

Abstract
Ketamine, a clinically relevant drug, has been shown to enhance opioid-induced analgesia and prevent hyperalgesia. However, the molecular mechanisms involved are not clearly understood. As previous studies found that activation of opioid receptors leads to the phosphorylation of mitogen-activated protein kinases, we investigated whether ketamine could modulate μ-opioid receptor (μOR)-mediated ERK1/2 phosphorylation. We find that acute treatment with ketamine enhances (~2- to 3-fold) the levels of opioid-induced ERK1/2 phosphorylation in recombinant as well as cells endogenously expressing μOR. Interestingly, we find that in the absence of ketamine ERK1/2 signaling is desensitized 10 min after opioid exposure whereas in its presence significant levels (~3-fold over basal) are detected. In addition, ketamine increases the rate of resensitization of opioid-mediated ERK1/2 signaling (15 min in its presence vs. 30 min in its absence). These results suggest that ketamine increases the effectiveness of opiate-induced signaling by affecting multiple mechanisms. In addition, these effects are observed in heterologous cells expressing μOR suggesting a non-NMDA receptor-mediated action of ketamine. Together this could, in part, account for the observed effects of ketamine on the enhancement of the analgesic effects of opiates as well as in the duration of opiate-induced analgesia.
Keywords: G-protein coupled receptors, ketamine, morphine, opioid receptors
Ketamine [2-(o-chlorophenyl)-2-(methylamino) cyclohexa-none hydrochloride] produces profound analgesia in man and animals when administered as an intravenous anesthetic (Stannard and Porter 1993; Eide et al. 1995; Mathisen et al. 1995). It blocks peripheral and CNS sodium channels (Scheller et al. 1996) and interacts with monoaminergic and voltage-sensitive Ca+2 channels (Hirota and Lambert 1996). Although it acts primarily as a non-competitive NMDA-receptor antagonist (Willets et al. 1990; Yamamura et al. 1990), ketamine also modulates the activity of a number of receptors including the nicotinic (Scheller et al. 1996) and muscarinic receptors (Hustveit et al. 1995) as well as the μ-, δ- and κ-opioid receptors (Smith et al. 1980; Finck and Ngai 1982; Hustveit et al. 1995; Hirota et al. 1999).
Activation of μOR by potent opioid analgesics leads to the development of acute tolerance as well as long-lasting hyperalgesia (Ziegelgänsberger and Tölle 1993; Larcher et al. 1998; Laulin et al. 1998; Celerier et al. 2000). This is thought to be due to the induction of NMDA-dependent central sensitization processes (Laulin et al. 1998; Celerier et al. 2000) because it has been shown that intrathecal morphine causes a large increase in glutamate release (Stiller et al. 1998). In addition, activation of μOR leads to enhanced glutamatergic neurotransmission via a protein kinase C-mediated removal of Mg+2 blockade of the NMDA-receptor channel (Chen and Huang 1992; Royblat et al. 1993). Previous studies have shown that subanesthetic doses of ketamine prevent opioid-induced hyperalgesia (Bristow and Orlikowski 1989; Royblat et al. 1993; Tverskoy et al. 1994; Hirota and Lambert 1996; Javery et al. 1996; Abdel-Ghaffar et al. 1998), inhibit the development of tolerance to morphine antinociception in rats (Trujillo and Akil 1994) and enhance the analgesic effects of opioids as well as the duration of analgesia (Joachimsson et al. 1986; Javery et al. 1996; Tverskoy et al. 1996; Wong et al. 1996; Stubhaug et al. 1997). However, the molecular mechanisms involved in these processes are not clearly understood.
A number of studies have shown that activation of opioid receptors can trigger acute signaling via heterotrimeric G-proteins leading to the inhibition of adenylyl cyclase, modulation of inwardly rectifying K+ or voltage-dependent Ca+2 channels and activation of the mitogen-activated protein kinase (MAPK) pathway (Fukuda et al. 1996; Li and Chang 1996; Gutstein et al. 1997; Trapaidze et al. 2000). This acute signaling is time-dependent because upon continued exposure to agonists it fades, a phenomenon known as receptor desensitization. Upon removal of agonists and after allowing the cells to recover, the receptors are able to undergo another wave of signaling events upon re-exposure to agonists. This is known as receptor resensitization. The ability of ketamine to increase the duration of opioid-induced analgesia suggests that it could also modulate opioid-induced signaling in such a way that these receptors remain in an active state for a longer time. This could be achieved by influencing the time course of desensitization and resensitization of signaling by opioid receptors. In this study, we used SK-N-SH (a human neuroblastoma cell line that endogeneously expresses μOR) and Chinese hamster ovary (CHO)-μ (a heterologous cell line that stably expresses recombinant μOR) to see the effect of ketamine on μOR signaling via the MAPK pathway. Our findings show that the ERK1/2 phosphorylation is significantly potentiated by ketamine and that this has a profound effect in the desensitization and resensitization of opioid-induced ERK1/2 signaling.
Materials and methods
Materials
Ketamine, morphine, fentanyl, anti-tubulin antibodies, protease and phosphatase inhibitor cocktails were obtained from Sigma-Aldrich (St. Louis, MO, USA). SK-N-SH cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Dulbecco’s modified Eagle’s medium, F12 media, fetal bovine serum, geneticin and streptopenicillin were obtained from Invitrogen (Carlsbad, CA, USA). Mouse anti-phospho ERK1/2 antibodies and rabbit total ERK1/2 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Odyssey blocking buffer, goat anti-mouse IR Dye 800 and goat anti-rabbit IR Dye 680 were obtained from Li-COR (Lincoln, NE, USA). All other reagents were obtained from Fisher Scientific (Sunawee, GA, USA).
Cell culture
SK-N-SH cells were grown to confluence in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 100 μg/mL streptopenicillin. CHO cells stably expressing Flag tagged mouse μOR were grown is F12 media containing 10% fetal bovine serum, 100 μg/mL streptopenicillin and 500 μg/mL geneticin. Cells were subcultured at a ratio of 1 : 5 with partial replacement of the media on the day before subculturing or harvested at day 5 (the characterization of this cell line is described in Trapaidze et al. 2000).
Binding assays
Saturation binding assays were carried out as described previously (Gomes et al. 2003) using SK-N-SH cells (2 × 105 cells) and different concentrations (0–10 nM) of either [3H]morphine or [3H]fentanyl in the absence or presence of ketamine (10 or 100 μM) in an assay buffer comprising 50 mM Tris–Cl, pH 7.4 containing containing 0.32 M sucrose. Non-specific binding was determined in the presence of 1 μM diprenorphine and was less than 5% of the total binding.
[35S]GTPγS binding assay
Membranes were prepared from SK-N-SH cells and subjected to a [35S]GTPγS binding assay as described previously (Gomes et al. 2003) using morphine or fentanyl (0–10 μM) in the absence or presence of ketamine (10 nM, 10 μM or 100 μM).
ERK1/2 phosphorylation
The level of phosphorylated ERK1/2 was determined as described previously (Trapaidze et al. 2000).
Effect of μ-opiod receptor agonists
Cells were treated with different concentrations (0–100 μM) of ketamine, morphine, fentanyl, morphine + ketamine (1 : 1), fentanyl + ketamine (1 : 1) for 5 min at 37°C. Cells were lysed with 2% sodium dodecyl sulfate in 50 mM Tris–Cl, pH 7.5 containing protease and phosphatase inhibitor cocktail and aliquots of lysates were subjected to western blot analysis with anti-phospho ERK1/2 antibodies (1 : 1000) as described previously (Gomes et al. 2003). Sample loading was confirmed with anti-tubulin antibodies (1 : 20 000). Cells not exposed to drug treatment were taken as control.
Effect of ketamine (0.01/10/100 μM) on μ-induced phosphorylation of MAPK
Cells were treated with different concentrations (0–100 μM) of morphine or fentanyl in the absence or presence of 0.01/10/100 μM ketamine for 5 min at 37°C.
Effect of μ-antagonist on potentiation of μ-induced phosphorylation of MAPK by ketamine
Cells were treated with morphine or fentanyl (100 nM) in the absence or presence of 10 μM of the μ-receptor antagonist, CTOP (D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2), for 5 min at 37°C. Western blot analysis was carried out with mouse anti-phospho ERK1/2 and rabbit total ERK1/2 antibodies (1 : 1000) diluted in Odyssey blocking buffer and goat anti-mouse IR Dye 800 and goat anti-rabbit IR Dye 680 (1 : 10 000). Bands were visualized using the Odyssey Imaging System (Li-COR) and quantitated as described (Gomes et al. 2003; Heimann et al. 2007).
Time course of ERK1/2 desensitization
Cells were treated with 10 μM morphine or fentanyl for different time periods (0–90 min) at 37°C in the absence or presence of ketamine (10 μM). Cells not exposed to drug treatment were taken as control.
Time course of ERK1/2 resensitization
Cells were treated for 30 min with 10 μM morphine or fentanyl at 37°C so as to induce the desensitization of MAPK signaling. Cells were washed free of opioids (three times with serum free media) and incubated for 5–90 min with media in the absence or presence of ketamine (10 μM). This was followed by a 5-min treatment with morphine or fentanyl to see whether signaling through the MAPK pathway had been re-established. Cells not exposed to drug treatment were taken as control. A schematic representation of the protocol used to assay for the effect of ketamine on the resensitization of ERK1/2 signaling by μOR agonists is shown in Fig. 4a.
Fig. 4.
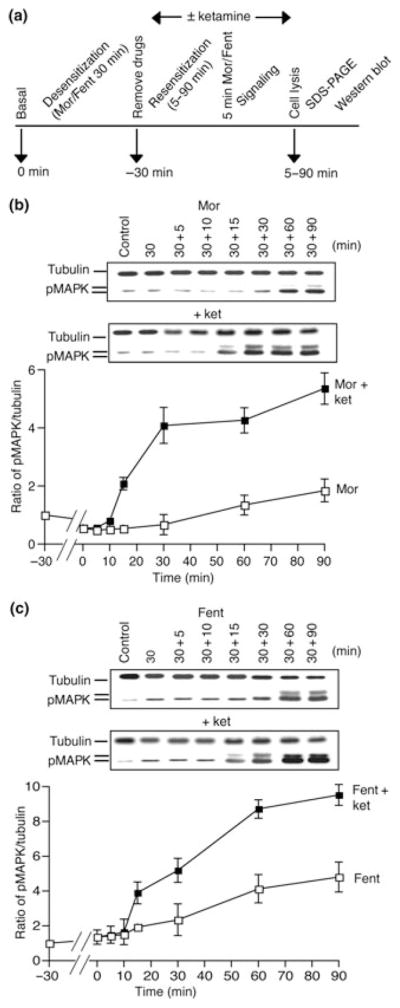
Effect of ketamine on resensitization of opioid-induced ERK1/2 phosphorylation. (a) Schematic representation of the assay protocol used to investigate the effect of ketamine on resensitization of opioid-induced ERK1/2 phosphorylation. SK-N-SH cells were treated for 30 min at 37°C with 10 μM of either morphine (b) or fentanyl (c). Cells were washed free of opioids and incubated for 5–90 min with media in the absence or presence of ketamine (10 μM). This was followed by a 5 min treatment with morphine or fentanyl (10 μM) following which cells were lysed and subjected to western blot analysis using anti-phospho ERK1/2 and anti-tubulin antibodies as described in Materials and methods section. Results are the mean ± SE of six independent experiments in triplicates.
Statistical analysis
Data obtained was analyzed using Dunnett’s test or Bonferroni’s multiple comparison tests. Values of p < 0.05 were considered to be significant.
Results
Phosphorylation of ERK1/2 by μOR agonists
We examined the effects of ketamine on μOR agonist-induced phosphorylation of ERK1/2. For this CHO cells, stably expressing μOR were treated with 10 μM of either ketamine, morphine, fentanyl alone or in combination [morphine + ketamine (1 : 1) or morphine + fentanyl (1 : 1)] for 5 min followed by lysis and western blot analysis. We find that ketamine on its own induces a ~3.5-fold stimulation in the phosphorylation of ERK1/2 (Fig. 1a and Figure S1a). Morphine or fentanyl alone induced a ~ 9.6- and a ~13.2-fold increase in the levels of phosphorylated ERK1/2 respectively (Fig. 1a and Figure S1a). Interestingly, when the cells were treated with a combination of ketamine and morphine (1 : 1) or ketamine and fentanyl (1 : 1) there was a dramatic increase in the levels of phosphorylated ERK1/2 which was greater than that obtained with either ketamine, morphine or fentanyl alone (~18.8 in the case of ketamine and morphine and ~28.6 in the case of ketamine and fentanyl) (Fig. 1a and Figure S1a). This robust increase in the levels of phosphorylated ERK1/2 appears to be specifically due to the presence of ketamine because treatment of cells with morphine + fentanyl (1 : 1) did not induce an additive increase in the levels of phosphorylated ERK1/2 (not shown). Next, we examined the effects of ketamine on μOR-mediated signaling in SK-N-SH cells that express endogenous receptors. We find that in these cells ketamine induces a dose-dependent increase in the levels of phosphorylated ERK1/2 with an EC50 of 7 ± 0.96 μM and an Emax of 2.7 ± 0.1 (Fig. 1b). Morphine or fentanyl alone also induced a dose-dependent increase in the levels of phosphorylated ERK1/2 with an EC50 of 5.8 ± 0.9 nM and an Emax of 4.4 ± 0.2 in the case of morphine and an EC50 of 68 ± 14 nM and an Emax of 8.1 ± 0.3 in the case of fentanyl. Interestingly, treatment with a combination of ketamine and morphine (1 : 1) or ketamine and fentanyl (1 : 1) caused a dramatic dose-dependent increase in the levels of phosphorylated ERK1/2 which was greater than that obtained with either ketamine, morphine or fentanyl alone. The combination of ketamine and morphine gave an EC50 of 5.9 ± 1 nM and an Emax of 13.8 ± 0. 3 whereas that of ketamine and fentanyl gave an EC50 of 0.5 ± 0.1 nM and an Emax of 13.9 ± 0. 2 (Fig. 1b).
Fig. 1.

Phosphorylation of ERK1/2 by μOR agonists. (a) CHO cells stably expressing Flag-tagged μOR were treated for 5 min at 37°C with 10 μM of either ketamine, morphine, fentanyl, morphine + ketamine (1 : 1) or fentanyl + ketamine (1 : 1). (b) SK-N-SH cells that endogenously express μOR were treated for 5 min at 37°C with different concentrations (0–100 μM) of ketamine, morphine, ketamine + morphine (1 : 1), fentanyl, fentanyl + ketamine (1 : 1). (c) CHO cells stably expressing Flag-tagged μOR were treated with morphine or fentanyl (100 nM) in the absence or presence of 10 μM of μ-receptor antagonist, CTOP, for 5 min at 37°C. Cell lysates from panels a–c were subjected to western blot analysis using anti-phospho ERK1/2 and, anti-total ERK1/2 or anti-tubulin antibodies as described in Materials and methods section. Values were normalized to control cells not subjected to drug treatment. Results are the mean ± SE of six independent experiments in triplicates; values that represent significant differences from control by Dunnett’s test are indicated **p < 0.001.
To see if the potentiation of opioid-induced ERK1/2 phosphorylation by ketamine was mediated specifically via μOR we examined the effect of a receptor selective antagonist, CTOP. As seen from Fig. 1c, 10 μM CTOP completely blocks the potentiation of morphine or fentanyl (100 nM)-induced ERK1/2 phosphorylation mediated by ketamine (10 μM). Taken together these results indicate that the potentiation of opioid-induced ERK1/2 phosphorylation by ketamine is mediated via μOR.
Effect of ketamine on morphine/fentanyl dose response curves
Next, we examined the effects of a single concentration of ketamine (10 μM) on the levels of phosphorylated ERK1/2 induced by different doses of morphine or fentanyl. We see a significant increase (p < 0.01) in the levels of phosphorylated ERK1/2 upon treatment with morphine (Fig. 2a) or fentanyl (Fig. 2b) in the presence of ketamine. Interestingly, ketamine effects were found to be biphasic; ketamine induced a greater increase in the phosphorylation of ERK1/2 at lower doses of morphine or fentanyl (0.1–1 nM) as compared with higher doses (0.01–100 μM) (Fig. 2). In the presence of ketamine, morphine (1 nM) caused a ~28.7-fold increase in the levels of phosphorylated ERK1/2 compared with ~2.6 seen in the absence of ketamine (Fig. 2a). Similarly, fentanyl (1 nM) caused a ~33.3-fold increase in the levels of phosphorylated ERK1/2 in the presence of ketamine compared with ~1.8 seen in the absence of ketamine (Fig. 2b). Similar observations were made when morphine or fentanyl dose response curves were carried out in the presence of 100 μM ketamine (Figure S1b and c) although the extent of ERK1/2 phosphorylation observed was slightly lower (~19.5 for 1 nM morphine and ~25.9 for fentanyl) than that observed with 10 μM ketamine. We also examined the effect of a very low dose (10 nM) of ketamine on morphine dose response curves. We find that a low dose of ketamine also has a biphasic effect on morphine dose response inducing a greater increase in the phosphorylation of ERK1/2 at lower morphine doses (0.1–100 nM) as compared with higher doses (1–100 μM) (Figure S2). In the presence of ketamine, morphine (100 nM) caused a ~8.9-fold increase in the levels of phosphorylated ERK1/2 compared with ~3.2 seen in the absence of ketamine (Figure S2).
Fig. 2.

Effect of ketamine on morphine/fentanyl-induced ERK1/2 phosphorylation. SK-N-SH cells were treated for 5 min at 37°C with different concentrations (0–100 μM) of morphine (a) or fentanyl (b) in the absence or presence of ketamine (10 μM) and subjected to western blot analysis using anti-phospho ERK1/2 and anti-tubulin antibodies as described in Materials and methods section. Results are the mean ± SE of six independent experiments in triplicate.
Next, we examined if ketamine affected the binding of μOR agonists by carrying out binding assays in SK-N-SH cells using [3H]morphine or [3H]fentanyl. Saturation binding data were fit by non-linear analysis of 1 or 2 binding site models using the Graph Prism software (San Diego, CA, USA). Analysis of the data shows that 10 μM ketamine had no effect on [3H]morphine binding whereas 100 μM ketamine led to significant changes in both the Kd and Bmax. [3H]Morphine binding in the absence of ketamine gave a Kd of 7.7 ± 2.3 nM and a Bmax of 313 ± 50 fmol/mg protein; the observed Kd was 10.3 ± 1.4 nM in the presence of 10 μM ketamine and 19.2 ± 3.6 (p < 0.05) in the presence of 100 μM ketamine whereas the Bmax was 376 ± 37 fmol/mg protein in the presence of 10 μM ketamine and 701 ± 94 (p < 0.01) in the presence of 100 μM ketamine. Similarly, [3H]fentanyl binding in the absence of ketamine gave a Kd of 0.9 ± 0.3 nM and a Bmax of 183 ± 11 fmol/mg protein; the observed Kd was 1.3 ± 0.3 nM in the presence of 10 μM ketamine and 3.1 ± 0.9 (p < 0.05) in the presence of 100 μM ketamine whereas the Bmax was 201 ± 13 fmol/mg protein in the presence of 10 μM ketamine and 297 ± 32 (p < 0.01) in the presence of 100 μM ketamine. These results suggest that the effects seen with 10 μM ketamine on morphine- or fentanyl-mediated increases in ERK1/2 phosphorylation are not caused by changes in the binding properties of μ receptors.
We also examined whether ketamine-modulated opioid-mediated signaling upstream of ERK1/2 phosphorylation by examining its effects on opioid-mediated increases in [35S]GTPγS binding in SK-N-SH cell membranes. We find that ketamine causes significant increases (p < 0.01) in the EC50 of morphine (from 25 to 299 nM) and fentanyl (from 6.7 to 61 nM) (Table S1). In addition, ketamine showed a trend towards decreasing maximal opioid-mediated increases in [35S]GTPγS binding although this was not statistically significant. Taken together these results indicate that ketamine differentially modulates the pathways leading to ERK1/2 phosphorylation by μOR agonists.
Time course of desensitization of opioid-induced ERK1/2 phosphorylation
To explore the mechanism of ketamine-mediated increase in μOR receptor-mediated ERK1/2 phosphorylation we determined the time point at which attenuation of morphine or fentanyl signaling via the ERK1/2 pathway occurred in the absence and presence of ketamine. We find that ketamine alone induced peak levels of phosphorylation of ERK1/2 (~2-fold above basal) between 2 and 5 min and the signal returned to basal levels by 10 min of exposure to ketamine (Fig. 3). In addition, morphine or fentanyl alone induced peak levels (~4- and ~7-fold above basal respectively) between 5 and 10 min after exposure to the drug and the signal returned to basal levels by 30 min (Fig. 3a and b). However, a combination of ketamine with morphine or fentanyl resulted in a robust increase in peak levels (~12-fold above basal) between 5 and 10 min which did not return to basal levels even after 90 min of exposure to the drugs. At this time point (90 min), the levels of phosphorylated ERK1/2 were found to be ~3-fold (over control) in the case of morphine + ketamine and ~4-fold (over control) for fentanyl + ketamine (Fig. 3). These results suggest that in addition to potentiating morphine or fentanyl-mediated phosphorylation of ERK1/2, ketamine delays the extent of desensitization of μOR.
Fig. 3.
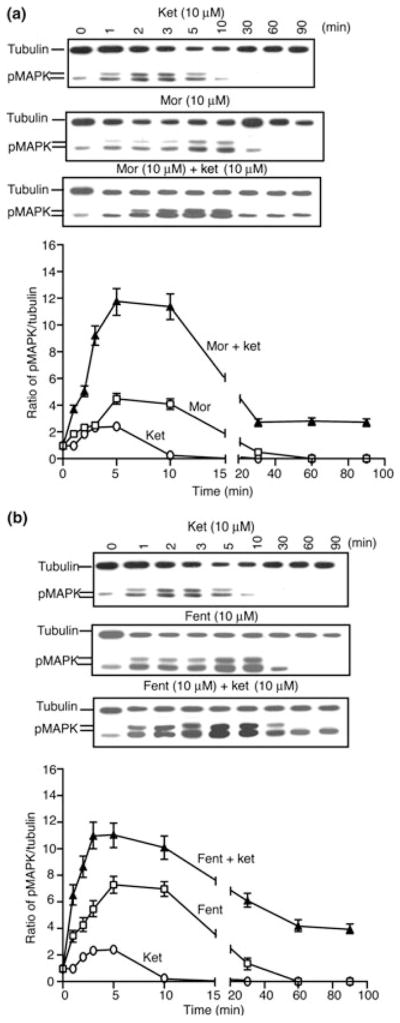
Time course of desensitization of opioid-induced ERK1/2 phosphorylation. SK-N-SH cells were treated with 10 μM of either morphine (a) or fentanyl (b) for different time periods (0–90 min) at 37°C in the absence or presence of ketamine (10 μM) and subjected to western blot analysis using anti-phospho ERK1/2 and anti-tubulin antibodies as described in Materials and methods section. Results are the mean ± SE of six independent experiments in triplicates.
Effect of ketamine on resensitization of μOR-induced ERK1/2 phosphorylation
Next, we examined if the presence of ketamine affected the rate of receptor resensitization (i.e. recovery of morphine- or fentanyl-mediated phosphorylation of ERK1/2). For this, the receptors were desensitized by treatment with either morphine or fentanyl (10 μM) for 30 min (see Fig. 3). Media containing the drugs were removed, cells were quickly washed and incubated in media in the absence or presence of ketamine (10 μM) for 5–90 min to allow for receptor resensitization. At the end of each time interval, cells were treated for 5 min with 10 μM morphine or fentanyl followed by western blot analysis. A schematic representation of the protocol used for these experiments is shown in Fig. 4a.
As seen from Fig. 4b and c in the absence of ketamine, exposure to a second 5 min pulse of either morphine or fentanyl leads to increases in phosphorylated ERK1/2 levels only after 30 min of withdrawal of the drugs from the media. However, in the presence of ketamine we observe a significant shift in the time required for the exposure to a second 5 min pulse of either morphine or fentanyl to induce an increase in phosphorylated ERK1/2 levels (15 min vs. ~30 min in the absence of ketamine) (Fig. 4b and c). In addition, exposure to a second pulse of morphine 90 min after drug withdrawal from the media, in the absence of ketamine, leads to ~2-fold increase in ERK1/2 phosphorylation and this is increased to ~5-fold in the presence of ketamine (Fig. 4b). Similar observations were made in the case of fentanyl, where in the absence of ketamine we see ~4-fold increase in ERK1/2 phosphorylation that is increased to ~10-fold in the presence of ketamine. These results suggest that ketamine is able to affect the rate of receptor resensitization of μ receptor-mediated ERK1/2 signaling.
Effect of ketamine on total ERK1/2 levels
Next, we examined whether opioid treatment in the absence or presence of ketamine changed total ERK1/2 levels. For this, we treated SK-N-SH cells or CHO cells stably expressing μOR with morphine or fentanyl (10 μM) in the absence or presence of ketamine (10 or 100 μM) for upto 4 h. We find that both in CHO cells stably expressing μOR (data not shown) or in SK-N-SH cells endogenously expressing μOR, total ERK1/2 levels are not significantly modulated following treatment with any of these drugs alone or in combination (Figure S3).
Discussion
In this study, we find a synergistic effect of ketamine on morphine/fentanyl-mediated signaling that appears to be selective to the ERK1/2 pathway. Although ketamine was shown to inhibit the formation of cAMP in a dose-dependent manner in CHO cells expressing recombinant μ-, δ- and κ-opioid receptors, this effect was found to be neither additive nor synergistic when ketamine was used in conjunction with opioid receptor agonists (Hirota et al. 1999). Interestingly, we find that ketamine is more effective in potentiating the phosphorylation of ERK1/2 by lower doses of opioid agonists than by higher doses. This correlates with studies showing that ketamine administration increases the effectiveness of opioids used in pain management (Abdel-Ghaffar et al. 1998).
Although ketamine acts primarily as a non-competitive NMDA-receptor antagonist (Willets et al. 1990; Yamamura et al. 1990), several studies have shown that it also exhibits non-NMDA-mediated effects by modulating the activity of nicotinic (Scheller et al. 1996), muscarinic (Hustveit et al. 1995), opioid (Smith et al. 1980; Finck and Ngai 1982; Hustveit et al. 1995; Hirota et al. 1999) as well as AMPA receptors (Li et al. 2010; Zarate et al. 2010). However, the molecular mechanisms underlying this are not clearly understood. A recent study found that intraperitoneal administration of ketamine led to activation of the mTOR and ERK pathways in synaptoneurosomes of prefrontal cortex and this was completely blocked by pretreatment with a selective AMPA receptor inhibitor (Li et al. 2010). In vitro studies examining intracellular signaling have shown that ketamine interacts stereoselectively with μOR and κOR (Finck and Ngai 1982; Hustveit et al. 1995). Studies examining the effects of ketamine on binding of opiate ligands find that high but clinically achievable concentrations of ketamine (> 100 μM) are able to displace a non-selective opioid receptor antagonist ([3H] diprenorphine) binding to recombinant μOR (Hirota et al. 1999). In these studies, ketamine was found to significantly increase the affinity of [3H]diprenorphine binding with no change in total binding. In this study, we find that ketamine significantly increases the affinity of [3H]morphine or [3H]fentanyl albeit only at high doses (100 μM) in cells endogenously expressing μ receptors. The fact that we find that ketamine significantly potentiates morphine- or fentanyl-mediated signaling at doses that do not affect binding supports a role for interactions at the level of intracellular signaling by these drugs.
An interesting observation made in the current study is that while ketamine had no significant effect on the potency of morphine (EC50 from 5.8 to 5.9 nM) in inducing ERK1/2 phosphorylation, it substantially increased the potency of fentanyl (EC50 from 68 to 0.5 nM). This could reflect that ketamine differentially modulates the pathways leading to ERK1/2 phosphorylation following μOR activation by morphine or fentanyl. Studies show that Gi/o coupled receptor-mediated ERK1/2 phosphorylation can occur via G-protein-dependent/-independent mechanisms (May and Hill 2008). In this context, a recent study showed that while morphine could induce ERK1/2 phosphorylation primarily via a G protein-dependent pathway through PKCε, fentanyl could activate both PKCε and β-arrestin-mediated pathways (Zheng et al. 2011). Given that fentanyl can activate ERK1/2 phosphorylation by multiple mechanisms compared with morphine, this would suggest that the robust effects observed with a combination of ketamine and fentanyl could be due to greater effects of ketamine on G protein-independent pathways of ERK1/2 phosphorylation. This is supported by our observation that ketamine did not potentiate opioid (morphine or fentanyl)-mediated signaling at the level of receptor associated G-proteins. Thus, our results suggest that ketamine stabilizes μOR conformations that promote its association with β-arrestin thereby potentiating G protein-independent signaling. Alternatively, ketamine may be changing the localization of the μOR from lipid to non-lipid rafts given the evidence showing that G-protein-mediated ERK1/2 phosphorylation requires μOR localization to lipid rafts whereas β-arrestin-mediated ERK1/2 phosphorylation requires its localization to non-lipid raft domains (Zheng et al. 2008). Further studies are needed to evaluate these possibilities.
The ability of ketamine to increase the duration of opioid-induced analgesia suggests that it could also modulate opioid-induced signaling in such a way that these receptors remained in an active state for a longer time. This could be achieved by influencing the time course of desensitization and resensitization of signaling by opioid receptors. Receptor desensitization which occurs upon short-term (sec to min) exposure of cells to agonists is mediated by uncoupling of activated receptors from G-proteins and this effectively terminates the signaling process. Receptor resensitization occurs after withdrawal of agonists from the media leading to a new round of signaling events following a second challenge with agonists. Our results demonstrate that ketamine delays the desensitization and improves the resensitization of ERK1/2 signaling. The overall effect of ketamine would thus appear to be in keeping opioid-induced ERK1/2 signaling active for a longer time period.
Interestingly, several studies have demonstrated the involvement of ERK1/2 in nociception. For example, noxius stimulus leads to an increase in ERK1/2 phosphorylation in DRG neurons (Ji et al. 1999; Huang et al. 2000) whereas administration of an inhibitor of ERK1/2 phosphorylation leads to pain alleviation (Ji et al. 1999; Karim et al. 2001). However, activation of μOR by opiates leads to ERK1/2 phosphorylation and antinociception (Fukuda et al. 1996; Li and Chang 1996; Gutstein et al. 1997; Trapaidze et al. 2000) and this can be potentiated by ketamine. This indicates that ERK1/2 plays a central role in pain perception that may be modulated by down-stream signaling events eventually leading to either nociception or antinociception.
In conclusion, our results implicate ERK1/2 signaling in ketamine–opioid interactions. They also suggest that events involved in ERK1/2 signaling may have a significant role in delineating the putative beneficial effects of ketamine in preemptive analgesia. As our results indicate that ketamine delays the desensitization and improves the resensitization of signaling via this pathway the overall effect would be in maintaining this pathway active for a longer time period. This could have profound effects on the physiologic responses that occur as a result of the activation of opioid receptors and could account for the observed effects of ketamine on the duration of opioid-induced analgesia.
Acknowledgments
We would like to thank Raphael Rozenfeld and Ittai Bushlin for critical reading of the manuscript. This work is supported in part by grants DA08863 and DA0019251 (to LAD).
Abbreviations used
- CHO
Chinese hamster ovary
- CTOP
D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2
- ERK1/2
extracellular signal-regulated kinase 1/2
- MAPK
mitogen-activated protein kinase
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article:
Figure S1. Effect of ketamine on the phosphorylation of ERK1/2 by μOR agonists.
Figure S2. Effect of a low dose of ketamine on morphine-induced ERK1/2 phosphorylation.
Figure S3. Effect of ketamine on total ERK1/2 levels.
Table S1. Effect of ketamine on agonist-mediated [35S]GTPγS binding.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Abdel-Ghaffar ME, Abdulatif MA, al-Ghamdi A, Mowafi H, Anwar A. Epidural ketamine reduces post-operative epidural PCA consumption of fentanyl/bupivacaine. Can J Anaesth. 1998;45:103–109. doi: 10.1007/BF03013246. [DOI] [PubMed] [Google Scholar]
- Bristow A, Orlikowski C. Subcutaneous ketamine analgesia: Postoperative analgesia using subcutaneous infusions of ketamine and morphine. Ann R Coll Surg Engl. 1989;71:64–66. [PMC free article] [PubMed] [Google Scholar]
- Celerier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, Simonnet G. Long-lasting hyperalgesia induced by fentanyl in rats: Preventive effect of ketamine. Anesthesiology. 2000;92:465–472. doi: 10.1097/00000542-200002000-00029. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang LY. Protein kinase C reduces Mg+2 block of NMDA receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Eide PK, Stubhaug A, Oye I, Breivik H. Continuous subcutaneous administration of the N-methyl-D-aspartic acid (NMDA) receptor antagonist ketamine in the treatment of post-herpetic neuralgia. Pain. 1995;61:221–228. doi: 10.1016/0304-3959(94)00182-E. [DOI] [PubMed] [Google Scholar]
- Finck AG, Ngai SH. Opiate receptor mediation of ketamine analgesia. Anesthesiology. 1982;56:124–127. doi: 10.1097/00000542-198204000-00011. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- Gomes I, Filipovska J, Devi LA. Opioid receptor oligomerization. Detection and functional characterization of interacting receptors. Methods Mol Med. 2003;84:157–183. doi: 10.1385/1-59259-379-8:157. [DOI] [PubMed] [Google Scholar]
- Gutstein HB, Rubie EA, Mansour A, Akil H, Woodgett JR. Opioid effects on mitogen-activated protein kinase signaling cascades. Anesthesiology. 1997;87:1118–1126. doi: 10.1097/00000542-199711000-00016. [DOI] [PubMed] [Google Scholar]
- Heimann AS, Gomes I, Dale CS, et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc Natl Acad Sci USA. 2007;104:20588–20593. doi: 10.1073/pnas.0706980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Lambert DG. Ketamine: its mechanism(s) of action and unusual clinical uses. Br J Anaesth. 1996;77:441–444. doi: 10.1093/bja/77.4.441. [DOI] [PubMed] [Google Scholar]
- Hirota K, Okawa H, Appadu BL, Grandy DK, Devi LA, Lambert DG. Stereoselective interaction of ketamine with recombinant μ, κ and δ opioid receptors expressed in Chinese hamster ovary cells. Anesthesiology. 1999;90:174–182. doi: 10.1097/00000542-199901000-00023. [DOI] [PubMed] [Google Scholar]
- Huang WJ, Wang BR, Yao LB, et al. Activity of p44/42 MAP kinase in the caudal subnucleus of trigeminal spinal nucleus is increased following perioral noxious stimulation in the mouse. Brain Res. 2000;861:181–185. doi: 10.1016/s0006-8993(00)02015-1. [DOI] [PubMed] [Google Scholar]
- Hustveit O, Maurset A, Oye I. Interaction of chiral forms of ketamine with opioid, phencyclidine, sigma and muscarinic receptors. Pharmacol Toxicol. 1995;77:355–359. doi: 10.1111/j.1600-0773.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- Javery KB, Ussery TW, Steger HG, Colclough GW. Comparison of morphine and morphine with ketamine for postoperative analgesia. Can J Anaesth. 1996;43:212–215. doi: 10.1007/BF03011736. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Joachimsson PO, Hedstrand U, Eklund A. Low-dose ketamine infusion for analgesia during postoperative ventilator treatment. Acta Anesthesiol Scand. 1986;30:439–446. doi: 10.1111/j.1399-6576.1986.tb02505.x. [DOI] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., IV Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher A, Laulin JP, Celerier E, Le Moal M, Simonnet G. Acute tolerance associated with a single opiate administration: involvement of N-methyl-D-aspartate-dependent pain facilitatory systems. Neuroscience. 1998;84:583–589. doi: 10.1016/s0306-4522(97)00556-3. [DOI] [PubMed] [Google Scholar]
- Laulin JP, Larcher A, Celerier E, Le Moal M, Simonnet G. Long-lasting increased pain sensitivity in rat following exposure to heroin for the first time. Eur J Neurosci. 1998;10:782–785. doi: 10.1046/j.1460-9568.1998.00083.x. [DOI] [PubMed] [Google Scholar]
- Li LY, Chang KJ. The stimulatory effects of opioids on mitogen-activated protein kinase in Chinese hamster ovary cells transfected to express mu-opioid receptors. Mol Pharmacol. 1996;50:599–602. [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathisen LC, Skjelbred P, Skoglund LA, Oye I. Effect of ketamine, an NMDA receptor inhibitor, in acute and chronic oroficial pain. Pain. 1995;61:215–220. doi: 10.1016/0304-3959(94)00170-J. [DOI] [PubMed] [Google Scholar]
- May LT, Hill SJ. ERK phosphorylation: spatial and temporal regulation of G-protein- coupled receptors. Int J Biochem Cell Biol. 2008;40:2013–2017. doi: 10.1016/j.biocel.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Royblat L, Korotkoruchko A, Katz J, Glazer M, Greemberg L, Fisher A. The effect of low-dose ketamine in addition to general anesthesia. Anesth Analg. 1993;77:1161–1165. doi: 10.1213/00000539-199312000-00014. [DOI] [PubMed] [Google Scholar]
- Scheller M, Bufler J, Hertle I, Schneck HJ, Franke C, Kochs E. Ketamine blocks currents through mammalian nicotinic acetylcholine receptor channels by interaction with both the open and the closed state. Anesth Analg. 1996;83:830–836. doi: 10.1097/00000539-199610000-00031. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Pekoe GM, Martin LL, Coalgate B. The interaction of ketamine with the opiate receptor. Life Sci. 1980;26:789–795. doi: 10.1016/0024-3205(80)90285-4. [DOI] [PubMed] [Google Scholar]
- Stannard CF, Porter GE. Ketamine hydrochloride in the treatment of phantom limb pain. Pain. 1993;36:227–230. doi: 10.1016/0304-3959(93)90214-A. [DOI] [PubMed] [Google Scholar]
- Stiller CO, Gustafsson H, Bergquist J. Morphine induced release of GABA, glycine and glutamate in the dorsal horn of the spinal cord in anaesthetized rats. Soc Neurosci Abstr. 1998;24:1867. [Google Scholar]
- Stubhaug A, Breivik H, Eide PK, Kreunen M, Foss A. Mapping of punctate hyperalgesia around a surgical incision demonstrates that ketamine is a powerful suppressor of central sesitization to pain following surgery. Acta Anaesthesiol Scand. 1997;41:1124–1132. doi: 10.1111/j.1399-6576.1997.tb04854.x. [DOI] [PubMed] [Google Scholar]
- Trapaidze N, Gomes I, Cvejic S, Bansinath M, Devi LA. Opioid receptor endocytosis and activation of MAP kinase pathway. Mol Brain Res. 2000;76:220–228. doi: 10.1016/s0169-328x(00)00002-4. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of opiate tolerance by noncompetitive N-methyl-D-aspartate receptor antagonists. Brain Res. 1994;633:178–188. doi: 10.1016/0006-8993(94)91538-5. [DOI] [PubMed] [Google Scholar]
- Tverskoy M, Oz Y, Isakson A, Finger J, Bradley ELJ, Kissin I. Preemptive effect of fentanyl and ketamine on postoperative pain and wound hyperalgesia. Anesth Analg. 1994;78:205–209. doi: 10.1213/00000539-199402000-00002. [DOI] [PubMed] [Google Scholar]
- Tverskoy M, Oren M, Vaskovivh M, Dashkovsky I, Kissin I. Ketamine enhances local anesthetic and analgesic effects of bupivacine by peripheral mechanism: a study in postoperative patients. Neurosci Lett. 1996;215:5–8. doi: 10.1016/s0304-3940(96)12922-0. [DOI] [PubMed] [Google Scholar]
- Willets J, Balster RL, Leander JD. The behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol Sci. 1990;11:423–428. doi: 10.1016/0165-6147(90)90150-7. [DOI] [PubMed] [Google Scholar]
- Wong CS, Liaw WJ, Tung CS, Su YF, Ho ST. Ketamine potentiates analgesic effect of morphine in postoperative epidural pain control. Reg Anesth. 1996;21:534–541. [PubMed] [Google Scholar]
- Yamamura Y, Harada K, Okamura A, Kemmotsu O. Is the site of action of ketamine anesthesia the N-methyl-D-aspartate receptor? Anesthesiology. 1990;72:704–710. doi: 10.1097/00000542-199004000-00021. [DOI] [PubMed] [Google Scholar]
- Zarate C, Jr, Machado-Vieira R, Henter I, Ibrahim L, Diazgranados N, Salvadore G. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18:293–303. doi: 10.3109/10673229.2010.511059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chu J, Qiu Y, Loh HH, Law PY. Agonist-selective signaling is determined by the receptor location within membrane domains. Proc Natl Acad Sci USA. 2008;105:9421–9426. doi: 10.1073/pnas.0802253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chu J, Zhang Y, Loh HH, Law PY. Modulating mu-opioid receptor phosphorylation swithches agonist-dependent signaling as reflected in PKCe activation and dendritic spine stability. J Biol Chem. 2011;286:12724–12733. doi: 10.1074/jbc.M110.177089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelgänsberger W, Tölle TR. The pharmacology of pain signaling. Curr Opin Neurobiol. 1993;3:611–618. doi: 10.1016/0959-4388(93)90063-5. [DOI] [PubMed] [Google Scholar]