

Small-molecule drugs often interact with more than one protein in vivo. Recent estimates indicate that multi-target engagement occurs in up to ~ 80% of current drugs[1,2]. A complete understanding of such binding interactions and their related kinetics (dose, time) is important for a number of reasons. First, the continued development of newer drugs that are either more selective or inhibit multiple targets (polypharmacology) requires an understanding of binding partners in vivo. Second, although many successful drugs are in routine clinical use, their exact mechanism of action is still often poorly understood[3]. A better understanding of targeted proteins could also lead to the development of new drug candidates or be used to reduce toxicities. The problem is further complicated in that current drug screens are often performed on isolated proteins, established cell lines or homogeneous mouse models rather than heterogeneous cells harvested directly from patients. Third, a more thorough understanding of cognate binding partners is important in the development of companion imaging agents and diagnostic drugs.
For the majority of drugs and molecular imaging agents there does not exist a proteome-wide understanding of their behavior. This is not entirely surprising, however, given the technical difficulties of such analyses, the scant amounts of many proteins, and the fast decay of isotope-based imaging agents. Nevertheless, having the ability to obtain such data could provide strong clues toward mechanisms, suggest potential unrecognized actions, and/or aid in the interpretation of data. Mass spectrometry-based methods have emerged as an ideal technique to pinpoint protein targets and off-target effects for a particular drug. Activity-based protein profiling methods typically rely on covalent linkage of the inhibitor of interest to the protein targets to identify active enzyme targets [4–6]. However, the covalent modification could significantly alter the properties of the original drug. Alternative methods rely on secondary target pull-down from cell lysates. Proteins from cell lysates may have altered conformation or become denatured and no longer bind to the drug of interest, leading to an unintended underrepresentation of the true number of secondary targets of a drug[7]. SILAC is another highly sensitive method to identify drug targets but it is low throughput and expensive[8–10]. In contrast to activity-based protein profiling, compound-centric approaches provide an unbiased method to identify protein targets, regardless of their activation status[2,11]. These techniques have been used for a variety of clinically relevant inhibitors, such as Gefitinib and Imatinib, to assess their promiscuity[12]. However, one potential limitation of these methods is the immobilization of the inhibitor on an agarose or sepharose matrix, which could lead to an underrepresentation of potential targets by confining the inhibitor to a particular orientation[2]. More recent techniques have used a copper-catalyzed bioorthogonal click-chemistry reaction to label the drug and have used affinity beads for purification of secondary protein targets from live cells[5,13]. One limitation of this technique is the use of copper-catalyzed chemistry, which can lead to cell toxicity and could affect secondary targets that are identified. Another important issue is the recovery of captured proteins on solid support after bioorthogonal ligation reactions. Efficient recovery of the target protein is often carried out under harsh and denaturing conditions, which can lead to contamination by nonspecific captured materials and the loss of protein partners, structural information, and protein function. Several cleavable linkers have been applied in order to circumvent this limitation[14]. What is thus still lacking for the field is a simple method of drug-protein isolation prior to mass spectrometry analysis.
We hypothesized that transcyclooctene-tagged drug conjugates can be used to efficiently pull down target proteins through the use of complementary tetrazine beads. Here, we describe a non-covalent protein pull-down method using a model system (Olaparib (AZD2281), a PARP inhibitor) to identify protein targets (Figure 1). First, Olaparib was synthesized with a trans-cyclooctene (TCO) moiety and incubated with live cells. Protein bound drug was then pulled out from cell lysates using cleavable tetrazine (Tz) beads. Released protein was then separated on a SDS-PAGE gel, excised, digested and analyzed by mass spectrometry (Figure 1). Using this method, we were able to recover not only the intended primary target of Olaparib, PARP1, but also over a dozen previously unsuspected possible secondary binding proteins.
Figure 1.
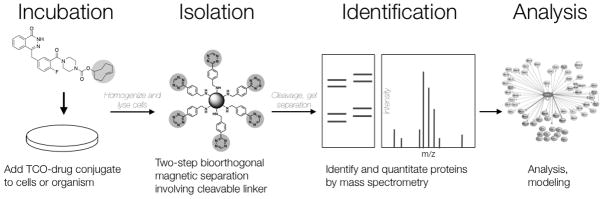
Overview of steps from drug administration to analysis involved in bioorthogonal proteomics. Live cells are incubated with a TCO-drug conjugate. Cell lysates are then prepared and TCO-drug bound to target protein is isolated using a Tz-labeled cleavable linker decorated on streptavidin magnetic beads. Protein is then run on an SDS-PAGE gel and desired bands are isolated and submitted for mass spectrometry analysis.
Olaparib (Scheme 1A) is an inhibitor of poly(ADP-ribose) polymerase 1 (PARP1), an important cellular protein that senses DNA damage and initiates the base excision repair pathway[15]. It has been shown that the 4-N-piperazine of Olaparib can be modified without significantly decreasing PARP1 binding affinity[16]. We therefore synthesized the 4-N-piperazine of Olaparib as described previously, with minor modification[17]. The TCO moiety was conjugated to the 4-N-piperazine position to generate Olaparib-TCO (Scheme 1B). To confirm that modification of Olaparib with TCO does not significantly alter the binding of the drug to PARP1, the inhibitory effect of Olaparib-TCO was evaluated against recombinant PARP1. Treatment with Olaparib-TCO resulted in an IC50 value of 35.8 nM, still in the nanomolar range but higher than the 7 nM IC50 obtained with unmodified Olaparib (Supplementary Figure 1A, Supplementary Table 1). To further confirm the specificity of the TCO-modified drug, we took advantage of the bioorthogonal chemistry and utilized carboxyfluorescein diacetate-tetrazine (CFDA-Tz) for localization of the drug by imaging. Olaparib-TCO, imaged with CFDA-Tz, localized to the nucleus (known location of PARP1) in MHH-ES1 Ewing’s sarcoma cells (Supplementary Figure 1B). In addition, an antibody against PARP1 showed similar nuclear localization (Supplementary Figure 1B). Additionally, Olaparib-TCO localized to some extent to the cytoplasm of these cells, indicating potential interaction with secondary Olaparib targets.
Scheme 1.

Synthetic scheme of Olaparib-TCO and the Tz-cleavable linker. (A) Olaparib for comparison (B) Olaparib-TCO and (C) Tz-cleavable linker. Key components shaded in grey.
To use bioorthogonal TCO/Tz chemistry for pull-down experiments, we designed and synthesized a cleavable enrichment linker 12 that contains a biotin affinity tag for enrichment on one end. The other end contained a Tz moiety for convenient scavenging of various TCO-labeled drugs through bioorthogonal chemistry. Between the two ends we incorporated a 2-(4′-hydroxy-2′-alkoxy phenylazo)benzoic acid as a cleavable site. This cleavable linker had previously been validated for protein pull down/release under very mild conditions[18]. For cleavage efficiency, the linker has a pegylated region to increase water solubility and a free ortho-carboxylic acid and free para-phenol group for reactivity (Scheme 1C). To synthesize the cleavable linker, a convergent approach was used to make the protected azo-arene 7 by a diazonium coupling between aniline 6 and resorcinol 3. The amine derivative 6 was obtained from the commercially available methyl 2-amino-5-bromo-benzoate. The bromoarene was first exchanged with a cyano group under reflux followed by hydrogenation, yielding the primary amine 5, which was then protected with an Fmoc group furnishing compound 6 in three steps. The Boc-protected resorcinol derivative 3 was prepared by coupling the resorcinol with a tetraethyleneglycol 2 spacer synthesized from the commercially available tetraethylene glycol monoamine. Diazotation of aniline 6 and reaction with phenol 3 gave the orthogonally protected linker 7 with 79% yield. The tetrazine reactive group was then introduced on one side by removing the Fmoc group using piperidine treatment, followed by ester hydrolysis and coupling of tetrazine-NHS on the free primary amine 9. Finally, the biotin enrichment tag was introduced on the other side by deprotecting the Boc-amino group, which was further coupled with biotin-NHS affording compound 12 in eleven steps. The cleavage kinetics of the final linker 12 was monitored by UV spectroscopy at 463 nm, which showed a half-life ≪1 s and a total cleavage time of 20 s with 1 mM dithionite solution. Under these conditions, no side products were observed and the total cleavage was confirmed by mass spectrometry (Supplementary Figure 2).
To test the utility of the Olaparib-TCO/Tz cleavable linker for protein pull-down, we used MHH-ES1 Ewing’s sarcoma cells that are sensitive to Olaparib and A2780 ovarian cancer cells that express high levels of PARP1. Live MHH-ES1 and A2780 cells were treated with Olaparib-TCO for 1 hour to allow for drug internalization and binding to its cellular primary and secondary targets. Negative control pull-down experiments were done on the same cell lines treated with DMSO. Cells were then washed with media to remove unbound drug, followed by cell lysis with a gentle lysis buffer. Lysates containing Olaparib-TCO labeled proteins were treated with streptavidin magnetic beads decorated with the biotin cleavable linker 12 (see Supporting Information). After 1 hour, small-molecule captured proteins were released from the beads by treatment with sodium dithionite, leaving the non-specifically bound proteins on the solid support. Analysis of non-specific cleavage was done by replacing the dithionite with buffer alone. Pull-down samples were then separated by SDS-PAGE followed by silver staining. Proteins specifically released by dithionite were excised from the gel, trypsinized and analyzed by LC/MS-MS for identification (Figure 2 and Supplementary Figure 3). We curated data by selecting hits that were i) repeatable during protein pull-down and ii) appeared in both tested cells lines (A2780 and MHH-ES1). We thus obtained a list of ~ a dozen proteins (Table 1). As expected, PARP1 was one of the top proteins that was identified in all experiments.
Figure 2.
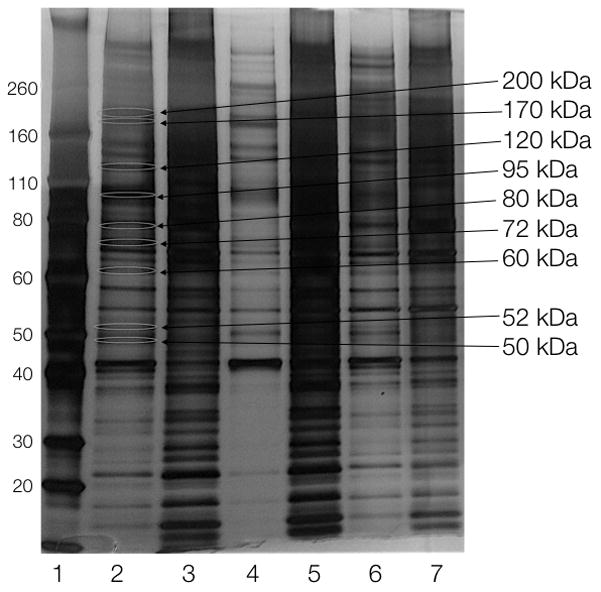
Silver-stained SDS-PAGE gel of the proteomics pull-down in A2780 cells. Lanes: 1) marker, 2) Olaparib-TCO, cleaved with 25 mM DT, 3) protein left on beads from 2, 4) Olaparib-TCO, cleaved with 0 mM DT, 5) protein left on beads from 4, 6) DMSO, cleaved with 25 mM DT, 7) protein left on beads from 6. Sizes on right indicate bands that were isolated for mass spectrometry analysis.
Table 1.
List of proteins identified in A2780 ovarian cancer cells (OV) and MHH-ES1 Ewing’s sarcoma cells (ES).
| Protein | Symbol | Confidence | Known Target | PARP1 complex | Cells | Molecular Function | |
|---|---|---|---|---|---|---|---|
| OV | ES | ||||||
| PARP1 | PARP1 | Very high | Yes | N/A | Yes | Yes | DNA binding/DNA damage repair |
| X-ray repair protein | XRCC5 | Very high | No | Yes | Yes | No | ATP/DNA/RNA-binding proteins (with PARP) |
| DNA topoisomerase 2 | TOP2B | Very high | No | Yes | Yes | No | DNA topological change |
| DNA topoisomerase 2 | TOP2A | Very high | No | No | Yes | No | DNA topological change |
| AP2 complex | AP2A1 | Very high | No | No | Yes | Yes | Transport protein (vesicles) |
| Terminal uridylyltransferase | TUT4 | Very high | No | No | No | Yes | miRNA biogenesis suppressor |
| YTH domain family protein | YTHD2 | Very high | No | No | Yes | No | signal transduction? |
| Tubulin | TBA1C | Very high | No | No | Yes | No | GTPase activity |
| Ras-GTP-ase | G3BP1 | High | No | No | Yes | No | ATP/DNA/RNA-binding proteins |
| Clip-associated protein | CLAP1 | High | No | No | Yes | No | Kinetochore binding |
| Clathrin heavy chain | CLH1 | High | No | No | Yes | No | Structural molecule activity |
| 60 S ribosomal protein | RL4 | High | No | No | Yes | Yes | RNA binding, constituent of ribosome |
| 60S ribosomal protein | RL5 | High | No | No | No | Yes | RNA binding, constituent of ribosome |
| Vimentin | VIME | High | No | No | Yes | No | Structural component of cytoskeleton |
| ATP synthase | ATPB | High | No | No | Yes | No | ATP synthesis |
| 78 kDa glucose reg protein | GRP78 | High | No | No | Yes | No | ATPase activity, ATP binding, ribosome binding |
| Lamina-associated polypeptide | LAP2A | High | No | No | No | Yes | Structural organization of the nucleus |
| Guanine nucleotide-binding protein | GBLP | High | No | No | No | Yes | Assembly and regulation of signaling molecules |
| Tropomyosin alpha-1 | TPM1 | High | No | No | No | Yes | Cytoskeleton |
| Alpha-enolase | ENOA | High | No | No | No | Yes | Glycolytic enzyme |
| Malate dehydrogenase, mito | MDHM | High | No | No | No | Yes | Citric acid cycle |
Beyond PARP1, little overlap was found when comparing the hits from the ovarian versus the Ewing’s sarcoma cell lines, which may arise from the differences in origin and protein expression between the two cell lines. Interestingly, neither cell line expresses PARP2, one of the other known PARPs targeted by Olaparib[19] (Supplementary Figure 4). We also identified several proteins predicted to interact with PARP1 based on previous work. For example XRCC5 and TOP2B were identified from the screen[20–21]. The remaining identified protein targets were grouped into categories based on the cellular function. The largest group of proteins were involved in maintaining cell structure (Vimentin, LAP2A, TBA1C, TPM1, CLH1, and CLAP1), while others were involved in the formation of signaling complexes (GBLP). Several proteins were involved in cellular metabolism (ATPB, GRP78, ENOA, and MDHM), which could affect tumor cell growth when inhibited by Olaparib. Finally, several proteins were involved in aspects of DNA or RNA binding (TOP2A, G3BP1, RL4, and RL5), which is where we began examining the identified secondary targets.
Validation of the identified targets requires biochemical analyses such as co-immunoprecipiation (to determine whether drugs are pulled out because of association with a protein complex), specific inhibitor assays or analyses in knock-in and knock-out models. For example, XRCC5 co-immunoprecipitated with PARP1 both in the absence and presence of Olaparib, suggesting that XRCC5 and PARP1 are present in a complex in A2780 cells, regardless of Olaparib treatment (Figure 3A). To further analyze one of the hits, we investigated topoisomerase (DNA) II alpha (TOP2A), an enzyme that controls and alters the topologic states of DNA during transcription. Immunoprecipitation experiments showed that the functional form of TOP2A (top band, Figure 3B) is not in a complex with PARP1 and thus may be a true secondary target of Olaparib (Figure 3B)[22]. However, additional experiments with a DNA relaxation assay[23] did not show any effects of Olaparib on the DNA unwinding enzymatic activity of TOP2A, as compared to the control (Figure 4). It is thus possible that Olaparib is bound to TOP2A, but does not alter its DNA unwinding activity. To explore this possibility, we performed surface plasmon resonance (SPR)[24] binding experiments with TOP2A (and PARP1 as a control) to determine the Kd of Olaparib-TCO binding. Using this method, we found that Olaparib-TCO does bind TOP2A, with an estimated Kd of 3.7 nM (PARP1 Kd is 22 nM, Supplementary Figure 5). These experiments demonstrate that targets can be individually worked up through classical biochemical assays. In the case of Olaparib, many of the identified targets do not yet have such functional assays firmly established (Table 1).
Figure 3.
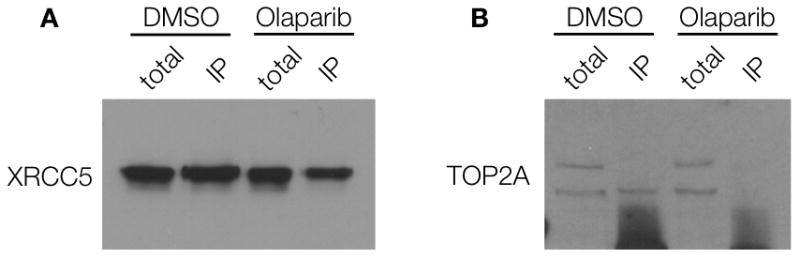
Co-immunoprecipitation (co-IP) in A2780 cells treated with 0.1% DMSO or 7 μM Olaparib for 1 hr. Cells were washed twice and incubated for 30 min to remove excess inhibitor. Cells were then lysed and incubated with PARP1 antibody-protein A magnetic beads. Following washing, protein complexes were eluted from the beads, boiled and run on a gel. Western blotting was done on 0.1% XRCC5 and 1% TOP2A total lysate or on the IP protein using antibodies against XRCC5 (A) or TOP2A (B).
Figure 4.
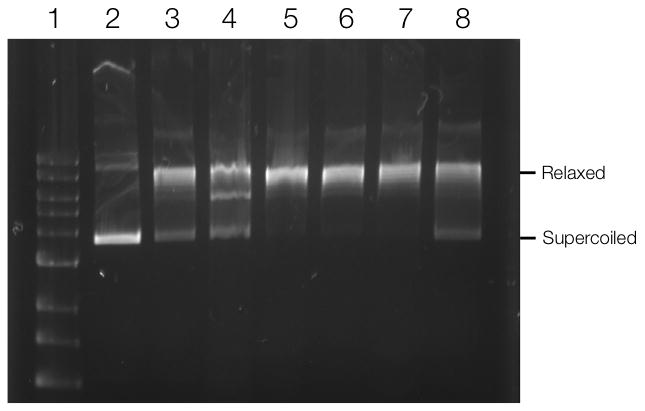
TOP2A DNA relaxation assay. TOP2A was incubated with pAcGFP1 DNA for 30 min at 37°C. The reaction was stopped using SDS and protein was digested by proteinase K. (1) 1kb DNA ladder; (2) pAcGFP1 DNA; (3) DNA incubated with TOP2A; DNA incubated with TOP2A and (4) 100 μM Etoposide; (5) 500 nM Olaparib, (6) 1 μM Olaparib; (7) 10 μM Olaparib; (8) 100 μM Olaparib.
The described method has a number of advantages. It is fast, sensitive, and relatively inexpensive to perform, as it does not use radioactivity or stable isotope labeling. It can be readily applied to live cells or whole organism[33], as the adducts are cell membrane permeable. While not specifically addressed here, work from others has shown that live cell compatibility of the bioorthogonal components may be important in certain cases when the inhibitor-binding ability of the target protein is different between live cells and cell lysates[7]. Previous work has also shown that the TCO reacts very rapidly and specifically with Tz, making bioorthogonal chemistry a suitable choice for proteomic pull-down assays[30].
The mild conditions used for pull-down and protein release in this method allow for capture of protein complexes, thus avoiding the use of photoaffinity-labeling methods and reducing non-specific labeling of proteins. While the precise Kd requirements for this method are not yet known, comparing this method with covalent-labeling methods in the future may provide an even more complete picture of the extremely weak to extremely tight binding secondary targets. Because of the simplicity of this method, it is easy to change variables (e.g. cell lines, doses, timing, modified compounds) to derive important biological data. Unlike drug screens against purified proteins, this method allows unbiased screens and focuses on proteins relevant in certain cells.
We anticipate that the described technique has a number of future applications. While cell based screens can result in a detailed picture of protein interaction in a clean model system (constant TCO source), it will be equally interesting to use the approach for in vivo screens. Such experiments would inform one on differences in drug binding between in vitro and in vivo settings (variable delivery, pharmacokinetics), and perhaps shed light on the validity of in vitro experiments to inform on in vivo behavior. In vivo screens would also allow drug binding to be profiled in different tissues (e.g. cancer versus normal organs (e.g. liver, kidney) in an effort to identify drug toxicities and off-target effects. Finally, the method may become useful in studying the biology of multi-target polypharmacologic drugs.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant number RO1CA164448 and P50CA86355, K.Y. was supported by a NIH grant T32-CA79443.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Jalencas X, Mestres J. Med Chem Commun. 2013;4:80. [Google Scholar]
- 2.Rix U, Superti-Furga G. Nat Chem Biol. 2009;5:616. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 3.Mitchison TJ. Mol Biol Cell. 2012;23:1. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cravatt BF, Sorensen EJ. Curr Opin Chem Biol. 2000;4:663. doi: 10.1016/s1367-5931(00)00147-2. [DOI] [PubMed] [Google Scholar]; Adam GC, Sorensen EJ, Cravatt BF. Mol Cell Proteomics. 2002;1:781. doi: 10.1074/mcp.r200006-mcp200. [DOI] [PubMed] [Google Scholar]
- 5.Yang PY, Liu K, Ngai MH, Lear MJ, Wenk MR, Yao SQ. J Am Chem Soc. 2010;132:656. doi: 10.1021/ja907716f. [DOI] [PubMed] [Google Scholar]
- 6.Krysiak JM, Kreuzer J, Macheroux P, Hermetter A, Sieber SA, Breinbauer R. Angew Chem Int Ed Engl. 2012;51:7035. doi: 10.1002/anie.201201955. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nomura DK, Dix MM, Cravatt BF. Nat Rev Cancer. 2010;10:630. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Speers AE, Cravatt BF. Chem Biol. 2004;11:535. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]; Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 8.Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, Mann M. Nat Protoc. 2011;6:147. doi: 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- 9.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 10.Ong SE, Mann M. Nat Protoc. 2006;1:2650. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 11.Schenone M, Dancik V, Wagner BK, Clemons PA. Nat Chem Biol. 2013;9:232. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brehmer D, et al. Cancer Res. 2005;65:379. [PubMed] [Google Scholar]; Bach S, et al. J Biol Chem. 2005;280:31208. doi: 10.1074/jbc.M500806200. [DOI] [PubMed] [Google Scholar]; Wan Y, et al. Chem Biol. 2004;11:247. doi: 10.1016/j.chembiol.2004.01.015. [DOI] [PubMed] [Google Scholar]; Bantscheff M, et al. Nat Biotechnol. 2007;25:1035. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]; Rix U, et al. Blood. 2007;110:4055. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]; Remsing Rix LL, et al. Leukemia. 2009;23:477. doi: 10.1038/leu.2008.334. [DOI] [PubMed] [Google Scholar]
- 13.Shi H, Zhang CJ, Chen GY, Yao SQ. J Am Chem Soc. 2012;134:3001. doi: 10.1021/ja208518u. [DOI] [PubMed] [Google Scholar]
- 14.Leriche G, Chisholm L, Wagner A. Bioorg Med Chem. 2012;20:571. doi: 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- 15.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. Nucleic Acids Res. 2003;31:5526. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menear KA, et al. J Med Chem. 2008;51:6581. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 17.Reiner T, Earley S, Turetsky A, Weissleder R. Chembiochem. 2010;11:2374. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budin G, Moune-Dimala M, Leriche G, Saliou JM, Papillon J, Sanglier-Cianferani S, Van Dorsselaer A, Lamour V, Brino L, Wagner A. Chembiochem. 2010;11:2359. doi: 10.1002/cbic.201000574. [DOI] [PubMed] [Google Scholar]
- 19.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell Å, Ekblad T, Öncü D. Nature biotechnology. 2012;30:283. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 20.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. Mol Cell Biol. 1998;18:3563. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ariumi Y, Masutani M, Copeland TD, Mimori T, Sugimura T, Shimotohno K, Ueda K, Hatanaka M, Noda M. Oncogene. 1999;18:4616. doi: 10.1038/sj.onc.1202823. [DOI] [PubMed] [Google Scholar]
- 21.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. Science Signaling. 2006;312:1798. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 22.Nitiss JL. Nat Rev Cancer. 2009;9:327. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gellert M. Annu Rev Biochem. 1981;50:879. doi: 10.1146/annurev.bi.50.070181.004311. [DOI] [PubMed] [Google Scholar]
- 24.Tassa C, Liong M, Hilderbrand S, Sandler JE, Reiner T, Keliher EJ, Weissleder R, Shaw SY. Lab Chip. 2012;12:3103. doi: 10.1039/c2lc40337d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budin G, Yang KS, Reiner T, Weissleder R. Angew Chem Int Ed Engl. 2011;50:9378. doi: 10.1002/anie.201103273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang KS, Budin G, Reiner T, Vinegoni C, Weissleder R. Angew Chem Int Ed Engl. 2012;51:6598. doi: 10.1002/anie.201200994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiner T, Lacy J, Keliher EJ, Yang KS, Ullal A, Kohler RH, Vinegoni C, Weissleder R. Neoplasia. 2012;14:169. doi: 10.1593/neo.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devaraj NK, Weissleder R, Hilderbrand SA. Bioconjug Chem. 2008;19:2297. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauer J, Mielert A, Lang D, Peter D. Chemische Berichte. 1965;98:1435. [Google Scholar]; Balcar J, Chrisam G, Huber FX, Sauer J. Tetrahedron Lett. 1983;24:1481. [Google Scholar]
- 30.Karver MR, Weissleder R, Hilderbrand SA. Bioconjug Chem. 2011;22:2263. doi: 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jewett JC, Bertozzi CR. Chem Soc Rev. 2010;39:1272. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Davis L, Chin JW. Nat Rev Mol Cell Biol. 2012;13:168. doi: 10.1038/nrm3286. [DOI] [PubMed] [Google Scholar]
- 32.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Nat Nanotechnol. 2010;5:660. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]; Haun JB, Castro CM, Wang R, Peterson VM, Marinelli BS, Lee H, Weissleder R. Sci Transl Med. 2011;3:71ra16. doi: 10.1126/scitranslmed.3002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T, Weissleder R. Nat Commun. 2013;4:1504. doi: 10.1038/ncomms2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.