

Abstract
Background
Autoimmunity to neuronal proteins occurs in several neurological syndromes, where cellular and humoral responses are directed to surface as well as intracellular antigens. Similar to myelin autoimmunity, pathogenic immune response to neuroaxonal components such as neurofilaments may contribute to neurodegeneration in multiple sclerosis.
Methods
We studied the immune response to the axonal protein neurofilament light (NF-L) in the experimental autoimmune encephalomyelitis animal model of multiple sclerosis. To examine the association between T cells and axonal damage, pathology studies were performed on NF-L immunized mice. The interaction of T cells and axons was analyzed by confocal microscopy of central nervous system tissues and T-cell and antibody responses to immunodominant epitopes identified in ABH (H2-Ag7) and SJL/J (H2-As) mice. These epitopes, algorithm-predicted peptides and encephalitogenic motifs within NF-L were screened for encephalitogenicity.
Results
Confocal microscopy revealed both CD4+ and CD8+ T cells alongside damaged axons in the lesions of NF-L immunized mice. CD4+ T cells dominated the areas of axonal injury in the dorsal column of spastic mice in which the expression of granzyme B and perforin was detected. Identified NF-L epitopes induced mild neurological signs similar to the observed with the NF-L protein, yet distinct from those characteristic of neurological disease induced with myelin oligodendrocyte glycoprotein.
Conclusions
Our data suggest that CD4+ T cells are associated with spasticity, axonal damage and neurodegeneration in NF-L immunized mice. In addition, defined T-cell epitopes in the NF-L protein might be involved in the pathogenesis of the disease.
Keywords: Neurofilament light, Axonal damage, Neurodegeneration, Experimental autoimmune encephalomyelitis, Multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic demyelinating and neurodegenerative disease of the central nervous system (CNS) widely considered due to aggressive, autoreactive T cells and antibodies to myelin [1-3]. However, accumulating evidence shows that immune responses to neuronal and axonal proteins are also present in a wide range of neurodegenerative disorders including MS [4-6]. That these responses may contribute to axonal and neuronal damage, pathological hallmarks of MS, is supported by observations that immunization with neuronal antigens and transfer of antibodies directed to neuronal and axonal proteins induce neuronal damage in animals [7-10].
The lack of expression of molecules of the major histocompatibility complex (MHC) class II on neurons indicates that neurons cannot activate CD4+ cells in an antigen-specific manner. However, neurons constitutively express or readily upregulate expression of MHC class I during inflammation, indicating that neurons may become targets for CD8+ T cells [11]. Conceptually, both CD4+ and CD8+ T cells could mediate attack on axons and neurons, either by direct contact via antigen-independent interactions or as a result of collateral damage [12]. Activated T cells in the CNS are reported to produce cytotoxic molecules as well as glutamate, nitric oxide and reactive oxygen species that could contribute to the damage and progressive neurodegeneration observed in MS and other neurodegenerative diseases in which inflammation has been described [13-15]. In addition, activation of B cells might lead to the production of specific antibodies to neuronal antigens that could also contribute to the damage and progressive neurodegeneration [16].
To examine the mechanisms of autoimmunity to neurons we have developed a model of autoimmune induced axonal and neuronal damage following immunization of mice with the neuronal cytoskeletal protein neurofilament light (NF-L) [7]. Whether T-cell responses to neuroaxonal components are pathogenic in MS is as yet unknown; although we have recently shown that NF-L is phagocytosed by MHC class II+ microglia/macrophages in MS brain lesions [17], indicating a potential source by which autoreactive T cells could become reactivated in MS. In mice, we have shown that autoimmunity to NF-L causes spasticity and neurodegeneration and that axonal damage is a direct consequence of such responses [7,8]. Infiltration of CD3+ T cells and B220+ cells mainly localized in the dorsal column of the spinal cord of NF-L immunized mice was also observed [8]. Likewise, immunoglobulin deposits were observed into the axons in mice immunized with NF-L protein [7].
In the present study, we characterized the T-cell infiltrates in the CNS of spastic mice. Both CD4+ and CD8+ T cells were found in close association to axons, although CD4+ T cells dominated the infiltrates in lesions. In addition, increased perforin expression and cells positive for granzyme B could be observed in the spinal cord of mice immunized with NF-L. Furthermore, NF-L peptides were screened for T-cell and B-cell responses in ABH (H2-Ag7) and SJL/J (H2-As) mice, and the pathogenic potential of these peptides and predicted binding motifs to H2-Ag7 present in the NF-L sequence were investigated.
In summary, our study reveals that T cells associated with the expression of cytotoxic molecules are present in lesions in the dorsal columns of spastic mice immunized with NF-L and we show, for the first time, that active immunization with defined NF-L peptides induced neurological disease in ABH mice.
Materials and methods
Mice
Male and female 10-week-old Biozzi ABH (H-2dq1) and SJL/J (H-2s) mice were obtained from Harlan (Bicester, UK) and Charles River laboratories (Kent, UK) or were bred at QMUL (London, UK). All procedures were performed in accordance with the UK Animals (Scientific Procedures) Act (1986) and approved by the local ethics committee. All procedures were performed following Institutional ethical review in accordance to the United Kingdom Animals (Scientific Procedures) Act (1986) and European Union Directive 2010/63/EU. Animals were housed and monitored consistent with the principles of the ARRIVE guidelines as described previously [18].
Antigens
Spinal cord homogenate (SCH) prepared from 60 ABH mice was lyophilized and reconstituted in PBS as described previously [18]. rmNF-L was prepared as described previously [17]. Overlapping 16-amino-acid peptides (Table 1) based on the mouse protein sequence (NCBI protein ID: NP035040) and myelin oligodendrocyte glycoprotein (MOG35–55) peptide (MEVGWYRSPFSRVVHLYRNGK) were synthesized as peptide amides (CONH2) (Cambridge Research Biochemicals Ltd, Billingham, UK).
Table 1.
Sequences of mouse neurofilament-light peptidesa
| Sequence | Amino acid | Sequence | Amino acid |
|---|---|---|---|
| 1 to 16 |
SSFGYDPYFSTSYKRR |
273 to 288 |
MQNAEEWFKSRFTVLT |
| 9 to 24 |
FSTSYKRRYVETPRVH |
281 to 296 |
KSRFTVLTESAAKNTD |
| 17 to 32 |
YVETPRVHISSVRSGY |
289 to 304 |
ESAAKNTDAVRAAKDE |
| 25 to 40 |
ISSVRSGYSTARSAYS |
297 to 312 |
AVRAAKDEVSESRRLL |
| 33 to 48 |
STARSAYSSYSAPVSS |
305 to 320 |
VSESRRLLKAKTLEIE |
| 41 to 56 |
SYSAPVSSSLSVRRSY |
313 to 328 |
KAKTLEIEACRGMNEA |
| 49 to 64 |
SLSVRRSYSSSSGSLM |
321 to 336 |
ACRGMNEALEKQLQEL |
| 57 to 72 |
SSSSGSLMPSLENLDL |
329 to 344 |
LEKQLQELEDKQNADI |
| 65 to 80 |
PSLENLDLSQVAAISN |
337 to 352 |
EDKQNADISAMQDTIN |
| 73 to 88 |
SQVAAISNDLKSIRTQ |
345 to 360 |
SAMQDTINKLENELRS |
| 81 to 96 |
DLKSIRTQEKAQLQDL |
353 to 368 |
KLENELRSTKSEMARY |
| 89 to 104 |
EKAQLQDLNDRFASFI |
361 to 376 |
TKSEMARYLKEYQDLL |
| 97 to 112 |
NDRFASFIERVHELEQ |
369 to 384 |
LKEYQDLLNVKMALDI |
| 105 to 120 |
ERVHELEQQNKVLEAE |
377 to 392 |
NVKMALDIEIAAYRKL |
| 113 to 128 |
QNKVLEAELLVLRQKH |
385 to 400 |
EIAAYRKLLEGEETRL |
| 121 to 136 |
LLVLRQKHSEPSRFRA |
393 to 408 |
LEGEETRLSFTSVGSI |
| 129 to 144 |
SEPSRFRALYEQEIRD |
401 to 416 |
SFTSVGSITSGYSQSS |
| 137 to 152 |
LYEQEIRDLRLAAEDA |
409 to 424 |
TSGYSQSSQVFGRSAY |
| 145 to 160 |
LRLAAEDATNEKQALQ |
417 to 432 |
QVFGRSAYSGLQSSSY |
| 153 to 168 |
TNEKQALQGEREGLEE |
425 to 440 |
SGLQSSSYLMSARSFP |
| 161 to 176 |
GEREGLEETLRNLQAR |
433 to 448 |
LMSARSFPAYYTSHVQ |
| 169 to 184 |
TLRNLQARYEEEVLSR |
441 to 456 |
AYYTSHVQEEQTEVEE |
| 177 to 192 |
YEEEVLSREDAEGRLM |
449 to 464 |
EEQTEVEETIEATKAE |
| 185 to 200 |
EDAEGRLMEARKGADE |
457 to 472 |
TIEATKAEEAKDEPPS |
| 193 to 208 |
EARKGADEAALARAEL |
465 to 480 |
EAKDEPPSEGEAEEEE |
| 201 to 216 |
AALARAELEKRIDSLM |
473 to 488 |
EGEAEEEEKEKEEGEE |
| 209 to 224 |
EKRIDSLMDEIAFLKK |
481 to 496 |
KEKEEGEEEEGAEEEE |
| 217 to 232 |
DEIAFLKKVHEEEIAE |
489 to 504 |
EEGAEEEEAKDESEDT |
| 225 to 240 |
VHEEEIAELQAQIQYA |
497 to 512 |
AKDESEDTKEEEEGGE |
| 233 to 248 |
LQAQIQYAQISVEMDV |
505 to 520 |
KEEEEGGEGEEEDTKE |
| 241 to 256 |
QISVEMDVSSKPDLSA |
513 to 528 |
GEEEDTKESEEEEKKE |
| 249 to 264 |
SSKPDLSAALKDIRAQ |
521 to 536 |
SEEEEKKEESAGEEQV |
| 257 to 272 |
ALKDIRAQYEKLAAKN |
529 to 544 |
ESAGEEQVAKKKD |
| 265 to 280 | YEKLAAKNMQNAEEWF |
aMouse neurofilament-light sequence (NCBI reference sequence: NP035040).
Induction of experimental autoimmune encephalomyelitis
Mice were injected subcutaneously with 1 mg SCH, 200 μg rmNF-L, 200 μg MOG35–55 or rmNF-L and MOG35–55 (1:1), or pools of 30 μg each peptide emulsified with incomplete Freund’s adjuvant (Difco Laboratories, (Detroit, Michigan, USA) supplemented with 48 μg Mycobacterium tuberculosis and 6 μg Mycobacterium butyricum (Difco Laboratories) on day 0 and day 7 as described previously [19]. Control mice were immunized with complete Freund’s adjuvant (CFA) only. All mice were injected with 200 ng Bordetella pertussis toxin (Sigma St. Louis, Missouri, USA) intraperitoneally, immediately after immunization and 24 hours later.
To identify encephalitogenic epitopes, four to six mice were immunized with rmNF-L, individual or pooled peptides. To optimize identification, sequences containing motifs that bind to or interact with H2-Ag7 were selected as described previously [19]. The Rankpep server was additionally used to predict binding to H2-Ag7[20].
Mice were monitored daily and scored according to a neurological scale: 0, normal; 0.5, partial loss of tail tone; 1, paralysis or spasticity of the tail; 2, impaired righting reflex; 3, paralysis or spastic paresis of one limb; 4, paralysis or spastic paresis of two limbs; and 5, moribund [7,18]. Mice were sacrificed by carbon dioxide inhalation and brains and spinal cords snap-frozen in liquid nitrogen or processed for pathology [7].
Immunohistochemistry
Sections (3 μm) from snap-frozen spinal cord tissues were fixed with acetone and incubated overnight at 4°C with mAb directed to CD4 (YTS 191.1.2), CD8 (YTS 169AG; ImmunoTools, Friesoythe, Germany), MHC-I antigens (HM1091; Hycult Biotech, Plymouth Meeting, PA, USA) or biotinylated MHC-II (OX 6, a kind gift of Jack van Horssen, VU University Medical Center) diluted in antibody diluent (Immunologic; Duiven, The Netherlands). After washing, endogenous peroxidase was blocked with 0.3% H2O2 in PBS. Sections stained for CD4, CD8 and MHC-I were incubated with biotinylated rabbit anti-rat Ig (Dako, Glostrup, Denmark) for 1 hour, followed by peroxidase-coupled avidin–biotin complex (ABC kit; Vector Laboratories, Burlingame, CA, USA). Sections stained with biotinylated MHC-II were incubated with streptavidin–horseradish peroxidase complex (Dako) for 1 hour. All secondary antibodies were visualized with 3,3′-diaminobenzidine. Antibodies were prescreened on brain, liver, lung, spleen and tonsil tissues and isotype control mAb served as negative control. The percentage of CD4+ and CD8+ T cells were counted at 25× objective at three levels of the spinal cord.
For immunofluorescence, sections were incubated with blocking solution (CleanVision IHC/ICC; Immunologic) containing 10% normal goat serum for 2 hours, washed in PBS and incubated with mAb to NF-L (10H9), SMI-32 (Covance, Princeton, NJ, USA) or NeuN (Merck Millipore; Darmstadt, Germany) and CD3 (CD3-12; Serotec, Oxford, UK), CD4 (YTS 191.1.2) or CD8 (YTS 169AG; ImmunoTools) overnight at 4°C. After washing in PBS, sections were incubated with goat anti-mouse IgG1 Alexa 594 or goat anti-rat IgG Alexa 488 (Invitrogen; Paisley, UK) for 60 minutes at room temperature. Sections were viewed using confocal laser scanning microscopy (Leica DMI6000; Rijswijk, The Netherlands). Image processing was performed using NIH Image J software [21].
Granzyme B staining was performed on paraffin-embedded sections (4 μm). In brief, sections were deparaffinized and rinsed in H2O. Subsequently, endogenous peroxidase was blocked as described above. After rinsing in PBS, antigen retrieval in Tris–ethylenediamine tetraacetic acid buffer (pH 9.0) was performed in a microwave followed by incubation with 10% normal goat serum. Sections were incubated overnight at 4°C with polyclonal rabbit anti-granzyme B (ab4059; Abcam; Cambridge, UK) in 1% BSA. Subsequently, sections were washed in PBS and incubated with secondary antibody Envision anti Rabbit labeled with horseradish peroxidase (K4002; Dako) for 30 minutes and visualized with 3,3′-diaminobenzidine.
Reverse transcriptase polymerase chain reaction
Spinal cords from control and NF-L immunized ABH mice were dissolved in lysis buffer (NucleoSpin RNA/Protein kit; Machery-Nagel GmbH, Düren, Germany) and homogenized with 1.4 mm ceramic beads (Precellys 24; Peqlab Biotechnologie GmbH, Erlangen, Germany) at 5000 rpm for 15 seconds. Subsequently, RNA was isolated using NuceloSpin (Macherey-Nagel) according to the manufacturer’s recommendations. Purity was confirmed using 260:280 OD ratios (Nano-Drop 1000; Peqlab Biotechnologie GmbH). RT reactions were performed with the MMLV RT-kit and random hexanucleotide primers (Invitrogen) and gene expression was measured using Taq-Polymerase (Biomol GmbH, Hamburg, Germany).
Primers for perforin amplification (sense, 5′-CTGCCACTCGGTCAGAATG-3′; antisense, 5′-CGGAGGGTAGTCACATCCAT-3′) were used at annealing temperature of 59°C, amplifying an 88-base-pair fragment. Expression levels of the reference gene hypoxanthine guanine phosphoribosyl transferase were used as control. Primers for hypoxanthine guanine phosphoribosyl transferase amplification (sense, 5′-GCTGGTGAAAAGGACCTCT-3′; antisense: 5′-CACAGGACTAGAACACCTGC-3′) were used at an annealing temperature of 60°C, amplifying a 248-base-pair fragment, and primers for 18sRNA amplification (sense, 5′-CGGCTACCACATCCAAGGAA-3′; antisense, 5′-GCTGGAATTACCGCGGCT-3′) were used at an annealing temperature of 60°C, amplifying a 187-base-pair fragment. Gene expression was performed using RT-PCR technology (Bio-Rad; Munich, Germany) with SYBR green (SensiMix™; Bioline; Luckenwalde, Germany), as published previously [22].
T-cell proliferation assays
ABH and SJL/J mice were immunized with rmNF-L in CFA or CFA only (n = 4 per group). Spleen cells were collected 10 days after priming and single-cell suspensions (3 × 105/ml) cultured in RPMI medium (Gibco, Invitrogen; Paisley, UK) with 5% FCS (Gibco, Invitrogen), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 5 mM Hepes, and 5 × 10–5 M 2-mercaptoethanol (Gibco, Invitrogen). Cells were incubated with NF-L peptides or rmNF-L for 72 hours. Proliferation was determined by incorporation of [3H]-thymidine (GE Healthcare, Uppsala, Sweden). Stimulation indices (SIs) were calculated as the proliferative response in the presence of antigen divided by the response in the absence of antigen. SI in the CFA control group ranged from 0.6 to 1.1. Positive stimulation was defined as SI >1.5.
Enzyme-linked immunosorbent assay
To identify B-cell epitopes in NF-L, Biozzi ABH and SJL/J mice (n = 4 per group) were immunized with rmNF-L protein in complete adjuvant and serum was collected on day 15 post immunization. The mouse immune sera were tested for their reactivity to NF-L overlapping peptides. Nunclon plates (Nunc, Roskilde, Denmark) were coated overnight at 4°C with 10 μg/ml mouse NF-L peptides or rmNF-L protein, in carbonate buffer. Plates were washed twice in PBS and blocked for 1 hour at 37°C with 2% BSA/PBS. After blocking, ABH or SJL/J immune sera, diluted 1:100 in 1% (BSA/PBS), were added and incubated for 1 hour at room temperature. After washing in PBS–Tween 0.1%, the plates were incubated for 1 hour at room temperature with horseradish peroxidase-conjugated rabbit anti-mouse Ig (Dako). The reaction product was developed with TMB substrate (Thermo Fisher Scientific; Loughborough, UK) and stopped by the addition of 2 M hydrochloric acid. The absorbance was measured at 450 nm, using a Synergy HT microplate reader (Bio-Tek instruments; Winooski, Vermont, USA). Background values were obtained by the reactivity of immune serum on peptide uncoated wells. An absorbance above the mean plus three standard deviations of the background reactivity was taken as positive.
Statistical analysis
For comparison of clinical experimental autoimmune encephalomyelitis (EAE) scores, significance between the groups was determined by nonparametric Mann–Whitney rank-sum test (SigmaStat; Systat Software Inc., San Jose, CA, USA). Data represent the mean ± standard error of the mean (**P <0.01, *P <0.05).
Results
CD4+ and CD8+ T cells are associated with neuronal/axonal damage in spastic mice
To examine the presence of T cells in the CNS during disease, CD4+ or CD8+ T cells within the lesions of NF-L immunized mice at the peak of disease were examined. Using immunohistochemistry, the ratio of CD4+ and CD8+ T cells in the lesions was determined (Figure 1A, B, respectively). We observed that significantly more CD4+ T cells were present within the lesions in a ratio of 9:1 CD4+:CD8+ T cells (data not shown). To support a role for CD8+ T cells we examined the expression of MHC class I in the spinal cord of diseased mice. In contrast to expression of MHC class I antigens on inflammatory cells in the lesion as well as perivascular cells (Figure 1C) and microglia in normal-appearing white matter (Figure 1D), there was no evidence of MHC-class I antigen expression on neurons or axons in the lesions (Figure 1E, F) or in normal-appearing tissues (data not shown). Likewise, while MHC class II was expressed on inflammatory cells in the blood vessels and in the parenchyma, being present on ramified cells resembling microglia (Figure 1G), no expression on neurons and axons was observed (Figure 1H, I), as has been shown before in normal or pathological tissues during EAE [23].
Figure 1.

T cells in lesions of spastic mice. Immunohistochemistry showing (A) CD8+ and (B) CD4+ T cells in the dorsal column of a spastic mouse. (C) to (F) Major histocompatibility complex (MHC) class I antigen staining expressed on (C) endothelial cells, and (D) microglia (E, F), but not on neurons. (G) to (I) MHC class II staining on ramified cells resembling microglia but not neurons (H, I). A to E, G to H, bar = 10 μm; F and I, bar = 5 μm.
To examine the interaction of T cells and axons we used confocal microscopy of CNS tissues from spastic mice. We observed a close association of CD3+ T cells aligning with possible damaged axons as indicated by SMI-32 staining (Figure 2A), in which there was close association between the T cell and the axon (Figure 2A, insert). In cross-sections of the lesion, CD4+ T cells were observed within areas of axonal damage (Figure 2B) and frequently observed between axons with increased immunoreactivity for SMI-32 and neurofilament, indicating axonal damage (Figure 2C, D). While CD8+ T cells were also observed between thickened axons in the lateral funiculus and adjacent to Neu-N+ neurons in the spinal cord (Figure 2E, F), these were rare as compared with CD4+ T cells.
Figure 2.
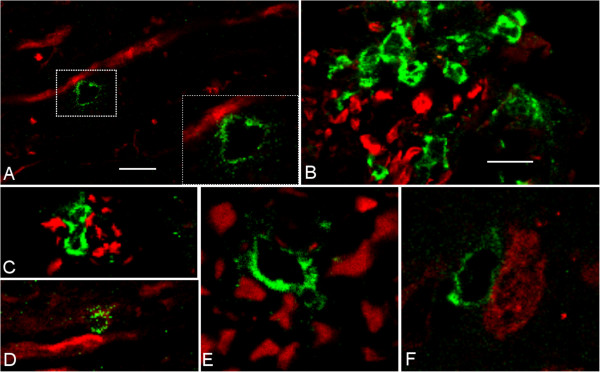
T cells associated with axonal damage in spastic mice. Confocal images of (A) CD3+ T cell (green) in contact with SMI32+ axon (red). (B, C) CD4+ T cells in area of axonal damage and (D) next to swollen NF-L+ axons. (E) CD8+ T cells between NF-L+ axons and (F) Neu-N+ neurons in the spinal cord. A, E, F, bar = 5 μm; B, C, D, bar = 25 μm.
Cytotoxic molecules are present in the spinal cord of NF-L immunized mice
To examine the potential mechanisms of T-cell-mediated axonal damage, the expression of the cytotoxic molecules granzyme B and perforin was examined in the spinal cord of spastic mice. Immunostaining revealed the expression of granzyme B in spinal cord lesions of mice immunized with NF-L (Figure 3A). Immunodetection of perforin in mouse CNS was not possible due to lack of specific mAbs. Instead, RT-PCR on spinal cord tissues was performed and showed that perforin mRNA levels were increased in rmNF-L immunized mice compared with controls (Figure 3B). In addition, granzyme B and perforin were expressed by peripheral CD4+ T cells from NF-L immunized mice compared with CD8+ T cells (data not shown).
Figure 3.
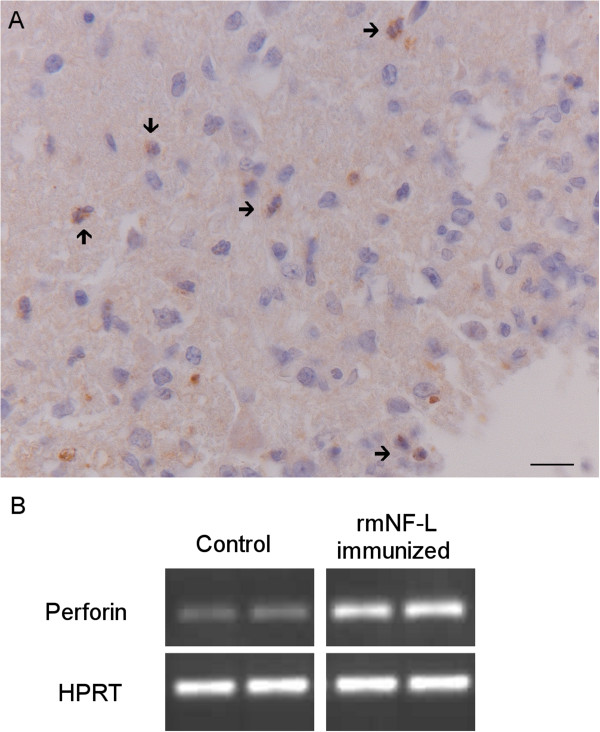
Production of cytotoxic molecules in spastic mice. (A) Granzyme B-positive staining (indicated by arrows) was performed on paraffin-embedded spinal cord sections: bar = 10 μm. (B) Gel electrophoresis of PCR products show perforin expression is increased in spastic mice (hypoxanthine guanine phosphoribosyl transferase (HPRT) = internal control). NF-L, neurofilament light.
Autoimmunity to NF-L exacerbates MOG35–55 experimental autoimmune encephalomyelitis in ABH mice
To increase the likelihood of exposing NF-L protein in the CNS, ABH mice were co-immunized with MOG35–55 to induce myelin damage (Figure 4). While SCH induced classical EAE about 11 days after immunization, co-immunization of rmNF-L with MOG35–55 induced significantly augmented disease in which spasticity was observed on days 16 and 17 (P = 0.005), indicating that myelin damage may first be necessary to expose neuronal antigens. Within this timeframe, mice immunized with rmNF-L only did not develop clinical disease as we have reported previously [7]. However, by day 21, NF-L immunized mice did develop neurological signs of disease with a mean day of onset 19.8 ± 1.6, EAE score 1.5 ± 1.0, group score 0.5 ± 0.4 and an EAE incidence of 50%.
Figure 4.

Augmentation of MOG35–55-induced experimental autoimmune encephalomyelitis by neurofilament light. ABH mice were immunized with spinal cord homogenate, MOG35–55, rmNF-L or rmNF-L and MOG35–55. **P <0.005; Mann–Whitney U test. NF-L, neurofilament light.
Immunodominant B-cell and T-cell epitopes of neurofilament light in mice
To determine immunodominant epitopes in the NF-L protein, antibody and T-cell responses to synthetic 16-mer overlapping peptides spanning the mouse NF-L sequence (Table 1) were screened in ABH and SJL/J mice immunized with rmNF-L in CFA [19]. Since we have seen before that immunization with NF-L in Biozzi mice can result in antibody production and binding to axons, it was of interest to screen the antibody responses to the overlapping peptides [7]. Antibody reactivity to NF-L peptides in both strains of mice revealed an immunodominant region that corresponds to the overlapping region (NF-L amino acids 169 to 208) and the single region (amino acids 169 to 184) in ABH and SJL/J mice respectively (Figure 5A, B). This region is located in the coil 1b domain of NF-L. In line with our observations, these motifs (amino acids 154 to 195) are likely to be linear B-cell epitopes according to the BCPred prediction algorithm [24]. In SJL/J mice, NF-L specific antibodies also reacted to the single peptides amino acids 41 to 56 and amino acids 345 to 360 (Figure 5B). In both strains, the mean absorbance values for antibody response to rmNF-L protein were within the optical density range of 1.21 to 1.34.
Figure 5.

B-cell responses to neurofilament light peptides in ABH and SJL/J mice. (A) ABH mice and (B) SJL/J mice were immunized with rmNF-L and serum antibody reactivity to NF-L peptides was measured on day 10. Mean absorbance (optical density (OD) >0.25) was considered positive.
Immunodominant T-cell responses to two dominant regions, amino acids 241 to 288 and 345 to 368, were observed in ABH mice (Figure 6A). In comparison, T-cell responses in SJL/J mice were observed to amino acids 241 to 288 (Figure 6B). In both strains, the SIs were lower than responses to rmNF-L alone (SI = 5 to 10). These results are in line with data using overlapping 15-mer peptides of NF-L in ABH mice (data not shown), indicating that immunodominant epitopes reside within the rod domain (linker 2 and coil 2B) of NF-L. No overlapping peptide induced significant proliferation in cells from CFA immunized mice.
Figure 6.

T-cell responses to NF-L peptides in ABH and SJL/J mice. (A) ABH mice and (B) SJL/J mice were immunized with rmNF-L and T-cell proliferation to NF-L peptides was measured on day 10. Stimulation index >1.5 was considered positive.
Pathogenicity of immunodominant epitopes
To determine the pathogenicity of immunodominant epitopes, ABH mice were immunized with individual NF-L peptides spanning the T-cell immunodominant region (amino acids 241 to 272) or with a pool of immunodominant B-cell epitopes. In contrast to SCH-induced EAE, immunization of ABH mice with amino acids 241 to 256, 249 to 264, 257 to 272, 345 to 360 and 353 to 368 induced spasticity of the tail and hind limbs in line with studies using rmNF-L (Table 2, panel A) [7]. Immunization with NF-L peptides also induced a delayed onset of clinical signs occurring 10 days later than SCH-induced EAE (Table 2, panel A). Rota-rod studies did not reveal additional signs of neurological disease (data not shown). SJL/J mice immunized with rmNF-L also developed very mild clinical disease of spasticity (data not shown). The pathogenic potential of NF-L peptides was also observed with some, but not all, peptides predicted to bind with H2-Ag7 (Table 2, panel B). No significant effect was observed after immunization with the immunodominant B-cell epitopes (amino acids 169 to 208) (Additional file 1).
Table 2.
Pathogenic neurofilament-light peptides in ABH micea
| Antigen/peptides | Number with EAE | Mean group scoreb | Mean EAE scorec | Mean day of onset |
|---|---|---|---|---|
|
Panel A |
|
|
|
|
| SCH |
4/4 |
4.0 ± 0.0* |
4.0 ± 0.0 |
15.3 ± 0.3 |
| rmNF-L |
4/5 |
0.7 ± 0.3* |
0.9 ± 0.2 |
31.0 ± 1.4 |
| 241 to 256 |
1/5 |
0.1 ± 0.1 |
0.5 ± N/A |
21.0 ± N/A |
| 249 to 264 |
5/5 |
0.9 ± 0.3* |
0.9 ± 0.3 |
25.6 ± 6.4 |
| 257 to 272 |
3/5 |
0.4 ± 0.2 |
0.7 ± 0.2 |
29.3 ± 0.6 |
| 265 to 280 |
0/5 |
N/A |
N/A |
N/A |
| 273 to 288 |
0/5 |
N/A |
N/A |
N/A |
| 345 to 360 |
3/5 |
0.7 ± 0.3 |
1.2 ± 0.3 |
29.3 ± 0.3 |
| 353 to 368 |
3/5 |
0.5 ± 0.2 |
0.8 ± 0.2 |
25.7 ± 4.4 |
|
Panel B |
|
|
|
|
| 129 to 144 |
2/5 |
0.2 ± 0.1 |
0.5 ± 0.0 |
18.5 ± 6.5 |
| 297 to 312 |
2/5 |
0.2 ± 0.1 |
0.5 ± 0.0 |
30.5 ± 0.5 |
| 313 to 328 |
0/5 |
N/A |
N/A |
N/A |
| 113 to 128, 281 to 296, 289 to 304, 321 to 336 (4) |
3/4 |
0.9 ± 0.4 |
1.2 ± 0.3 |
28.7 ± 0.7 |
| 201 to 216, 377 to 392, 457 to 472 (3) |
0/5 |
N/A |
N/A |
N/A |
|
Panel C |
|
|
|
|
| CFA | 0/5 | N/A | N/A | N/A |
aAnimals were injected with (panel A) 200 μg spinal cord homogenate (SCH), rmNF-L or single immunodominant NF-L peptides in CFA or with (panel B) peptides in Complete Freund’s adjuvant (CFA) predicted to bind H2-Ag7 or with (panel C) CFA alone.
bMean ± standard error of the mean (SEM) of the maximum experimental autoimmune encephalomyelitis (EAE) score of animals in the group.
cMean ± SEM of the maximum EAE score of affected animals.
N/A, not applicable. *P <0.05, compared with CFA immunized mice.
Additionally, the capacity of NF-L-specific T cells to transfer spasticity was tested in ABH mice. Lymph node and spleen cells from rmNF-L-immunized ABH mice were collected 12 days after immunization and stimulated for 10 days in vitro with rmNF-L. Activated cells were adoptively transferred into irradiated naïve recipients and followed until day 25. Mild signs of disease (score 0.5) were observed in two of seven mice revealing the pathogenic potential of T cells to NF-L (data not shown).
Discussion
Accumulating evidence indicates that as well as myelin-specific T cells, neuronal-specific T cells may gain access to the CNS and contribute to neurodegeneration. Immune responses to neurons are reported in Rasmussen’s encephalitis [25], Alzheimer’s disease [26], Parkinson’s disease [27] and paraneoplastic disorders [5], underscoring the potential role of pathogenic neuronal-reactive T cells in MS in which axonal damage correlates with disability. To examine the role of T cells in neurodegeneration, we have studied autoimmune-meditated neurodegeneration in mice immunized with NF-L in which spasticity and paralysis, clinical features characteristic of MS, are observed [7,8]. In previous studies, infiltration of CD3+ and B220+ cells and immunoglobulin deposits were observed in spinal cord lesions of NF-L immunized mice [7].
In this study, we aimed to further investigate the cellular and humoral immune response in the mouse model of NF-L-induced neurodegeneration. Immunohistochemistry revealed the association of both CD4+ and CD8+ T cells with neuronal damage and the expression of cytotoxic molecules in the CNS. We also identify immunodominant regions and encephalitogenic epitopes on the NF-L protein. Together, our data support the accumulating evidence that T cells play a role in neuronal and axonal damage in neurodegenerative disorders. Adoptive transfer experiments to test the ability of NF-L peptide-specific T cells to induce disease would be of interest in future studies.
A pre-requisite for antigen-specific CD8+ cytotoxic damage of neurons is the expression of MHC class I molecules. Strong evidence exists of MHC class I on neurons, neurites and axons, revealing differential expression depending on neuronal subtype, development stage and the inflammatory stimulus. MHC class I expression is highly upregulated on neurons following IFNγ treatment but not TNFα [28]. Intriguingly, CD8+ T cells directed to NF-L generated from spastic mice produce high levels of IFNγ [4], indicating that T cells to NF-L could trigger progressive neurodegeneration. Further support for T-cell-mediated neuronal damage comes from the finding that, in EAE, myelin-reactive T cells are activated by neurofilament peptides [29]. Such interaction between T cells and neurons may induce antigen-specific lysis of neurons [30] or dysfunction by impairing electrical signaling [31] and induction of rapid microtubule axonal destabilization [32]. In the spastic model it is unlikely that antigen-specific CD8+ T-cell activation leads to neuronal damage since MHC class I was not present on neurons, either in the lesions or in normal appearing tissue. While it is unlikely that CD4+ T cells induce neuronal death in an antigen-specific fashion, as neurons do not express MHC class II, pathogenic CD4+ T cells expressing NKG2C could injury neurons and axons expressing HLA-E, as reported for oligodendrocytes in MS [33].
T-cell-mediated neuronal damage may alternatively occur via antigen-independent interactions involving Fas–FasL, TRAIL, CD11a and CD40. In culture, polyclonally activated CD4+ and CD8+ T cells are cytotoxic to human neurons [30,32], underscoring the potential pathogenic role of both CD4+ and CD8+ T cells. In line with these studies, our results show that granzyme B and perforin are present in the lesions of spastic mice, indicating that neuronal damage is more likely due to antigen-independent mechanisms. The predominance of CD4+ T cells in the lesions of NF-L immunized mice may be due to selective recruitment or selective depletion of CD8+ T cells [34]. Moreover, the ratio between the T-cell subsets in the lesions in our model is similar to myelin-induced EAE [35]. Further studies on the expression of cytotoxic molecules in T-cell subpopulations in the CNS of NF-L immunized mice will be necessary.
Our observations that co-immunization with MOG and NF-L leads to exacerbated disease is pertinent to what might occur in MS during myelin damage, in which neurons may be more vulnerable to immune responses to neuronal antigens. In this way, the course of neuronal degeneration might be accelerated in the presence of NF-L reactive T cells [4] and antibodies to NF in MS [36].
EAE studies have been instrumental in identifying the role of T cells and antibodies to myelin in disease. These have revealed important information about peptide:MHC:T-cell receptor interactions crucial for development of tolerance regimes using altered peptide ligands or tolerogenic routes for delivery of pathogenic peptides to modulate myelin-specific T cells [37,38]. These approaches have proved effective in chronic EAE models, preventing the clinical relapses, but do not control progressive disease [37], indicating that mechanisms other than myelin autoimmunity, such as autoimmunity to neuronal proteins, are involved in neurodegeneration in MS.
In ABH mice, immunization with MOG and proteolipid protein readily induces neurological disease in which flaccid paralysis and preferential myelin loss is observed. In contrast, immunization with NF-L protein induces a predominantly spastic disease in which neuronal damage is the primary pathological feature. To determine the pathogenic potential of NF-L peptide, we used a peptide mapping approach and identified a peptide core within the NF-L protein that contains similar elements to previously defined H2-Ag7 motifs within myelin basic protein, MOG and proteolipid protein (Table 3) [19]. This supports many of the predictions made by computer modeling of peptide H2–Ag7 interactions [39]. Similar to the finding following immunization with myelin antigens [19], several encephalitogenic epitopes were identified in NF-L immunized ABH mice. One should note that when using overlapping peptides, inappropriate sequence alignment could mask encephalitogenic epitopes or induce peptide epitopes to become tolerogenic [40]. In addition, neurofilament modulates oligodendrocyte proliferation and differentiation [41], thereby masking the possible pathogenic impact of these proteins. Peptide mapping approaches have identified immunodominant epitopes of myelin basic protein in mice and humans and prompted tolerance strategies to myelin basic protein-specific T cells in MS. Current studies are underway to identify T-cell responses to NF-L epitopes in MS patients who respond to the NF-L protein [4]. These studies may reveal whether such responses are present in subtypes of MS or correlate with extent of cognitive changes or progressive disease. Such findings may be key to the development of personal therapeutic approaches using tolerance regimens.
Table 3.
Pathogenic peptides associated with neurological disease in ABH mice
| Protein and peptide residues | Amino acid sequence | Reference |
|---|---|---|
| PLP 54 to 76 |
DYEYLINVIHAFQYVIGASF |
[19] |
| MBP 12 to 35 |
LATASTMDHARHGFLPRHRDTSGI |
[19] |
| MOG 1 to 23 |
GQFRVIGPGYPIRALVGDEQED |
[19] |
| αB crystallin 52 to 61 |
FFLRAPSWI |
[42] |
| NF-L 129 to 144 |
SEPSRFRALYEQEIRD |
This study |
| NF-L 257 to 272 | ALKDIRAQYEKLAAKN | This study |
MBP myelin basic protein, MOG myelin oligodendrocyte glycoprotein, NF-L neurofilament light, PLP proteolipid protein.
In conclusion, we show that peptide epitopes of NF-L induce neurological disease and that potentially pathogenic CD4+ T cells dominate the lesions of NF-L immunized mice, a model for immune-mediated neurodegeneration. Our data suggest that, similar to peptide therapies targeting myelin responses [43], immune therapies targeting neuronal-specific T cells could thus be beneficial in reducing neurodegeneration in inflammatory disorders such as MS.
Consent
Written informed consent was obtained from the patient for the publication of this report and any accompanying images.
Abbreviations
BSA: Bovine serum albumin; CNS: Central nervous system; CFA: Complete Freund’s adjuvant; EAE: Experimental autoimmune encephalomyelitis; mAb: Monoclonal antibody; MHC: Major histocompatibility complex; MOG: Myelin oligodendrocyte glycoprotein; MS: Multiple sclerosis; NF-L: Neurofilament light; PBS: Phosphate-buffered saline; PCR: Polymerase chain reaction; RT: Reverse transcriptase; SCH: Spinal cord homogenate; SI: Stimulation index.
Competing interests
The authors have no competing interests.
Authors’ contributions
FP, BJvdS, and SA designed research, performed research, analyzed data, and wrote the manuscript. MV, MK, CB, RP-B, KU, GP, WG, RH, AR, PvdV, and DB performed research, analyzed data, and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ information
Prof. David Baker and Prof. Sandra Amor share the senior authorship.
Supplementary Material
Figure showing the pathogenicity of immunodominant NF-L epitopes. ABH mice (n = 5 per group) were immunized with NF-L peptides spanning the immunodominant regions (amino acids 241 to 288 and 169 to 208). Mice were immunized subcutaneously with 200 μg rmNF-L protein or NF-L peptides emulsified in complete Freund’s adjuvant containing Mycobacterium tuberculosis. Plots show the mean ± standard error of the mean daily clinical score.
Contributor Information
Fabiola Puentes, Email: f.puentes@qmul.ac.uk.
Baukje J van der Star, Email: b.vanderstar@vumc.nl.
Marion Victor, Email: mvictor@ukaachen.de.
Markus Kipp, Email: mkipp@ukaachen.de.
Cordian Beyer, Email: cbeyer@ukaachen.de.
Regina Peferoen-Baert, Email: r.peferoenbaert@vumc.nl.
Kimberley Ummenthum, Email: k.ummenthum@vumc.nl.
Gareth Pryce, Email: g.pryce@qmul.ac.uk.
Wouter Gerritsen, Email: w.gerritsen@vumc.nl.
Ruth Huizinga, Email: r.huizinga@erasmusmc.nl.
Arie Reijerkerk, Email: a.reijerkerk@vumc.nl.
Paul van der Valk, Email: p.vandervalk@vumc.nl.
David Baker, Email: david.baker@qmul.ac.uk.
Sandra Amor, Email: s.amor@vumc.nl.
Acknowledgements
The authors thank Bert van het Hof for technical assistance. This work was supported by Stichting MS Research, The Netherlands, grant number 07–627, the DANA foundation and by the MS Society of Great Britain and Northern Ireland, grant number NSCG-1F7R.
References
- Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162:1–11. doi: 10.1111/j.1365-2249.2010.04143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort JM, Bsibsi M, Gerritsen WH, van der Valk P, Bajramovic JJ, Steinman L, Amor S. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2010;69:694–703. doi: 10.1097/NEN.0b013e3181e4939c. [DOI] [PubMed] [Google Scholar]
- Huizinga R, Hintzen RQ, Assink K, van Meurs M, Amor S. T-cell responses to neurofilament light protein are part of the normal immune repertoire. Int Immunol. 2009;21:433–441. doi: 10.1093/intimm/dxp011. [DOI] [PubMed] [Google Scholar]
- Iorio R, Lennon VA. Neural antigen-specific autoimmune disorders. Immunol Rev. 2012;248:104–121. doi: 10.1111/j.1600-065X.2012.01144.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Da RR, Guo W, Ren HM, Hilgenberg LG, Sobel RA, Tourtellotte WW, Smith MA, Olek M, Gupta S, Robertson RT, Nagra R, Van Den Noort S, Qin Y. Axon reactive B cells clonally expanded in the cerebrospinal fluid of patients with multiple sclerosis. J Clin Immunol. 2005;25:254–264. doi: 10.1007/s10875-005-4083-5. [DOI] [PubMed] [Google Scholar]
- Huizinga R, Heijmans N, Schubert P, Gschmeissner S, Hart BA T, Herrmann H, Amor S. Immunization with neurofilament light protein induces spastic paresis and axonal degeneration in Biozzi ABH mice. J Neuropathol Exp Neurol. 2007;66:295–304. doi: 10.1097/nen.0b013e318040ad5c. [DOI] [PubMed] [Google Scholar]
- Huizinga R, Gerritsen W, Heijmans N, Amor S. Axonal loss and gray matter pathology as a direct result of autoimmunity to neurofilaments. Neurobiol Dis. 2008;32:461–470. doi: 10.1016/j.nbd.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Beltran E, Hernandez A, Lafuente EM, Coret F, Simo-Castello M, Bosca I, Perez-Miralles FC, Burgal M, Casanova B. Neuronal antigens recognized by cerebrospinal fluid IgM in multiple sclerosis. J Neuroimmunol. 2012;247:63–69. doi: 10.1016/j.jneuroim.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Derfuss T, Parikh K, Velhin S, Braun M, Mathey E, Krumbholz M, Kumpfel T, Moldenhauer A, Rader C, Sonderegger P, Pöllmann W, Tiefenthaller C, Bauer J, Lassmann H, Wekerle H, Karagogeos D, Hohlfeld R, Linington C, Meinl E. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci U S A. 2009;106:8302–8307. doi: 10.1073/pnas.0901496106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. 2004;14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel K, Melzer N, Herrmann AM, Schuhmann MK, Bittner S, Ip CW, Hunig T, Meuth SG, Wiendl H. Collateral neuronal apoptosis in CNS gray matter during an oligodendrocyte-directed CD8(+) T cell attack. Glia. 2010;58:469–480. doi: 10.1002/glia.20938. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Glutamate excitotoxicity – a mechanism for axonal damage and oligodendrocyte death in multiple sclerosis? J Neural Transm Suppl. 2000. pp. 375–385. [DOI] [PubMed]
- Woodroofe MN. Cytokine production in the central nervous system. Neurology. 1995;45:S6–S10. doi: 10.1212/wnl.45.6_suppl_6.s6. [DOI] [PubMed] [Google Scholar]
- Witherick J, Wilkins A, Scolding N, Kemp K. Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Dis. 2010;2011:164608. doi: 10.4061/2011/164608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, Reynolds R, Aloisi F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130:1089–1104. doi: 10.1093/brain/awm038. [DOI] [PubMed] [Google Scholar]
- Huizinga R, van der Star BJ, Kipp M, Jong R, Gerritsen W, Clarner T, Puentes F, Dijkstra CD, van der Valk P, Amor S. Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia. 2012;60:422–431. doi: 10.1002/glia.22276. [DOI] [PubMed] [Google Scholar]
- Al-Izki S, Pryce G, O'Neill JK, Butter C, Giovannoni G, Amor S, Baker D. Practical guide to the mouse induction of relapsing progressive experimental autoimmune encephalomyelitis in the Biozzi ABH. Mult Scler Relat Disord. 2012;1:29–38. doi: 10.1016/j.msard.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Amor S, O'Neill JK, Morris MM, Smith RM, Wraith DC, Groome N, Travers PJ, Baker D. Encephalitogenic epitopes of myelin basic protein, proteolipid protein, myelin oligodendrocyte glycoprotein for experimental allergic encephalomyelitis induction in Biozzi ABH (H-2Ag7) mice share an amino acid motif. J Immunol. 1996;156:3000–3008. [PubMed] [Google Scholar]
- Rankpep. [ http://imed.med.ucm.es/Tools/rankpep.html]
- Image J. http://rsb.info.nih.gov/ij/index.html]
- Braun A, Dang J, Johann S, Beyer C, Kipp M. Selective regulation of growth factor expression in cultured cortical astrocytes by neuro-pathological toxins. Neurochem Int. 2009;55:610–618. doi: 10.1016/j.neuint.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Butter C, O'Neill JK, Baker D, Gschmeissner SE, Turk JL. An immunoelectron microscopical study of the expression of class II major histocompatibility complex during chronic relapsing experimental allergic encephalomyelitis in Biozzi AB/H mice. J Neuroimmunol. 1991;33:37–42. doi: 10.1016/0165-5728(91)90032-3. [DOI] [PubMed] [Google Scholar]
- BCPred. [ http://ailab.cs.iastate.edu/bcpreds/]
- Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, Schramm J, Elger CE, Lassmann H. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol. 2002;51:311–318. doi: 10.1002/ana.10100. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR. Evidence linking neuronal cell death to autoimmunity in Alzheimer's disease. Brain Res. 2003;982:19–30. doi: 10.1016/S0006-8993(03)02881-6. [DOI] [PubMed] [Google Scholar]
- Benkler M, Agmon-Levin N, Hassin-Baer S, Cohen OS, Ortega-Hernandez OD, Levy A, Moscavitch SD, Szyper-Kravitz M, Damianovich M, Blank M, Chapman J, Shoenfeld Y. Immunology, autoimmunity, and autoantibodies in Parkinson's disease. Clin Rev Allergy Immunol. 2012;42:164–171. doi: 10.1007/s12016-010-8242-y. [DOI] [PubMed] [Google Scholar]
- Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, Lassmann H, Dornmair K, Kurschus FC, Liblau RS, Wekerle H. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med. 2009;15:626–632. doi: 10.1038/nm.1975. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Goodyer CG, Antel JP, Yong VW. Vulnerability of human neurons to T cell-mediated cytotoxicity. J Immunol. 2003;171:368–379. doi: 10.4049/jimmunol.171.1.368. [DOI] [PubMed] [Google Scholar]
- Meuth SG, Herrmann AM, Simon OJ, Siffrin V, Melzer N, Bittner S, Meuth P, Langer HF, Hallermann S, Boldakowa N, Herz J, Munsch T, Landgraf P, Aktas O, Heckmann M, Lessmann V, Budde T, Kieseier BC, Zipp F, Wiendl H. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci. 2009;29:15397–15409. doi: 10.1523/JNEUROSCI.4339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller N, Shriver LP, Bodiga VL, Ray A, Basu S, Ahuja R, Jana A, Pahan K, Dittel BN. Lymphocytes with cytotoxic activity induce rapid microtubule axonal destabilization independently and before signs of neuronal death. ASN Neuro. 2013;5:e00105. doi: 10.1042/AN20120087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaguia F, Saikali P, Ludwin S, Newcombe J, Beauseigle D, McCrea E, Duquette P, Prat A, Antel JP, Arbour N. Cytotoxic NKG2C + CD4 T cells target oligodendrocytes in multiple sclerosis. J Immunol. 2013;190:2510–2518. doi: 10.4049/jimmunol.1202725. [DOI] [PubMed] [Google Scholar]
- Zangi L, Zlotnikov Klionsky Y, Yarimi L, Bachar-Lustig E, Eidelstein Y, Shezen E, Hagin D, Ito Y, Takai T, Reich-Zeliger S, Lask A, Milstein O, Jung S, Shinder V, Reisner Y. Deletion of cognate CD8 T cells by immature dendritic cells: a novel role for perforin, granzyme A, TREM-1, and TLR7. Blood. 2012;120:1647–1657. doi: 10.1182/blood-2012-02-410803. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Baker D, O'Neill JK, Davison AN, Turk JL. Isolation and characterization of cells infiltrating the spinal cord during the course of chronic relapsing experimental allergic encephalomyelitis in the Biozzi AB/H mouse. Cell Immunol. 1993;146:335–350. doi: 10.1006/cimm.1993.1031. [DOI] [PubMed] [Google Scholar]
- Silber E, Semra YK, Gregson NA, Sharief MK. Patients with progressive multiple sclerosis have elevated antibodies to neurofilament subunit. Neurology. 2002;58:1372–1381. doi: 10.1212/WNL.58.9.1372. [DOI] [PubMed] [Google Scholar]
- Pryce G, O'Neill JK, Croxford JL, Amor S, Hankey DJ, East E, Giovannoni G, Baker D. Autoimmune tolerance eliminates relapses but fails to halt progression in a model of multiple sclerosis. J Neuroimmunol. 2005;165:41–52. doi: 10.1016/j.jneuroim.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Smith PA, Morris-Downes M, Heijmans N, Pryce G, Arter E, O'Neill JK, Hart TB, Baker D, Amor S. Epitope spread is not critical for the relapse and progression of MOG 8–21 induced EAE in Biozzi ABH mice. J Neuroimmunol. 2005;164:76–84. doi: 10.1016/j.jneuroim.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Liu GY, Baker D, Fairchild S, Figueroa F, Quartey-Papafio R, Tone M, Healey D, Cooke A, Turk JL, Wraith DC. Complete characterization of the expressed immune response genes in Biozzi AB/H mice: structural and functional identity between AB/H and NOD A region molecules. Immunogenetics. 1993;37:296–300. doi: 10.1007/BF00187458. [DOI] [PubMed] [Google Scholar]
- Miller A, Al-Sabbagh A, Santos LM, Das MP, Weiner HL. Epitopes of myelin basic protein that trigger TGF-beta release after oral tolerization are distinct from encephalitogenic epitopes and mediate epitope-driven bystander suppression. J Immunol. 1993;151:7307–7315. [PubMed] [Google Scholar]
- Fressinaud C, Eyer J. Axoskeletal proteins prevent oligodendrocyte from toxic injury by upregulating survival, proliferation, and differentiation in vitro. Neurochem Int. 2012;62:306–313. doi: 10.1016/j.neuint.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Thoua NM, van Noort JM, Baker D, Bose A, van Sechel AC, van Stipdonk MJ, Travers PJ, Amor S. Encephalitogenic and immunogenic potential of the stress protein alphaB-crystallin in Biozzi ABH (H-2A(g7)) mice. J Neuroimmunol. 2000;104:47–57. doi: 10.1016/S0165-5728(99)00246-5. [DOI] [PubMed] [Google Scholar]
- Bar-Or A, Vollmer T, Antel J, Arnold DL, Bodner CA, Campagnolo D, Gianettoni J, Jalili F, Kachuck N, Lapierre Y, Niino M, Oger J, Price M, Rhodes S, Robinson WH, Shi FD, Utz PJ, Valone F, Weiner L, Steinman L, Garren H. Induction of antigen-specific tolerance in multiple sclerosis after immunization with DNA encoding myelin basic protein in a randomized, placebo-controlled phase 1/2 trial. Arch Neurol. 2007;64:1407–1415. doi: 10.1001/archneur.64.10.nct70002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure showing the pathogenicity of immunodominant NF-L epitopes. ABH mice (n = 5 per group) were immunized with NF-L peptides spanning the immunodominant regions (amino acids 241 to 288 and 169 to 208). Mice were immunized subcutaneously with 200 μg rmNF-L protein or NF-L peptides emulsified in complete Freund’s adjuvant containing Mycobacterium tuberculosis. Plots show the mean ± standard error of the mean daily clinical score.