

Abstract
The primary structure of the opioid receptors have revealed that many of the structural features that are conserved in other G protein-coupled receptors are also conserved in the opioid receptors. Upon exposure to agonists, some G protein-coupled receptors internalize rapidly, whereas other structurally homologous G protein-coupled receptors do not. It is not known whether opioid receptors are regulated by rapid endocytosis. In transfected Chinese hamster ovary cells expressing the epitope-tagged wild type δ opioid receptor, exposure to 100 nM [D-Ala2,D-Leu5]enkephalin causes internalization of the receptor within 30 min as determined by confocal microscopy. The rate of internalization of the wild type receptor is rapid with a half-maximal reduction by about 10 min, as determined by the reduction in mean surface receptor fluorescence intensity measured using flow cytometry. In contrast, the cells expressing receptors lacking the C-terminal 15 or 37 amino acids exhibit a substantially slower rate of internalization. Furthermore, the cells expressing receptors with point mutations of any of the Ser/Thr between Ser344 and Ser363 in the C-terminal tail exhibit a significant reduction in the rate of receptor internalization. These results suggest that a portion of the C-terminal tail is involved in receptor internalization. Agents that block the formation of clathrin-coated pits considerably reduce the extent of agonist-mediated internalization of the wild type receptor. Taken together, these results suggest that the wild type opioid receptor undergoes rapid agonist-mediated internalization via a classic endocytic pathway and that a portion of the C-terminal tail plays an important role in this internalization process.
Exposure to opiates causes decreased sensitivity to the drug. This receptor desensitization is a process whereby continuous or repeated exposure to a high concentration of the opiates results in a reduced cellular response (1). The mechanism of desensitization of the opiate receptor is thought to be similar to the mechanism of desensitization of the well explored prototypic β2-adrenergic receptor (β2AR)1 (2). Rapid desensitization is thought to result from the alterations in the receptor conformation that interferes with its coupling to a G protein and by sequestration and internalization of the receptor into intracellular compartments. Longer desensitization is thought to be due to the receptor down-regulation with a net loss of binding sites within the cell (3). The cellular mechanisms involved in this desensitization process of the opioid receptors or the intracellular compartments involved in this process have not been well established.
The molecular cloning of the cDNA encoding opioid receptors has made the studies to address the mechanism of receptor desensitization feasible. The primary structure of the opioid receptors has revealed that they are members of “G protein-coupled receptor (GPCR) family” (4, 5). Many of the structural features that are conserved in other GPCRs are found in the opioid receptors; these include consensus N-linked glycosylation sites near the N terminus, a palmitoylation site in the C-terminal tail, disulfide bonds in the extracellular loop between the third and fourth transmembrane domain, and sites for phosphorylation in the C-terminal tail and in the first and third intracellular domain (6). In the case of other GPCRs, C-terminal tail of the receptor is shown to be phosphorylated, and this is implicated in receptor desensitization.
Mutational analysis of many members of the GPCR family provide evidence that the third cytoplasmic loop and the C-terminal tail are involved in the coupling of the membrane receptors with intracellular G proteins (2, 3). The same regions are shown to be involved in the control of receptor sequestration and internalization. A consensus sequence in the receptor for G protein coupling or for receptor internalization have not yet been clearly identified among the cloned GPCRs. It is thought that the membrane proximal regions of the third intracellular loop of these receptors are required for coupling, and multiple regions in the cytoplasmic loops and C-terminal tail are involved in the receptor internalization (7). The domain involved in the G protein coupling or the receptor sequestration/internalization of the opioid receptor is not known.
In order to address the questions regarding agonist-induced internalization of the opioid receptor, we have used mutants of the δ opioid receptor cDNA stably expressed in Chinese hamster ovary (CHO) cells. We have generated two types of mutant receptors, (i) deletion mutants lacking various portions of C-terminal tail or (ii) point mutants of the various Ser or Thr in the C-terminal tail. We used antisera against the epitope-tagged receptor and immunocytochemical techniques to examine the internalization process of the opioid receptor. We find that the opioid receptor undergoes rapid agonist-induced internalization, and a portion of the C-terminal tail is involved in this process. Moreover, mutation of any of the Ser/Thr between Thr352 and Ser363 in the C-terminal tail substantially reduces the agonist-induced internalization. Interestingly, many of these mutations (except for Thr353 → Ala) do not substantially affect the agonist-induced down-regulation. Taken together, these data suggest that a region in the C-terminal tail of the receptor plays an important role in the internalization process and provides evidence that overlapping but distinct regions are involved in the rapid agonist-induced internalization and the slow agonist-induced down-regulation of the receptor.
EXPERIMENTAL PROCEDURES
Generation of Mutants of the δ Opioid Receptor
Flag epitope-tagged mouse δ opioid receptor cDNA subcloned into pCDNA3 was used to generate the deletion mutants, ΔC15 and ΔC37 as described previously (8). ΔC7 is generated by using a polymerase chain reaction to amplify regions of the receptor from Thr211 to Gly365; amino acid numbering is adopted from the numbering of mouse δ opioid receptor (4). The point mutations were generated by oligonucleotide-directed mutagenesis using an Altered Sites-II in vitro mutagenesis kit from Promega (Madison, Wi) according to the manufacturer’s directions. The nucleotide sequence was confirmed by double-stranded DNA sequencing (9). The resulting C-terminal truncations and point mutations are shown in Fig. 1.
Fig. 1. Schematic representation of the C-terminal tail of wild type and mutant δ opioid receptors.
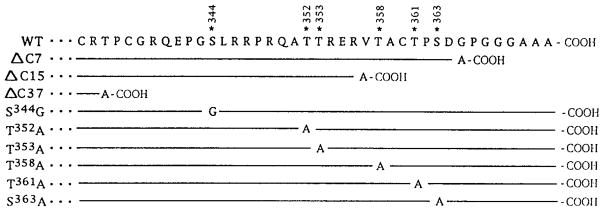
The C-terminal tail residues 333–372 of the wild type receptor is in a single-letter amino acid code. The asterisks point to the residues selected for generating mutants, and the numbers indicate the amino acid positions; the numbering is according to Evans et al. (4). The amino acid sequence of the mutants identical to the wild type are represented by a line and the changes are as indicated.
Approximately 3 × 105 CHO cells were transfected with the Qiagen-purified plasmid DNA using Lipofectin as described previously (8). Approximately 24–48 stable colonies expressing varying levels of the mutant receptors were selected. The determination of the receptor expression by binding of [3H]diprenorphine to whole cells was carried out as described previously (8). Specific binding is defined as the difference between the radioactivity bound to the cells in the absence and in the presence of 10 μM diprenorphine.
Binding Assay
Approximately 106 cells were incubated with [3H]di-prenorphine for 20 min in 0.5 ml of Krebs-Ringer-HEPES buffer, pH 7.4, at 37 °C or at 4 °C without or with the unlabeled diprenorphine. The cells were collected on Whatman GF-B filters and washed extensively with 50 mM Tris-Cl, pH 7.4. The radioactivity on the filters were determined after an overnight incubation of filters in Biosafe scintillation fluid (Beckman). Kd and Bmax values were determined by Scatchard analysis using the Ligand program.
Cell Staining and Immunofluorescence Microscopy
CHO cells stably transfected with wild type or mutant receptors were grown on coverslips and were treated without or with 100 nM DADLE for 30 min. Following incubation, the cells were washed with ice-cold 20 mM Tris-Cl, pH 7.5, containing 150 mM NaCl and 1 mM CaCl2 (TBS), fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS). Fixed cells were washed with TBS, permeabilized, and blocked with 0.1% Triton X-100 in Blotto (3% nonfat dry milk in 50 mM Tris-Cl, pH 7.5). Cells were incubated for 1 h at room temperature with 10 μg/ml primary antibody (anti-FLAG M1, Kodak/IBI) diluted in Blotto, washed with TBS, incubated for 30 min with 2 μg/ml fluorescein isothiocyanate-conjugated goat anti-mouse IgG (Vector Laboratories) diluted in Blotto, washed with TBS, and mounted on glass slides using Permount. Cells were examined using a Nikon 60× NA1.4 oil-immersion objective and standard fluorescein epifluorescence optics. Confocal fluorescence microscopy was performed using a laser scanning microscope fitted with the same objective.
Internalization of the Receptor
For flow cytometry, 1–2 × 105 cells/ well were plated onto a 24-well plate. After 24 h, the wells were treated with Dulbecco’s modified Eagle’s medium alone or with various doses of DADLE for 30 min or with 100 nM DADLE for various times at 37 °C. At the end of incubation the cells were chilled to 4 °C, washed with PBS, and incubated with 10 μg/ml primary antiserum (anti-FLAG M1 antiserum) in 50% fetal bovine serum, PBS for 1 h. The cells were washed twice with PBS and incubated with 10 μg/ml fluorescein isothiocyanate-conjugated goat anti-mouse IgG. The cells were collected from the wells with 5 mM EDTA and analyzed on a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems, Inc.). Live cells were gated by light scatter or exclusion of propidium iodide, and 5,000–10,000 cells were acquired for each time point. Mean fluorescence of all live cells, minus mean fluorescence of cells stained only with fluorescein isothiocyanate-conjugated second antibody, was used for calculations (10).
Sequestration and Down-regulation of the Receptor
For these studies, 1–2 × 106 cells were treated with 100 nM DADLE for various time periods. Following treatment, the cells were extensively washed with Krebs-Ringer HEPES buffer. The binding assay was carried out on whole cells at 4 °C using [3H]diprenorphine as the radioligand and 1 μM diprenorphine or 1 μM DADLE as the displacers. Sequestration is defined as the difference between the [3H]diprenorphine binding displaceable by the membrane impermeable ligand, DADLE, and the [3H]diprenorphine binding displaceable by the lipophilic ligand, di-prenorphine. For down-regulation studies, the cells were treated with 100 nM DADLE for various periods of time up to 24 h. Following treatment, the cells were extensively washed with Krebs-Ringer HEPES buffer. The binding assay was carried out on whole cells or cell homogenates at 4 °C using [3H]diprenorphine as the radioligand and six to eight concentrations of diprenorphine as the displacer. Kd and Bmax values were determined by Scatchard analysis using the Ligand program.
RESULTS
To visualize and characterize the internalization of the opioid receptors, we generated CHO cell lines stably expressing epitope-tagged wild type and mutant opioid receptor. The monoclonal antibody “M1” specifically binds to the epitope and thus allows the detection of the receptor. Immunofluorescence microscopy of the wild type receptor-expressing cell lines stained with M1 antibody shows that a majority of the opioid receptors are primarily localized to the plasma membrane (Fig. 2A). Similarly, a majority of the truncated receptors are also localized to the cell surface (Fig. 2C). Within 30 min after the addition of 100 nM DADLE to the wild type receptor-expressing cells, the receptor fluorescence is primarily visible within the cytoplasm, suggesting that the receptors are redistributed to a compartment within the cells (Fig. 2B). In contrast, a similar treatment to the cells expressing the ΔC15 receptor has very limited, if any, effect on the plasma membrane localization of the receptor (Fig. 2D), suggesting that this receptor (lacking the C-terminal 15 amino acids) does not undergo rapid agonist-mediated redistribution. These data suggest that the wild type δ opioid receptor undergoes rapid endocytosis, and the C-terminal tail is involved in this process.
Fig. 2. Confocal immunofluorescence microscopy of epitope-tagged wild type and mutant δ opioid receptors expressed in CHO cells.
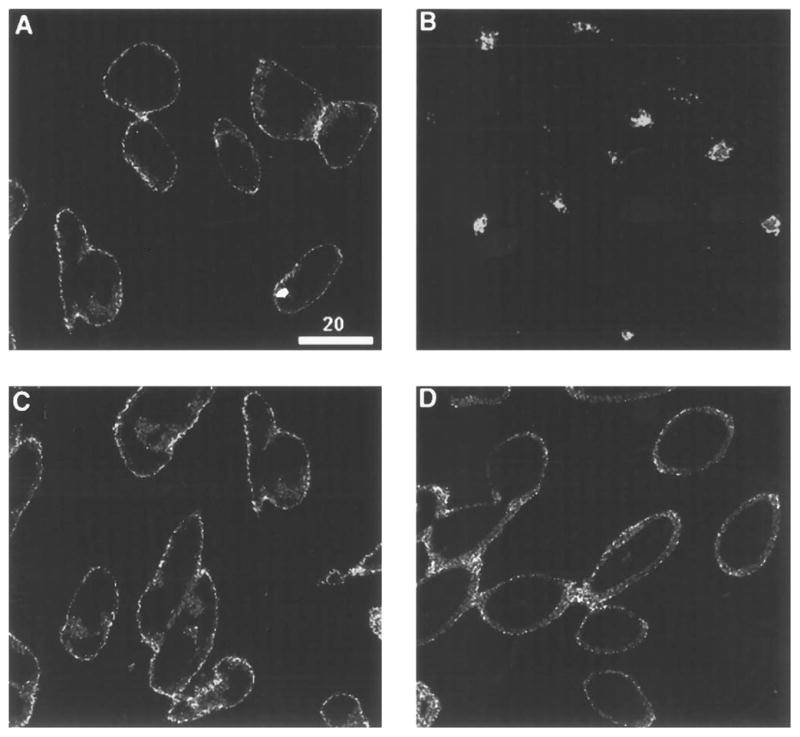
CHO cells expressing wild type (A and B) or ΔC15 receptor (C and D) were incubated in the absence (A and C) or presence of 100 nM DADLE (B and D) for 30 min. Fixation, permeabilization, and immunofluorescence staining of the receptors with the monoclonal antibody against the epitope tag are as described under “Experimental Procedures.” Cells were imaged by confocal fluorescence microscopy, using a plane of focus adjusted 3– 6 mm above the surface of the coverslip. This produces a cross-section through the center of the cell. Bright staining of the plasma membrane is apparent in A, C, and D, while prominent intracellular staining that appears as punctate staining within the cytoplasm is seen in C.
Next we examined the extent of internalization of the wild type receptor from the cell surface using flow cytometry. The extent of loss of receptors from the cell surface was determined by measuring the reduction in mean surface fluorescence intensity in the nonpermeabilized cells treated with 100 nM DADLE for 30 min. We find that this treatment causes a considerable reduction in fluorescence intensity (of about 75%) due to the loss of receptors from the cell surface (Table I). Treatment of the cells with 1 μM naloxone (an opioid antagonist) alone does not alter the level of internalization; treatment with 100 nM DADLE along with 1 μM naloxone antagonizes the DADLE-induced internalization (Table I). These results suggest that the rapid internalization of the receptor is an agonist-mediated process.
Table I. Effect of agonist and/or antagonist treatment on δ opioid receptor internalization.
The mean fluorescence of the cells not treated with DADLE is taken as control (100%). The data represent the mean ± SEM of three independent determinations.
| Treatment | Fluorescence |
|---|---|
| % | |
| No treatment | 100 ± 9 |
| 100 nM DADLE | 25 ± 5 |
| 1 μM Naloxone | 115 ± 19 |
| 100 nM DADLE + 1 μM naloxone | 127 ± 23 |
The CHO cells expressing the wild type receptor exhibit a rapid decrease in cell surface receptor fluorescence with more than 50% of the receptor lost from the surface within 10 min of treatment (Fig. 3). Similarly, HEK-293 cells expressing μ or δ opioid receptors also exhibit a rapid loss of receptors from the cell surface following treatment with high affinity agonists (11). More than 90% reduction in the δ opioid receptor fluorescence was achieved by about 30 min of DADLE treatment (Fig. 3); this is consistent with the loss of receptors from the cell surface as seen by confocal microscopy (Fig. 2) (11). The cells expressing ΔC7 receptor exhibits a time course of inhibition similar to that of the wild type with a half-maximal loss by about 10 min (Fig. 3). In contrast, the cells expressing the ΔC15 receptor shows a marked reduction in the rate of internalization (Fig. 3). These cells exhibit only about a 15% loss of receptor fluorescence after 30 min of treatment; these results are consistent with the detection of substantial amount of these receptors on the cell surface as seen by confocal microscopy. We find that the cells expressing ΔC37 also follow the kinetics of internalization similar to that of the cells expressing ΔC15 receptor supporting the notion that a portion of the C-terminal tail is involved in the rapid agonist-mediated internalization of the δ opioid receptor.
Fig. 3. Kinetics of receptor internalization.

The cells expressing wild type (□), ΔC7 (●), ΔC15 (■), or ΔC37 (○) were treated with 100 nM DADLE at 37 °C for the times indicated, stained, and analyzed by flow cytometry as described under “Experimental Procedures.” Internalization of the receptors is indicated by a reduction in the fluorescence measured in the cell population. The mean fluorescence after subtracting autofluorescence of cells (stained with second antibody alone) without DADLE treatment is taken as 100%. Percent internalization is defined as the percent reduction in mean fluorescence following various times of treatment with DADLE. The data represent the mean ± S.E. of three to four experiments. The data for cells expressing 1–2 × 105 receptors/cell are presented; similar dose-response curves were observed with additional transfected cultures expressing different numbers of receptors.
In order to examine if higher concentration of the agonist would increase the extent of internalization in the receptors lacking portions of the C-terminal tail, the cells were treated with higher doses of DADLE for 30 min and the extent of internalization was compared to the extent of internalization of the wild type receptors. The IC50 for the internalization of wild type receptor is about 3 nM; maximal internalization was observed at 100 nM (Fig. 4). The cells expressing the ΔC7 receptor also exhibit maximal internalization at 100 nM. In contrast, treatment of the cells expressing ΔC37 or ΔC15 receptors with as much as 1 μM DADLE does not substantially increase the level of internalization beyond the 20 or 30%, respectively (Fig. 4). Taken together, these results suggest that the rapid agonist-mediated internalization of the wild-type receptor requires relatively low amounts of DADLE, and that the mutants that are truncated by 15 or more amino acids of the C-terminal tail do not efficiently internalize even with higher doses of the agonist, further supporting the role played by the C-terminal tail in the agonist-mediated internalization.
Fig. 4. Dose response for the DADLE-induced internalization.

The cells expressing wild type (□), ΔC7 (●), ΔC15 (■), or ΔC37 (○) were treated with various concentrations of DADLE for 30 min at 37 °C for the times indicated, stained, and analyzed by flow cytometry as described under “Experimental Procedures.” Internalization of the receptors is indicated by a reduction in the fluorescence measured in a cell population. The mean fluorescence after subtracting autofluorescence of cells is taken as 100%. Percent internalization is defined as the percent reduction in mean fluorescence following various doses of treatment with DADLE. The data represent the mean ± S.E. of three to four experiments. The data for cells expressing 1–2 × 105 receptors/cell are presented; similar dose-response curves were observed with additional transfected cultures expressing different numbers of receptors.
In order to examine the role of individual Ser/Thr within this region of the C terminus in the rapid internalization process, we generated point mutants of the receptor at Ser344, Thr352, Thr353, Thr358, Thr361, and Ser363. The amino acid sequence of the C-terminal tail of the wild type and mutated receptor is presented in Fig. 1. When examined for the time course of internalization with 100 nM DADLE using flow cytometry, we find that majority of the point mutations result in a substantially slower rate and lower extent of internalization; approximately 30–40% loss of receptor fluorescence by about 30 min of treatment (Fig. 5). Longer exposure to the agonist does not substantially increase the rate of internalization (not shown). In contrast to the these point mutants, the Ser344 → Ala mutant exhibits a internalization kinetics similar to that of wild type receptor (Fig. 5). Taken together, these data imply that a region between Ser344 and Gly365 is involved in agonist-mediated rapid internalization and suggest that individual Ser/Thr play a critical role in the internalization process.
Fig. 5. Kinetics of the mutant receptor internalization.

The cells expressing wild type (□——□), Ser344 →Gly (●·····●), Thr352 →Ala (○- - - - -○), Thr353 → Ala (■– – –■), Thr358 → Ala (×–·–·–×), Thr361 → Ala (▲– – –▲), or Ser363 →Ala (•·····•) were treated with 100 nM DADLE at 37 °C for the times indicated, stained, and analyzed by flow cytometry as described under “Experimental Procedures.” Internalization of the receptors is indicated by a reduction in the fluorescence measured in the cell population. Percent internalization is defined as the percent reduction in mean fluorescence following various times of treatment with DADLE; mean fluorescence of cells without DADLE treatment is taken as (control) 100%. The data for cells expressing 1–2 × 105 receptors/cell are presented; similar dose-response curves were observed with additional transfected cultures expressing different numbers of receptors.
Prolonged treatment of cells with DADLE causes down-regulation of the wild type receptor and the C-terminal tail plays an important role in this process (8). In order to determine whether the point mutants that show altered internalization also exhibit altered down-regulation, we examined the rate and extent of the net loss of receptors from cells upon prolonged agonist treatment. We found that a majority of these mutant receptors exhibit a gradual and considerable decrease in the level of receptors over a 24-h treatment period (Fig. 6); the extent of down-regulation in these mutants is due to a change in the receptor number and not affinity of the receptor as determined by Scatchard analyses (data not shown). The extent of down-regulation of the mutant receptors is not substantially different from that of the cells expressing the wild type receptor. This is in contrast to the Thr353 → Ala mutant receptor that exhibits a substantial lack of agonist-induced down-regulation (8). Taken together, these results suggest that the region of the C-terminal tail governing the agonist-mediated rapid endocytosis is not identical to the region governing the agonist-mediated down-regulation of the receptor.
Fig. 6. Time course of down-regulation in cell lines expressing mutant receptors.

The cells expressing Ser344 → Gly (●·····●), Thr352 → Ala (○- - - - -○), Thr353 → Ala (■– – –■), Thr358-Ala (×–·–·–×), Thr361 → Ala (▲– – –▲), and Ser363 → Ala (•·····•) were treated with 100 nM DADLE for various periods of time, and the diprenorphine binding was carried out as described under “Experimental Procedures.” The receptor number in cells treated for 1–3 min with DADLE is taken as “control” (100%). The data represent mean ± S.E. of three to four experiments. The data for cells expressing 1–2 × 105 receptors/cell are presented; similar dose-response curves were observed with additional transfected cultures expressing different numbers of receptors.
In order to explore the mechanism of internalization, the effect of various agents that have previously been shown to modulate internalization of other receptors was examined on the extent of DADLE-induced receptor internalization. Pre-treatment of the cells for 15 min with 0.4 M sucrose or medium depleted with K+ is thought to disrupt clathrin-mediated endocytic pathway. We find that both treatments significantly affect DADLE-induced internalization of the wild type receptor as seen by the attenuation in the reduction in receptor fluorescence (Fig. 7). In contrast, treatment of the cells with NH4Cl or chloroquine does not have a substantial effect on internalization (Fig. 7). Also, 30-min pretreatment of the cells with modulators of protein kinase A, protein kinase C, or phosphatases had no substantial effect on the constitutive internalization or the DADLE-mediated internalization of the receptor. Taken together, these results suggest that the opioid receptor is internalized via a clathrin-coated endocytic pathway and that agents that increase intracellular pH, or modulators of phosphorylation, have no substantial effects on the internalization process.
Fig. 7. Effect of various agents on the DADLE-induced rapid internalization.

The cells expressing the wild type receptors were treated with various agents at indicated concentrations 15 or 30 min prior to the agonist treatment. The wells were treated with 100 nM DADLE for 30 min at 37 °C, chilled, washed, stained, and analyzed by flow cytometry as described under “Experimental Procedures.” The mean fluorescence of the cells not treated with DADLE is taken as control (100%). The data represent mean ± S.E. of three to four experiments. DA, DADLE; TPA, 12-O-tetradecanoylphorbol-13-acetate; Cal-phos; calphostin C; Stauro, staurosporin; OAG, 1-oleoyl-2-acetyl-sn-glycerol; H89, N-[2((3-(4-bromophenyl)-2-propenyl)-amino)ethyl]-5-isoquinoline sulfonamide; Okadaic, okadaic acid.
DISCUSSION
Exposure of the cells containing opioid receptors to opiates causes attenuation of response to the drugs by a process of desensitization. It is thought that the changes in secondary structure and/or the location of the receptor are involved in this process. The removal of the receptor from the cell surface by a process of cellular trafficking of the receptor (involving internalization and degradation) is thought to bring about the short term and long term desensitization. Despite the importance of this phenomenon, very little is known regarding the process of desensitization of the opioid receptors and the domains of the receptor involved in this process.
We have used the δ subtype of opioid receptors to study the agonist-mediated internalization and down-regulation. The availability of the opioid receptor cDNA has made it possible for the addition of the epitope tag and deletion and/or mutagenesis of the receptor. Previously we have shown that, when expressed in CHO cells, the wild type epitope-tagged receptor behaves essentially as the receptors in the NG108-15 cells (8). Upon prolonged exposure to opioids, the wild type receptor undergoes agonist-induced down-regulation with a time course similar to that of the NG108-15 cells (8). Using mutant receptors of the C-terminal tail, we have also shown that the C-terminal tail plays an important role in this process.
In this study, we have explored the involvement of the C-terminal tail in the early events in receptor trafficking. Using immunofluorescence microscopy of cells stained with a selective antibody to the epitope tag, the distribution of the receptor was visualized in CHO cells expressing the wild type and mutant receptors. We find that the δ opioid receptors predominantly reside in the plasma membrane. This is consistent with the ultrastructural studies using immunoelectron microscopy that have shown that the δ receptors are localized to both the presynaptic and postsynaptic membranes and to discrete regions within the synapse (12). In addition to the receptors on the plasma membrane, we find a small percentage of the receptors within the intracellular pool. The presence of the intracellular receptors is further supported by our finding from the peptide displacement studies. DADLE, a membrane-impermeable peptide ligand, is able to displace only about 70 – 80% of the diprenorphine binding (diprenorphine is a relatively lipophilic ligand that is able to enter the cells). These data suggest that, although majority of the receptors are on the cell surface, a small percent of the receptor is found intracellularly; this is true for both wild type and the mutant receptors expressed in CHO cells. The presence of intracellular receptors is consistent with the findings from immunohistochemical studies in rat brain where a fraction of the δ receptors are reported to be within the intracellular milieu (13).
Studies with the epitope-tagged μ or δ opioid receptor expressed in HEK-293 cells show that the receptor is also predominantly localized to the plasma membrane (11, 14). Addition of the peptide agonist causes internalization of these receptors. The agonist-induced redistribution of the μ or δ receptor depends on the nature of the agonist, for example, treatment of the cells with a peptide agonist causes internalization of the receptors, whereas treatment with an alkaloid agonist, morphine, does not (11, 14). The observation that morphine is able to induce receptor signaling without inducing rapid endocytosis underscores the importance of studying opioid receptor trafficking in the regulation of receptor function by morphine and other narcotic drugs.
We find that exposure of the wild type receptor to DADLE causes internalization within 30 min. This rapid internalization process is distinct from the slower process of receptor down-regulation. This rapid internalization occurs without a decrease in receptor number, suggesting receptor degradation is not involved. In support of this, we find that the agents, such as ammonium chloride or chloroquine, that modify the intracellular pH of endosomes/lysosomes and thus affect the functioning of these organelles, do not affect the agonist-induced internalization. These findings support the lack of involvement of organelles with acidic internal pH in the rapid-internalization process.
Consistent with the recent findings with the internalization of the other GPCRs, the predominant path for opioid receptor internalization is the classical endocytic pathway, initiated at the level of entry into classic clathrin-coated pits (11) (this study). Depletion of intracellular K+ or hypertonic media inhibits the receptor-mediated endocytosis of a number of receptors by blocking the clathrin-coated pit formation (15, 16). The fact that these treatments affect the internalization of the opioid receptors suggest that the opioid receptors are internalized by the classic pathway. It is possible that the opioid receptors are internalized in a noncoated pit-mediated pathway; the finding that K+ depletion does not completely inhibit the receptor internalization supports such a possibility. Other neuropeptide receptors, such as the cholecystokinin receptor, have been shown to be internalized by both the clathrin-coated and non-clathrin-coated pit-mediated pathways (17).
The receptor lacking the C-terminal 7 amino acids has the rapid internalization kinetics as that of the wild type receptor, suggesting that the internalization signal does not include the C-terminal 7 amino acids. In contrast, the receptor lacking the C-terminal 15 amino acids exhibits a lack of this rapid internalization kinetics, suggesting that a portion of the C-terminal tail between Arg356 and Gly365 is involved in modulating a signal and/or a conformation that is important for internalization. The internalization studies with the mutant receptors that have single amino acid changes show that mutation of any one of the Ser or Thr within this region reduces the rate and extent of internalization. It should be pointed out that the decrease in the rate of internalization with any one of these point mutants is not as substantial as the decrease in the rate of internalization with the receptor lacking the C-terminal 15 amino acids. These results suggest that the binding of an agonist could mediate changes in the secondary structure of this portion of the C-terminal tail and that could affect the rapid internalization process.
A search for a common motif involved in internalization of other GPCRs has shown that the NPXXY, conserved in many GPCRs, could be involved in this process (18). However, mutations of this region in some GPCRs do not affect internalization, suggesting that this motif might not be sufficient for internalization in those receptors; for example, mutation of the Tyr in the NPXXY motif in gastrin-releasing peptide receptor or in type 1 angiotensin II receptor does not alter the extent of internalization (19, 20). Recent studies with the chimeric receptors between the β2AR (that exhibits efficient internalization) and the β3AR (that does not exhibit internalization) have shown that the presence of both C-terminal tail and the intracellular loops (first and second) are needed for efficient internalization (7). A similar phenomenon could be true for the opioid receptor, since we find that mutation of any one of the Ser or Thr between Ser352 and Ser363 results in a reduction in the rapid internalization, and deletion of the C-terminal 15 amino acids results in substantially higher effect.
The molecular mechanisms responsible for the agonist-mediated internalization of the opioid receptor are not clearly understood. The observation that the truncated receptor is able to inhibit adenylate cyclase (8) but not able to internalize (this study) indicates that the coupling with a second messenger system is not sufficient to induce internalization. Similar observations were made by the internalization studies carried out with the C-terminally deleted neurotensin receptor (21). Although previous studies with β2AR had implied a requirement of functional coupling for efficient internalization (22), recent studies with a number of GPCRs, that lack internalization but are able to functionally couple, and the studies with avian β2AR, that internalizes well but is unable to promote second messenger responses (23–26), suggest that receptor-mediated activation of a G protein leading to the modulation of the second messenger levels is not sufficient for the receptor internalization.
It is not known if phosphorylation of the opioid receptors is involved in its internalization, endocytosis or down-regulation of these receptors. Both μ and δ opioid receptors expressed in heterologous cell lines have been shown to be phosphorylated and agonist treatment promotes further phosphorylation of the receptor (14, 27). Surprisingly, we find that the modulators of protein kinase A or protein kinase C do not alter the level of internalization of the δ receptor suggesting that phosphorylation by these kinases do not modulate receptor internalization. The possibility that phosphorylation of the receptor by β2AR kinase or related GPCR kinases may be involved in modulating receptor internalization can not be excluded. Studies with β2AR, where β2AR kinase is known to be involved in the regulation of receptor function, it has been shown that β-arrestin binds to the phosphorylated β2AR and contributes to it’s internalization (28, 29). β-Arrestin and Dynamin (a GTPase that regulates the formation and internalization of clathrin-coated vesicles) have recently been shown to modulate the internalization of GPCRs (29); it is possible that similar proteins are involved in the internalization of the opioid receptors.
We find that many of the mutant opioid receptors that show impaired internalization kinetics do not have substantially impaired down-regulation kinetics when examined for their long term exposure to agonist. We have previously shown that the receptor lacking the C-terminal 37 amino acids does not down-regulate, whereas the receptor lacking the C-terminal 15 amino acids does (8). Also, a single point mutation of Thr353 → Ala exhibits a substantial loss of down-regulation. In this study we find that many of the other Ser/Thr mutants exhibit a near normal agonist-mediated down-regulation but impaired rapid internalization, suggesting that short term agonist-induced internalization and the long term agonist-induced down-regulation are not directly dependent phenomena. Support for this also comes from studies with mutant β2AR that have shown that the internalization and down-regulation are independent phenomena (30).
In conclusion, we have demonstrated that the δ opioid receptor undergoes agonist-mediated rapid internalization. C-terminal deletion of the mutant receptors results in attenuation of this rapid-internalization. Using point mutations of Ser/Thr in C-terminal tail of the opioid receptor we have narrowed down the region involved in this rapid endocytosis to a domain in the middle of the C-terminal tail. Also, we have shown that the internalization is via a classic endocytic pathway and that conformational change in the receptor possibly via phosphorylation could play a role in this process.
Acknowledgments
We thank Dr. M. von Zastrow for the pCDNA3-FDOR1 plasmid. Flow cytometric analysis was performed in the Jonsson Comprehensive Cancer Center at UCLA and in the Kaplan Comprehensive Cancer Center at NYU School of Medicine.
Footnotes
This work is supported in part by National Institutes of Health Grants DA08863 and NS1788 (to L. A. D) and DA05010 and DA00218 (to C. J. E).
The abbreviations used are: β2AR, β2-adrenergic receptor; GPCR, G protein-coupled receptor; CHO, Chinese hamster ovary; DADLE, [D-Ala2,D-Leu5]enkephalin; PBS, phosphate-buffered saline.
References
- 1.Herz A. Opioids. Vol. 1. Springer-Verlag; Berlin, FRG: 1993. [Google Scholar]
- 2.Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Annu Rev Biochem. 1991;60:653–688. doi: 10.1146/annurev.bi.60.070191.003253. [DOI] [PubMed] [Google Scholar]
- 3.Benovic JL, Bouvier M, Caron MG, Lefkowitz RJ. Annu Rev Cell Biol. 1988;4:405–428. doi: 10.1146/annurev.cb.04.110188.002201. [DOI] [PubMed] [Google Scholar]
- 4.Evans CJ, Keith DE, Morrison H, Magendzo K, Edwards RH. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 5.Kieffer BL, Befort K, Gaveriaux-Ruff C, Hirth CG. Proc Natl Acad Sci U S A. 89:12048–12052. doi: 10.1073/pnas.89.24.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uhl GR, Childers S, Pasternak G. Trends Neurosci. 1994;17:89–93. doi: 10.1016/0166-2236(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 7.Jockers R, Da Silva A, Strosberg AD, Bouvier M, Marullo S. J Biol Chem. 1996;271:9355–9362. doi: 10.1074/jbc.271.16.9355. [DOI] [PubMed] [Google Scholar]
- 8.Cvejic S, Trapaidze N, Cyr C, Devi LA. J Biol Chem. 1996;271:4073–4076. doi: 10.1074/jbc.271.8.4073. [DOI] [PubMed] [Google Scholar]
- 9.Petanceska S, Devi L. J Biol Chem. 1992;267:26038–26043. [PubMed] [Google Scholar]
- 10.Schmid I, Schmid P, Giorgi JV. Cytometry. 1988;9:533–538. doi: 10.1002/cyto.990090605. [DOI] [PubMed] [Google Scholar]
- 11.Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- 12.Cheng PY, Svingos AL, Wang H, Clarke CL, Jenab S, Beczkowska IW, Inturissi CE, Pickel VM. J Neurosci. 1995;15:5976–5988. doi: 10.1523/JNEUROSCI.15-09-05976.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elde R, Arvidsson U, Riedl M, Vulchanova L, Lee JH, Dado R, Nakano A, Chakrabarti S, Zhang X, Loh HH, Law PY, Hokfelt T, Wessendorf Ann N Y Acad Sci. 1995;757:390–404. doi: 10.1111/j.1749-6632.1995.tb17497.x. [DOI] [PubMed] [Google Scholar]
- 14.Arden JR, Segredo V, Wang Z, Lameh J, Sadee W. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- 15.Heuser JE, Anderson RGW. J Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larkin JM, Brown MS, Goldstein JL, Anderson RGW. Cell. 1983;33:273–285. doi: 10.1016/0092-8674(83)90356-2. [DOI] [PubMed] [Google Scholar]
- 17.Roettger BF, Rentsch RU, Pinon D, Holicky E, Hadac E, Larkin JM, Miller LM. J Cell Biol. 1995;128:1029–1041. doi: 10.1083/jcb.128.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barak LS, Tiberi M, Freedman NJ, Kwatra MM, Lefkowitz RJ, Caron MG. J Biol Chem. 1994;269:2790–2795. [PubMed] [Google Scholar]
- 19.Slice LW, Wong HC, Sternini C, Grady EF, Bunnett NW, Walsh JH. J Biol Chem. 1994;269:21755–21762. [PubMed] [Google Scholar]
- 20.Hunyady L, Bor M, Baukal AJ, Balla T, Catt KJ. J Biol Chem. 1995;270:16602–16609. doi: 10.1074/jbc.270.28.16602. [DOI] [PubMed] [Google Scholar]
- 21.Hermans E, Octave JN, Malotreaux JM. Mol Pharmacol. 1996;49:365–372. [PubMed] [Google Scholar]
- 22.Cheung AH, Sigal IS, Dixon RAF, Strader CD. Mol Pharmacol. 1988;34:128–132. [PubMed] [Google Scholar]
- 23.Yu SS, Lefkowitz RJ, Hausdorff WP. J Biol Chem. 1993;268:337–341. [PubMed] [Google Scholar]
- 24.Nantel F, Bonin H, Emorine LJ, Zilberfarb V, Strosberg AD, Bouvier M, Marullo S. Mol Pharmacol. 1993;43:548–555. [PubMed] [Google Scholar]
- 25.Suzuki T, Nguyen CT, Nantel F, Bonin H, Valiquette M, Frielle T, Bouvier M. Mol Pharmacol. 1991;41:542–548. [PubMed] [Google Scholar]
- 26.von Zastrow M, Link R, Daunt D, Barsh G, Kobilka B. J Biol Chem. 1993;268:763–766. [PubMed] [Google Scholar]
- 27.Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Mol Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- 28.Ferguson SS, Downey WE, Colapietro AM, Barak LS, Menard L, Caron MG. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Ferguson SSG, Barak LS, Menard L, Caron MG. J Biol Chem. 271:18302–18305. doi: 10.1074/jbc.271.31.18302. [DOI] [PubMed] [Google Scholar]
- 30.Hausdorff WP, Campbell PT, Ostrowski J, Yu SS, Caron MG, Lefkowitz RJ. Proc Natl Acad Sci U S A. 1991;88:1979–2983. doi: 10.1073/pnas.88.8.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]