

Abstract
Objective
Recent postmortem studies reveal degenerative changes, including Purkinje cell (PC) loss, in most brains from individuals with essential tremor (ET). Heterotopic PCs (HPCs) (ie, PC bodies displaced into the molecular layer) may be found in neurodegenerative diseases with PC loss. HPCs have been observed in ET but no quantitative case control analysis has been performed.
Methods
HPCs were counted in 35 ET brains and 32 control brains (including 21 non-diseased controls and 11 diseased controls with progressive supranuclear palsy (PSP)) using a standard 20×25 mm cerebellar cortical section stained with a modified Bielscholwsky method.
Results
The median number of HPCs per section was three times higher in 35 ET cases (median 3, mean±SD 3.8±3.6, range 0–14) versus 32 controls (median 1, mean±SD 1.6±1.7, range 0–5) (p=0.007). The number of HPCs was similarly low in the 21 non-diseased controls and 12 PSP brains (median 1 in each group) (p=0.04 and p=0.01 compared with ET). In ET, the number of HPCs was inversely related to the number of PCs (Spearman's rho −0.36, p=0.038) (ie, cases with more HPCs had fewer PCs).
Conclusion
PC heterotopia, which occurs in cerebellar degenerative disorders, is also a feature of ET. These findings further contribute to our understanding of the postmortem changes in this common neurological disease.
Introduction
Essential tremor (ET) is one of the most common neurological diseases.1,2 Clinical and neuroimaging data suggest a cerebellar abnormality in ET.3,4 In recent controlled postmortem studies, ET cases had degenerative structural changes in the cerebellum, including a 6–7-fold increase in Purkinje cell (PC) axonal swellings (torpedoes) and a 30–40% reduction in the number of PCs.5,6
In the normal human cerebellum, PCs form a monolayer between the molecular and granular layers. The proper anatomical location of different cell types within each layer is required for normal cerebellar function. Heterotopic PCs (HPCs) are PCs where the cell body is mislocalised in the molecular layer. The significance of HPCs is unknown but HPCs are typically observed in either neurodevelopmental or neurodegenerative disorders.7–9 In cerebellar degenerative disorders such as spinocerebellar ataxia type 1 or 6, HPCs occur along with PC loss and torpedoes.8,9 HPCs have been observed in ET cases,5 but no systematic quantitative case control analysis has been performed so far. As ET could be a neurodegenerative disorder,10 we performed these controlled analyses to determine whether increased numbers of HPCs might be another hallmark of neurodegeneration in ET.
Methods
Brain repository and study subjects
The study was conducted at the Essential Tremor Centralised Brain Repository (ETCBR), Columbia University, New York. Postmortem cerebellar tissue was obtained from 36 ET cases and compared with both diseased and non-diseased controls (n=35). The diseased controls (n=12) had progressive supranuclear palsy (PSP). We specifically chose PSP brains for several reasons. Firstly, PSP is another neurodegenerative disorder and secondly, PSP is also characterised by structural changes in the cerebellum, including PC loss. All brains had complete neuropathological assessment as previously described.5
The clinical diagnosis of ETwas made by treating neurologists and confirmed by a neurologist specialising in movement disorders (EDL) using ETCBR clinical criteria.5 Non-diseased control brains were from individuals followed at the Alzheimer's Disease (AD) Research Centre or the Washington Heights Inwood Columbia Aging Project. They were followed prospectively with serial neurological examinations and were clinically free of AD, ET, Parkinson's disease, diffuse Lewy body disease, or PSP. PSP diagnoses were confirmed at postmortem based on National Institute of Neurological Disorders and Stroke criteria for PSP.11
We selected all available ET brains for study. Rather than matching by age or gender, non-diseased controls were selected to maximise the number with low PC counts (ie, to make them as similar as possible to the cases with regards to PC count) because we hypothesised that the presence of HPCs would co-vary with PC counts. The 12 available PSP brains were all selected.
Tissue processing and quantification of HPCs
A standard 3×20×25 mm parasagittal neo-cerebellar cortex tissue block was harvested from each brain. Paraffin sections (7 μm thick) were stained with Luxol Fast Blue haematoxylin and eosin or with a modified Bielschowsky silver method, as described previously.5 Each brain had standardised measurement of brain weight (grams), postmortem interval (hours between death and placement of brain in a cold room or on ice), Braak and Braak AD stage for neurofibrillary tangles12 and Consortium to Establish a Registry for AD (CERAD) ratings for neuritic plaques.13 PCs were counted as described previously5; briefly, PCs in five 100× fields (Luxol Fast Blue haematoxylin and eosin) were counted and summed. As in prior postmortem studies,7–9 a HPC was identified as a PC whose cell body was completely surrounded by the molecular layer without contacting the granular layer (figure 1). A trained physician (SHK), who was blinded to the clinical and diagnostic data, quantified HPCs using the standard Biel-schowsky stained section. In each case and control, the total number of HPCs was counted in the standard section.
Figure 1.
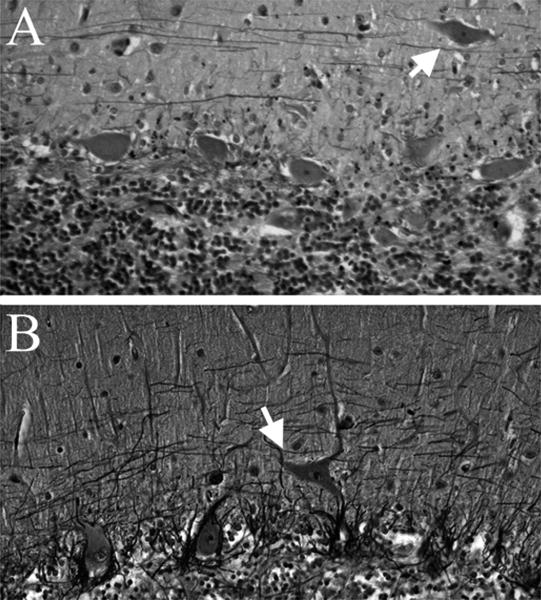
Heterotopic Purkinje cell (HPC) bodies (white arrows) are located in the molecular layer in two essential tremor brains. Orthotopic PCs are seen in the PC layer, below the molecular layer. Bielschowsky stained cerebellar cortical sections, 200× (A, B).
Data analyses
Analyses were performed in SPSS (V.17.0). There were four outliers (one ET, two non-diseased controls and one PSP) whose HPC counts (50, 33, 41 and 31) were ≥9 SDs above the mean of the remaining 67 brains. These four outliers were excluded from the primary analyses. In a secondary analysis, we included these four outliers to confirm the results of our primary analyses.
Demographic and clinical characteristics of ET cases and controls were compared using the Student's t and χ2 tests. The number of HPCs did not follow a normal statistical distribution (Kolmogorov–Smirnov test=2.51, p<0.001) even after exclusion of outliers (Kolmogorov–Smirnov test=1.86, p=0.002); therefore, medians were presented, and scores were compared using a non-parametric approach (Mann–Whitney test, Spearman's rho). Linear regression analyses were not possible because the number of HPCs did not follow a normal statistical distribution. Therefore, to assess the effects of possible confounders (age, gender, Braak AD stage, brain weight, postmortem interval and CERAD plaque score,), stratified analyses were performed.
Results
There were 36 ET cases and 35 controls (including 23 non-diseased controls and 12 PSP). After removing the four outliers, there were 35 ET cases and 32 controls (21 non-diseased controls and 11 PSP).
The remaining 35 ET cases and 32 controls differed with respect to age, gender, Braak AD stage and PC count but not brain weight, postmortem interval or CERAD plaque score (table 1). Importantly, the number of HPCs was not associated with age, gender, brain weight, postmortem interval, Braak AD stage or CERAD plaque score, so that case control differences in these variables could not have accounted for the observed case control difference in the HPC count and, furthermore, matching based on these variables was not necessary.
Table 1. Demographic characteristics of essential tremor cases and controls.
| ET | All controls (non-diseased controls and PSP) | Non-diseased controls | Diseased controls (PSP) | |
|---|---|---|---|---|
| N‡ | 35 | 32 | 21 | 11 |
| Age (years) | 84.9±6.5 | 74.5±13.6† | 74.9±16.2* | 73.7±7.1† |
| Female gender | 22 (62.9) | 11 (35.5)* | 7 (33.3)* | 4 (36.4) |
| Brain weight (g) | 1205±146 | 1245±176 | 1301±178* | 1137±117* |
| Postmortem interval (h) | 5.9±3.0 | 8.5±7.9 | 8.7±8.4 | 8.2±7.5 |
| Braak AD stage | 2.1±1.1 | 1.0±1.3† | 0.8±1.4† | 1.3±0.9† |
| CERAD plaque score§ | 0.9±1.1 | 0.4±0.7 | 0.4±0.8 | 0.4±0.7 |
| PC count | 35.1±11.7 | 43.2±17.8* | 47.9±17.9† | 32.3±12.2 |
| HPCs | 3.8±3.6, 3 (0–14) | 1.6±1.7, 1 (0–5)† | 1.9±1.8, 1 (0–5)* | 1.1±1.2, 1 (0–4)* |
Mean±SD and frequency (%) are reported.
p <0.05.
p <0.01, comparing each group to ET cases.
Four outliers were excluded.
CERAD plaque scores were converted from letters to numbers: 0 indicates absent; 1, sparse; 2, moderate; and 3, severe.
AD, Alzheimer's disease; CERAD, Consortium to Establish a Registry for AD; ET, essential tremor; HPC, heterotopic Purkinje cell; PC, Purkinje cell; PSP, progressive supranuclear palsy.
The median number of HPCs was three times higher in ET cases (median 3, mean±SD 3.8±3.6, range 0–14) than controls (median 1, mea±SD 1.6±1.7, range 0–5) (Mann–Whitney, p=0.007) (table 1). Including the four outliers produced similar results (medians 3.5 vs 1.0, Mann–Whitney p=0.039). The number of HPCs was similarly low in both non-diseased controls and PSP (median 1 in each group) (in Mann–Whitney comparison of ET cases to non-diseased controls, p=0.04; and in Mann–Whitney comparison of ET cases to PSP, p=0.01). In strata defined by age, gender, brain weight, postmortem interval, Braak AD stage and CERAD plaque score, the median number of HPCs was higher in ET cases than controls in each stratum (data not shown), further indicating that these factors did not confound the observed association between number of HPCs and diagnosis.
In ET cases, the number of HPCs was inversely related to the number of PCs (Spearman's rho −0.36, p=0.038) (ie, cases with higher HPC counts had fewer PCs).
In ET cases, we assessed whether the number of HPCs was associated with a range of clinical features. HPC numbers did not correlate with presence/absence of head tremor (Mann–Whitney z=0.04, p=0.98), rest tremor (Mann–Whitney z=0.06, p=0.95), family history of ET in a first degree relative (Mann–Whitney z=0.74, p=0.48), age of tremor onset (Spearman's r=−0.03, p=0.89) or tremor duration (Spearman's r=0.07, p=0.74). ET cases with voice tremor had a marginally higher HPC number than ET cases without voice tremor (respective medians 5.0 vs 2.0, Mann–Whitney z=2.01, p=0.048). We also assessed whether cancer or exposure to chemotherapeutic agents was associated with HPC number, finding that HPC number was similar among (1) ET cases with cancer who had been treated with chemotherapy (n=5), (2) ET cases with cancer but no exposure to these agents (n=10) and (3) ET cases without cancer (Kruskal–Wallis, p=0.32).
Discussion
We observed more PC heterotopias in ET cases than in non-diseased control brains and PSP brains. PCs form a monolayer between the granular and molecular layers of the cerebellar cortex. Occasionally, PC bodies are displaced into the molecular layer.7–9 These HPCs can be either a neurodevelopmental or neurodegenerative phenomenon. In the setting of defective neurodevelopment, HPCs usually occur in large clusters (ie, groupings of numerous PCs which we did not observe in our brains).7 However, in a neurodegenerative process, HPCs do not form clusters and can be observed with other degenerative changes, including torpedoes and PC loss.8,9 Neurodegenerative disorders such as spinocerebellar ataxia 1 or 6 are characterised by PC heterotopia.8,9 In murine models of PC death, weaver and staggerer mice both exhibit PC loss with heterotopia.14
The mechanism underlying PC heterotopia in the setting of neurodegeneration is not clear. The cerebellar molecular layer is occupied by PC dendrites, climbing fibres from the inferior olivary nucleus, parallel fibres from granule cells, interneurons, Bergmann glia and glia. Each of these structures is dynamically regulated. Climbing fibres can change innervation patterns dramatically in response to PC loss. Parallel fibres and PC dendrites may also regress during neurodegeneration. Astrocytes and microglia undergo morphological and functional changes during neurodegenerative processes and contribute to non-autonomous neuronal death. These changes can result in the remodelling of cerebellar structures, leading to defective PC body localisation.
It is interesting that the number of HPCs was increased in ET but not in PSP, considering both diseases are characterised by degenerative changes in the cerebellum, including PC loss. ET is a slowly progressive disorder, and we hypothesise that there may be a longstanding reorganisation of structures in the cerebellum in response to PC loss, which is important in tremor development. The fact that the number of HPCs correlates inversely with the number PCs further supports such a model in ET. In contrast, PSP patients have rapid deterioration and perhaps a less significant reorganisation of structures in the cerebellum. PC heterotopia is one possible hallmark of the reorganisation process of the cerebellum in ET. Future work should focus on the reorganisation of the molecular layer, including parallel fibres, glial cells and interneurons, to elucidate the mechanisms of ET.
Acknowledgments
Funding R01 NS42859 from the National Institutes of Health (Bethesda, Maryland), the Arlene Bronstein Essential Tremor Research Fund (Columbia University) and the Claire O'Neil Essential Tremor Research Fund (Columbia University).
Footnotes
Competing interests None.
Ethics approval This study was conducted with the approval of Columbia University.
Contributors Statistical analyses were conducted by EDL.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Dogu O, Sevim S, Camdeviren H, et al. Prevalence of essential tremor: door-to-door neurologic exams in Mersin Province, Turkey. Neurology. 2003;61:1804–6. doi: 10.1212/01.wnl.0000099075.19951.8c. [DOI] [PubMed] [Google Scholar]
- 2.Benito-Leon J, Louis ED. Essential tremor: emerging views of a common disorder. Nat Clin Pract Neurol. 2006;12:666–78. doi: 10.1038/ncpneuro0347. [DOI] [PubMed] [Google Scholar]
- 3.Colebatch JG, Findley LJ, Frackowiak RS, et al. Preliminary report: activation of the cerebellum in essential tremor. Lancet. 1990;336:1028–30. doi: 10.1016/0140-6736(90)92489-5. [DOI] [PubMed] [Google Scholar]
- 4.Jenkins IH, Bain PG, Colebatch JG, et al. A positron emission tomography study of essential tremor: evidence for overactivity of cerebellar connections. Ann Neurol. 1993;34:82–90. doi: 10.1002/ana.410340115. [DOI] [PubMed] [Google Scholar]
- 5.Louis ED, Faust PL, Vonsattel JP, et al. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain. 2007;130:3297–307. doi: 10.1093/brain/awm266. [DOI] [PubMed] [Google Scholar]
- 6.Axelrad JE, Louis ED, Honig LS, et al. Reduced Purkinje cell number in essential tremor: a postmortem study. Arch Neurol. 2008;65:101–7. doi: 10.1001/archneurol.2007.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura R, Kurita K, Kawanami T, et al. An immunohistochemical study of Purkinje cells in a case of hereditary cerebellar cortical atrophy. Acta Neuropathol. 1999;97:196–200. doi: 10.1007/s004010050974. [DOI] [PubMed] [Google Scholar]
- 8.Yamada M, Sato T, Tsuji S, et al. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:71–86. doi: 10.1007/s00401-007-0287-5. [DOI] [PubMed] [Google Scholar]
- 9.Gomez CM, Thompson RM, Gammack JT, et al. Spinocerebellar ataxia type 6: gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann Neurol. 1997;42:933–50. doi: 10.1002/ana.410420616. [DOI] [PubMed] [Google Scholar]
- 10.Louis ED. Essential tremors: a family of neurodegenerative disorders? Arch Neurol. 2009;66:1202–8. doi: 10.1001/archneurol.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–19. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer's disease: a commentary. Neurobiol Aging. 1997;18:S91–4. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong C, Hawkes R. Selective Purkinje cell ectopia in the cerebellum of the weaver mouse. J Comp Neurol. 2001;439:151–61. doi: 10.1002/cne.1339. [DOI] [PubMed] [Google Scholar]