains only one Spiroplasma read) form a single clade with short distances among each other and high divergences with other samples. When the Spiroplasma reads were excluded (Figure 6 panels B and D), individual samples of the same type do not cluster together. Additionally, the branches that separate samples of the same type are similar in length compared to those separating samples of different types.
Figure 6.

Hierarchical clustering analysis. Labels for sample ids: Ps#_AMG = P. schultei anterior midgut, Ra#_AMG = R. artemis anterior midgut, Ra#_PMG = R. artemis posterior midgut; the number indicates the individual id within each species (e.g., Ra6_AMG and Ra6_PMG represent the anterior midgut and the posterior midgut of individual #6 from R. artemis). Panels (A) and (B): Phylogeny-dependent clustering based on Fast UniFrac; Panels (C) and (D): Phylogeny-independent clustering based on PVCLUST.
Discussion
The overabundance of chloroplast contamination greatly reduced the amount of bacterial reads available for analysis. Starving the phasmids for two days was insufficient to clear their digestive tracts. For future study, a longer time period of starvation (e.g., five to ten days) would be preferable. While 16S primers that select against chloroplasts do exist, they also bias the results of broad range PCR and are not suitable replacements for “universal” primers when studying total microbial community. Despite the relatively low abundance of bacterial reads, the use of next generation sequencing techniques still provided thousands of usable reads of mostly uncultivated microbes. This data set provides at least one order of magnitude more reads than would be available using clone libraries or denaturing gradient gel electrophoresis.
The low number of bacterial reads compared to chloroplast reads suggests the abundance of bacterial cells in the phasmid gut is relatively low. These results, together with the lack of bacterial DNA in the fat body and salivary glands and the lack of bacteriocytes in the tissue slices, do not support the hypothesis that phasmids have obligate microbial symbioses. Had such mutualist endosymbionts existed in these samples, they would have been detected at similar levels to the Spiroplasma infestation. While lab-reared phasmids will likely have different gut microbiota from wild specimens, they are expected to retain any symbiotic microbes essential for survival. Symbiont absence in the lab suggests absence in the field as well, but this remains to be demonstrated. The data suggests that most of the microbes isolated were environmental, picked up by the phasmids from their diet or housing [18,45].
The lack of beneficial symbiotic microbes in phasmids is unsurprising given the aforementioned restrictions on their digestive system due to their distinct body shape. The phasmid gut lacks the space to develop the significantly modified sections seen in symbiont-housing organisms, such as the enlarged hindguts of termites. Given these restrictions, one would expect a phasmid to compensate for lack of symbionts either with modified feeding behavior (such as increased consumption and/or slower gut transit time) or through endogenous production of digestive enzymes such as cellulases, which research predicts the phasmids can produce [12,21,46]. No research on phasmid enzymes has been published yet, however. If phasmids did depend on gut bacteria to breakdown their leafy diet, one would expect a gut microbiota consisting predominantly of Bacteroidetes and Firmicutes, as one sees in the roaches and termites [2]. The abundance of Proteobacteria resembles the mostly transient microbes found in Orthoptera [18], which supports the closer phylogenetic relationship between that order and the Phasmatodea [47].
The nature of R. artemis’ Spiroplasma infestation needs investigation. Of the 162 OTUs, 32 were closely related to Spiroplasma sp. ‘Gent’ (GenBank AY569829), and another eight were closely related to Spiroplasma ixodetes (GenBank GU585671). The infection’s localization to the PMG suggests the bacteria may be colonizing the PMG’s enigmatic appendices, but histological data did not confirm this (Figure 2D). Spiroplasma infestation may also explain why R. artemis can be parthenogenetic in cultures, as some Spiroplasma species are known male-killing parasites [48] similar to Wolbachia[49]. Several other phasmid species, most notably the laboratory stick insect, Carausius morosus, are also parthenogenetic [50], so checking these cultures for Spiroplasma infestations may be revealing. DiBlasi et al. [22] concluded that the Spiroplasma is transmitted in phasmids by mites, yet the cultures used here were mite-free, suggesting vector-less transmission.
The most common non-Spiroplasma microbe isolated was identified as a member of Sphingobium, which may be involved in the degradation of aromatic hydrocarbons [51]. It may perform this role for the phasmid, aiding in digestion by degrading plant defensive compounds, but whether it is a true symbiont transmitted between individuals, an environmental microbe that can colonize the gut, or an allochthonous (transient) microbe found on the leaves and just passing through is unknown.
Conclusions
This paper marks the first attempt to catalog the microbial diversity of the Phasmatodea (Additional file 1: Table S1). While a clade of symbiont-dependent phasmids may exist, the likelihood is low. All evidence suggests the only heritable symbionts, allegedly ubiquitous in the Insecta [6], in the Phasmatodea are reproduction-manipulators like Spiroplasma. Still, the phasmid gut microbial community is diverse and merits further investigation. The possibility of fungal or other eukaryotic symbionts in the gut remains. The microbial community may function together to benefit the phasmid in ways akin to their functions on leaf surfaces, such as secondary chemical detoxification [52]. Lastly, if phasmid digestion is truly microbe-independent, then the enzymology of the gut demands further analysis given the potential for finding novel lignocellulases and other compounds of possible human industrial applications.
Availability of supporting data
A supplementary table (Table S1) the lists the taxonomic assignment OTUs is included as Additional file 1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MS and CHK conceived of the study. MS and WSL carried out the experiments. MS and CHK performed the data analysis. MS, LSK, and CHK wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Taxonomic assignment of the OTUs based on RDP CLASSIFIER and BLASTN sequence similarity search against the NCBI nt database.
Contributor Information
Matan Shelomi, Email: mshelomi@ucdavis.edu.
Wen-Sui Lo, Email: d101041011@mail.nchu.edu.tw.
Lynn S Kimsey, Email: lskimsey@ucdavis.edu.
Chih-Horng Kuo, Email: chk@gate.sinica.edu.tw.
Acknowledgements
We thank the curation staff at the Bohart Museum of Entomology insect culture care, Dr. Robert Kimsey for histology advising, and the staff at Academia Sinica for assistance in DNA sequencing (Ms. Mei-Jane Fang at the Institute of Plant and Microbial Biology for Sanger sequencing and Dr. Shu-Yun Tung at the Institute of Molecular Biology for 454 pyrosequencing). MS was supported by the USA National Science Foundation’s East Asia and Pacific Summer Institutes (EAPSI) program (NSF proposal number 1209449) and the co-sponsorship by the Taiwanese National Science Council’s Summer Institutes in Taiwan (SIT) program. Funding for this work was provided by research grants from Academia Sinica to CHK, and the University of California Davis and Humanities Graduate Research Fellowship.
References
- Buchner P. Endosymbiosis of Animals with Plant Microorganisms. New York: Interscience Publishers; 1965. [Google Scholar]
- Ishikawa H. In: Insect Symbiosis. Bourtzis K, Miller TA, editor. Boca Raton: CRC Press; 2003. Insect symbiosis: an introduction; pp. 1–21. [Google Scholar]
- Douglas AE. In: Insect Symbiosis. Bourtzis K, Miller TA, editor. Boca Raton: CRC Press; 2003. Buchnera bacteria and other symbionts of aphids; pp. 23–38. [Google Scholar]
- Lo N, Eggleton P. In: Biology of Termites: A Modern Synthesis. Bignell DE, Roisin Y, Lo N, editor. London: Springer; 2011. Termite phylogenetics and co-cladogenesis with symbionts; pp. 27–50. [Google Scholar]
- Douglas AE. Lessons from studying insect symbioses. Cell Host Microbe. 2011;10:359–367. doi: 10.1016/j.chom.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Dharne M, Patole M, Shouche YS. Microbiology of the insect gut: tales from mosquitoes and bees. J Biosci. 2006;31:293–295. doi: 10.1007/BF02704100. [DOI] [PubMed] [Google Scholar]
- Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One. 2012;7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda E, Pedrola L, Pereto J, Martinez-Blanch JF, Montagud A, Navarro E, Urchueguia J, Ramon D, Moya A, Porcar M. Microbial diversity in the midguts of field and lab-reared populations of the European corn borer Ostrinia nubilalis. PLoS One. 2011;6:e21751. doi: 10.1371/journal.pone.0021751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf ME, Tartar A. Termite digestomes as sources for novel lignocellulases. Biofuels, Bioprod Biorefin. 2008;2:540–552. doi: 10.1002/bbb.107. [DOI] [Google Scholar]
- Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernández M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Tokuda G. Cellulolytic systems in insects. Annu Rev Entomol. 2010;55:609–632. doi: 10.1146/annurev-ento-112408-085319. [DOI] [PubMed] [Google Scholar]
- Shelomi M, Perkins LE, Cribb BW, Zalucki MP. Effects of leaf surfaces on first instar Helicoverpa armigera (Hübner)(Lepidoptera: Noctuidae) behaviour. Australian J Entomol. 2010;49:289–295. doi: 10.1111/j.1440-6055.2010.00766.x. [DOI] [Google Scholar]
- Wilson DB. In: Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy. Himmel ME, editor. Oxford: Blackwell Publishing Ltd; 2008. Aerobic microbial cellulase systems; pp. 374–392. [Google Scholar]
- Moran NA, Plague GR, Sandstrom JP, Wilcox JL. A genomic perspective on nutrient provisioning by bacterial symbionts of insects. Proc Natl Acad Sci U S A. 2003;100(Suppl 2):14543–14548. doi: 10.1073/pnas.2135345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd PF. In: Microbial Mediation of Plant-Herbivore Interactions. Barbosa P, Krischik VA, Jones CG, editor. New York City: John Wiley & Sons, Inc; 1991. Symbiont-mediated detoxification in insect herbivores; pp. 411–440. [Google Scholar]
- Sabree ZL, Kambhampati S, Moran NA. Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc Natl Acad Sci U S A. 2009;106:19521–19526. doi: 10.1073/pnas.0907504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idowua AB, Edemaa MO, Oyedepo MT. Extracellular enzyme production by microflora from the gut region of the variegated grasshopper Zonocerus variegatus (Orthoptera: Pyrgomorphidae) Int J Trop Insect Sci. 2009;29:229–235. doi: 10.1017/S1742758409990312. [DOI] [Google Scholar]
- Kaufman MG, Klug MJ. The contribution of hindgut bacteria to dietary carbohydrate utilization by crickets (Orthoptera: Gryllidae) Comp Biochem Physiol. 1991;98A:117–123. [Google Scholar]
- Mead LJ, Khachatourians GG, Jones GA. Microbial ecology of the gut in laboratory stocks of the migratory grasshopper, Melanoplus sanguinipes (Fab.) (Orthoptera: Acrididae) Appl Environ Microbiol. 1988;54:1174–1181. doi: 10.1128/aem.54.5.1174-1181.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazemier AE, den Camp HJMO, Hackstein JHP, Vogels GD. Fibre digestion in arthropods. Comp Biochem Physiol. 1997;118A:101–109. [Google Scholar]
- DiBlasi E, Morse S, Mayberry JR, Avila LJ, Morando M, Dittmar K. New Spiroplasma in parasitic Leptus mites and their Agathemera walking stick hosts from Argentina. J Invertebr Pathol. 2011;107:225–228. doi: 10.1016/j.jip.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Cameron AE. Structure of the alimentary canal of the stick-insect, Bacillus rossii Fabr.; with a note on the parthenogenesis of this species. Proc Zool Soc London. 1912;82:172–182. [Google Scholar]
- Chopard L. In: Traité de Zoologie: Anatmie, Systématique, Biologie. Grassé P-P, editor. Paris: Masson; 1949. Ordre des Chéleutoptères; pp. 594–617. [Google Scholar]
- Lasker R, Giese AC. Cellulose digestion by the silverfish Ctenolepisma Lineata. J Exp Biol. 1940;33:542–553. [Google Scholar]
- Coupland RE. Observations on the normal histology and histochemistry of the fat body of the locust (Schistocera gregaria) J Exp Biol. 1957;34:290–296. [Google Scholar]
- Gurr E. A Practical Manual of Medical and Biological Staining Techniques. New York: Interscience Publishers, Inc.; 1953. [Google Scholar]
- Ramsay JA. The excretory system of the stick insect Dixippus morosus (Orthoptera, Phasmidae) J Exp Biol. 1955;32:183–199. [Google Scholar]
- Chou HH, Holmes MH. DNA sequence quality trimming and vector removal. Bioinformatics. 2001;17:1093–1104. doi: 10.1093/bioinformatics/17.12.1093. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drancourt M, Raoult D. Sequence-based identification of new bacteria: a proposition for creation of an orphan bacterium repository. J Clin Microbiol. 2005;43:4311–4315. doi: 10.1128/JCM.43.9.4311-4315.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45:2761–2764. doi: 10.1128/JCM.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Worobey M, Kuo C-H, Ndjango J-BN, Peeters M, Hahn BH, Hugenholtz P. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 2010;8:e1000546. doi: 10.1371/journal.pbio.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden T. BLAST+: architecture and applications. BMC Bioinforma. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucl Acids Res. 2012;40:D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- Gasparich GE, Whitcomb RF, Dodge D, French FE, Glass J, Williamson DL. The genus Spiroplasma and its non-helical descendants: phylogenetic classification, correlation with phenotype and roots of the Mycoplasma mycoides clade. Int J Syst Evol Microbiol. 2004;54:893–918. doi: 10.1099/ijs.0.02688-0. [DOI] [PubMed] [Google Scholar]
- Regassa LB, Gasparich GE. Spiroplasmas: evolutionary relationships and biodiversity. Front Biosci. 2006;11:2983–3002. doi: 10.2741/2027. [DOI] [PubMed] [Google Scholar]
- Thakuria D, Schmidt O, Finan D, Egan D, Doohan FM. Gut wall bacteria of earthworms: a natural selection process. ISME J. 2010;4:357–366. doi: 10.1038/ismej.2009.124. [DOI] [PubMed] [Google Scholar]
- Sun J-Z, Scharf ME. Exploring and integrating cellulolytic systems of insects to advance biofuel technology. Insect Sci. 2010;17:163–165. doi: 10.1111/j.1744-7917.2010.01348.x. [DOI] [Google Scholar]
- Flook PK, Rowell CHF. Inferences about orthopteroid phylogeny and molecular evolution from small subunit nuclear ribosomal DNA sequences. Insect Mol Biol. 1998;7:163–178. doi: 10.1046/j.1365-2583.1998.72060.x. [DOI] [PubMed] [Google Scholar]
- Jiggins FM, Hurst GD, Jiggins CD, Schulenburg JH VD, Majerus ME. The butterfly Danaus chrysippus is infected by a male-killing Spiroplasma bacterium. Parasitol. 2000;120:439–446. doi: 10.1017/S0031182099005867. [DOI] [PubMed] [Google Scholar]
- Huigens ME, Stouthamer R. In: Insect Symbiosis. Bourtzis K, Miller TA, editor. Boca Raton: CRC Press; 2003. Parthenogenesis associated with Wolbachia; pp. 247–266. [Google Scholar]
- More E. Parthenogenesis explained. Phasmid Studies. 1996;52:62–69. [Google Scholar]
- Singh A, Lal R. Sphingobium ummariense sp. nov., a hexachlorocyclohexane (HCH)-degrading bacterium, isolated from HCH-contaminated soil. Int J Syst Evol Microbiol. 2009;59:162–166. doi: 10.1099/ijs.0.65712-0. [DOI] [PubMed] [Google Scholar]
- Ohmart CP, Thomas JR, Bubela B. Surfactant-producing microorganisms isolated from the gut of a Eucalyptus-feeding sawfly, Perga affinis affinis. Oecologia. 1988;77:140–142. doi: 10.1007/BF00380937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Taxonomic assignment of the OTUs based on RDP CLASSIFIER and BLASTN sequence similarity search against the NCBI nt database.