

Significance
Understanding how to optimize lipid-loading onto CD1d molecules is important to better harness invariant natural killer T (iNKT) cells’ central role at the interface between innate and adaptive immunity. We report that the lipid transfer proteins saposins play an essential role in modulating human iNKT cell autoreactivity to antigen-presenting cells activated by inflammatory stimuli. Lipid-loading occurs in an endo-lysosomal compartment, where saposins work as “lipid editors,” capable of fine-tuning loading and unloading of CD1d molecules and increasing the off-rate of CD1d-bound lipids.
Keywords: autoreactivity, inflammation, innate immunity, lipid binding proteins
Abstract
Lipid transfer proteins, such as molecules of the saposin family, facilitate extraction of lipids from biological membranes for their loading onto CD1d molecules. Although it has been shown that prosaposin-deficient mice fail to positively select invariant natural killer T (iNKT) cells, it remains unclear whether saposins can facilitate loading of endogenous iNKT cell agonists in the periphery during inflammatory responses. In addition, it is unclear whether saposins, in addition to loading, also promote dissociation of lipids bound to CD1d molecules. To address these questions, we used a combination of cellular assays and demonstrated that saposins influence CD1d-restricted presentation to human iNKT cells not only of exogenous lipids but also of endogenous ligands, such as the self-glycosphingolipid β-glucopyranosylceramide, up-regulated by antigen-presenting cells following bacterial infection. Furthermore, we demonstrated that in human myeloid cells CD1d-loading of endogenous lipids after bacterial infection, but not at steady state, requires trafficking of CD1d molecules through an endo-lysosomal compartment. Finally, using BIAcore assays we demonstrated that lipid-loaded saposin B increases the off-rate of lipids bound to CD1d molecules, providing important insights into the mechanisms by which it acts as a “lipid editor,” capable of fine-tuning loading and unloading of CD1d molecules. These results have important implications in understanding how to optimize lipid-loading onto antigen-presenting cells, to better harness iNKT cells central role at the interface between innate and adaptive immunity.
In addition to MHC-peptide complexes, subsets of T cells are capable of recognizing lipids in the context of CD1 molecules (1). Of the five CD1 molecules, CD1d restricts the activity of a family of cells known as invariant natural killer T (iNKT) cells, because of their semi-invariant T cell receptor (TCR) use (2). Over the years, it has been shown that lipid-specific iNKT cells play an important immunoregulatory role in cancer, autoimmunity and infections (3). Furthermore, iNKT cells efficiently modulate adaptive immune responses through their cross-talk with dendritic cells (DC) and B cells, resulting in DC maturation and B-cell activation. This property sets iNKT cell agonists as promising powerful immune adjuvants (4), and understanding the molecular mechanisms of lipid antigen presentation and the requirements for iNKT cell activation is essential for clinical harnessing of this population.
Because lipids are water-insoluble in nature, lipid transfer proteins play a crucial role in assisting their extraction from membranes, solubilization for transport and processing, before loading onto CD1 molecules. In the endoplasmic reticulum (ER), the microsomal triglyceride transfer protein has been shown to assist loading of nascent CD1d molecules with self-phospholipids (5, 6). In the lysosomal compartment, lipid transfer proteins belonging to the saposin family have been shown to play an important role in loading lipids onto CD1d and CD1b molecules (7–11). Saposins are a group of four, small, heat-stable glycoproteins required for hydrolysis of sphingolipids by lysosomal hydrolases and membrane remodeling (12). The four saposins A–D derive from the lysosomal processing of the 68- to 73-kDa precursor protein prosaposin by a mechanism not completely understood, but requiring at least cathepsin D function (13). The physiological significance of the saposins in sphingolipid metabolism is underscored by the severe and fatal sphingolipidoses in humans with loss-of-function mutations in the prosaposin gene (14). It is also known that saposins play a crucial role in iNKT cell positive selection by cortical thymocytes, as prosaposin-deficient mice fail to select iNKT cells (9), thus highlighting their pivotal immunological function (15). However, it remains unclear whether saposins can facilitate loading of endogenous iNKT cell agonists in the periphery during inflammatory responses. Indeed, prosaposin-deficient mice have very few residual iNKT cells, and it has not been possible to investigate the role of saposins in modulating presentation of self-lipids by peripheral antigen-presenting cells (APCs) upon infection or during inflammation. We decided, therefore, to address the above question by investigating the role of saposins in CD1d lipid presentation in human APCs in combination with functional and kinetic studies with human recombinant saposins, which have provided insights into the molecular mechanisms of saposin-assisted lipid-loading and lipid exchange.
Results
Presentation of Endogenous CD1d Antigenic Lipids Is Prosaposin-Dependent.
A feature of CD1d-restricted T cells is weak activation triggered by recognition of CD1d molecules loaded with self lipids (16). This basal recognition of self-lipid complexes can be further amplified by innate signals and cytokine-driven signals (17–19), leading to iNKT cell activation during microbial infections, even upon exposure to pathogens not known to contain iNKT cell antigens (20). To study the importance of prosaposin in controlling iNKT cell autoreactivity to activated human APCs, we silenced prosaposin expression in the myelomonocytic cell line THP-1 (Fig. S1). After establishing the extent of prosaposin knockdown with four different shRNA lentiviruses, we used the cell line with the highest level of knockdown (90%) for all subsequent cellular experiments. A scramble shRNA lentivirus-transduced cell line was used as a control for nonspecific off-target silencing effects. In initial experiments with synthetic iNKT cell agonists, we confirmed that presentation of the α-galactosyl ceramide (α-GalCer) analog C20:2 was saposin-independent, as this lipid is preferentially loaded extracellularly (21) (Fig. S2A). Conversely, presentation of α-GalCer, of the nonglycosidic derivative threitol ceramide [ThrCer (22)] and of the glycolipid Gal(α1→2)GalCer (hereafter referred to as GalGalCer), which requires processing of the terminal galactose by α-galactosidase A (23), was severely affected by the lack of prosaposin (Fig. S2A). The Gal(α1→2) derivative of ThrCer (GalThr) was also saposin-dependent (Fig. S2A).
CD1d surface expression was not affected by lack of prosaposin (Fig. S2B), suggesting that the lower iNKT cell stimulatory capacity of prosaposin-deficient THP-1 was likely to be because of reduced loading of CD1d molecules with iNKT cell agonists. Indeed, prosaposin-deficient THP-1 cells showed a decreased staining with a fluorescent tetrameric soluble iNKT TCR (Fig. 1A), which could be corrected by transducing prosaposin deficient cells with a prosaposin retrovirus (Fig. 1A, Bottom lines).
Fig. 1.

Presentation of endogenous lipids is saposin-dependent. (A) THP-1–CD1d cells transduced with control (Top), prosaposin shRNA (Middle), and the latter reconstituted with a prosaposin retrovirus (Bottom) were pulsed overnight with decreasing concentrations (green, red, and blue lines) of α-GalCer (300, 100, 30 ng/mL); C20:2 (300, 100, 30 ng/mL), ThrCer (1,000, 300, 100 ng/mL), or GalGalCer (1,000, 500, 250 ng/mL). Flow cytometry of cells stained with soluble iNKT TCR tetramers is shown. Data are representative of five independent experiments. Gray dotted lines represent vehicle-pulsed cells. (B) THP-1–CD1d cells transduced with control or prosaposin shRNA were infected with L. monocytogenes at the indicated MOI and incubated with iNKT cells, in the presence or absence of the anti-CD1d blocking antibody. IFN-γ secreted in the supernatant at 36 h was measured by ELISA (mean ± SD). The response of seven other iNKT cell lines is shown in Fig S3. (C) THP-1–CD1d cells transduced with control (Top), prosaposin (C), or UGCG shRNA (Bottom) were infected with L. monocytogenes (Left), E. faecalis (Center), or K. aerogenes (Right), at the indicated MOI (colored histograms). Flow cytometry of cells stained with soluble iNKT TCR tetramers is shown. Data are representative of three to four independent experiments (cumulative results are shown in Fig. S2C). Gray lines represent uninfected cells and cyan lines represent cells stained with a control tetramer. (D) THP-1–CD1d cells transduced with control (Left), prosaposin shRNA (Center), and the latter reconstituted with a prosaposin retrovirus (Right) were pulsed overnight with the indicated concentrations of β-GlcCer (colored histograms). Flow cytometry of cells stained with soluble iNKT TCR tetramers is shown. Data are representative of three independent experiments (cumulative results are shown in Fig. S2C). Gray dotted lines represent vehicle-pulsed cells.
Having optimized an assay to study the importance of prosaposin in controlling iNKT cell activation, we next infected prosaposin-competent and -deficient THP-1 cells with the Gram-positive bacterium Listeria monocytogenes, because it is known that iNKT cells are critical in vivo for the development of protective immunity against Listeria (24), despite the absence of known iNKT cell antigens encoded by this microbe (20). We observed that iNKT cells displayed strong CD1d-dependent reactivity in response to Listeria-infected THP-1, which was reduced in the absence of prosaposin (Fig. 1). We observed comparable results with six of eight different iNKT cell lines, but with two iNKT lines, the reactivity against saposin-competent and -deficient cells was almost comparable (Fig. S3). The autoreactive iNKT cell response to bacterial infected THP-1 cells was dominated by IFN-γ secretion, and no significant increase in IL-4 secretion was detected (Fig. S3). iNKT cell activation in response to APCs infected with microbes depends on CD1d-mediated triggering of the TCR and is further modulated by cytokines, such as IL-12, released by APC in response to pattern-recognition receptor triggering (17). To investigate the contribution of CD1d-self lipids toward TCR-mediated iNKT cell activation in the absence of the confounding cytokine signals, we stained Listeria-infected prosaposin-competent and -deficient THP-1 cells with the soluble iNKT TCR, which we have previously shown to be able to detect up-regulation of CD1d-self lipid complexes at the cell surface of Toll-like receptor (TLR) agonist-matured THP-1 cells (19). We demonstrated that Listeria-infected prosaposin-deficient THP-1 cells displayed fewer CD1d–lipid complexes at the cell surface, compared with control THP-1 cells (Fig. 1C, Top and Middle rows, and Fig. S2C). Secretion of comparable amounts of IL-8 by prosaposin-competent and -deficient cells exposed to Listeria suggested equal infection of the two cell types (Fig. S4C). In addition, we observed similar results upon infection with other bacteria, both Gram-positive (Enterococcus faecalis) and Gram-negative (Klebsiella aerogenes) (Fig. 1C, Top and Middle rows, and Fig. S2C), suggesting a common mechanism of iNKT cell activation.
Because no antigenic lipids have been identified from Listeria extracts (20), we explored the possibility of iNKT cell activation by self-antigens. It has recently been shown that an abundant endogenous lipid, β-d-glucopyranosylceramide (β-GlcCer), is a potent iNKT cell self-antigen in mice and humans, contributing to iNKT cell activation following myeloid cell infection and in response to TLR agonists (25). We therefore silenced with shRNA β-glucosylceramide synthase (UGCG) to assess the contribution of β-GlcCer to iNKT cell autoreactivity in our experimental system. shRNA silencing of UGCG in THP-1 cells completely abrogated detection of CD1d–lipid complexes upon bacterial infection (Fig. 1C, Bottom). UGCG-silenced cells expressed comparable levels of CD1d to control cells, presented equally well GalGalCer, and released similar amounts of IL-8 upon bacteria infection (Fig. S4), excluding nonspecific effects on antigen presentation because of gene silencing. As an extension of the above results, we showed that recognition of synthetic β-GlcCer by human iNKT cells is prosaposin-dependent, as β-GlcCer pulsed prosaposin-deficient THP-1 cells displayed fewer CD1d–lipid complexes at the cell surface than wild-type controls (Fig. 1D and Fig. S2C). Defective β-GlcCer presentation by prosaposin-deficient cells was corrected following transduction with a prosaposin-encoding retrovirus (Fig. 1D).
Taken together, these results underscore an important role for saposins in facilitating loading of β-GlcCer–derived self-antigens up-regulated by THP-1 cells during bacterial infection for CD1d-dependent recognition by human iNKT cells.
Prosaposin-Dependent Presentation of Endogenous iNKT Cell Agonists Requires an Intact CD1d Cytoplasmic Domain.
Reactivity of human and murine iNKT cells to synthetic agonists depends on CD1d trafficking through the endolysosomal compartment. Conversely, although it is established that reactivity of murine iNKT cells to CD1d loaded with endogenous iNKT cell agonists requires trafficking of CD1d molecules through the lysosome (26–29), the contribution of the endolysosomal environment to human iNKT cell autoreactivity upon infection is unclear. To address this question, we overexpressed in THP-1 cells CD1d molecules deleted of the last 10 amino acids of the cytoplasmic tail [TD-CD1d (30)]. Upon infection with L. monocytogenes, THP-1 cells expressing TD-CD1d elicited lower iNKT cell activation (Fig. 2A) and displayed fewer CD1d–lipid complexes than cells expressing WT CD1d (Fig. 2B). Moreover, in the absence of prosaposin expression, CD1d–lipid complexes were no longer detectable at the cell surface of infected THP-1 cells transduced with TD-CD1d molecules (Fig. 2B). Loading of TD-CD1d molecules with exogenous synthetic iNKT cell agonists was also reduced (Fig. S5).
Fig. 2.

Reactivity to self-lipids requires trafficking of CD1d molecules through the endolysosomal compartment. (A) THP-1 expressing physiological levels of CD1d or transduced with WT or TD-CD1d molecules were infected with L. monocytogenes (MOI 150) and incubated with human iNKT cells. IFN-γ secreted in the supernatant at 36 h was measured by ELISA (mean ± S.D.). Data are representative of five independent experiments. (B) Staining with soluble iNKT TCR tetramers of THP-1 transduced with control or prosaposin shRNA and expressing WT or TD-CD1d molecules upon infection with L. monocytogenes at the indicated MOI. Staining of untransduced THP-1 is also shown as a control. Gray lines: uninfected cells. Data are representative of three independent experiments.
Taken together, these results indicate that presentation of self-lipids to human iNKT cells by bacteria-infected human APCs requires trafficking of CD1d molecules through the lysosomal compartment and saposin-assisted loading. Furthermore, these results are consistent with the known role of the cytoplasmic tail of murine CD1d in modulating trafficking of CD1d molecules and their loading with endogenous iNKT cell agonists (26, 28, 29).
Lipid-Loaded Saposin B Mediates Lipid Transfer onto CD1d Molecules and Accelerates Dissociation of CD1d-Bound Lipids.
The crystal structure of saposin B has revealed the presence of a large hydrophobic binding site capable of accommodating a broad range of different lipids (31). Although it is accepted that lipid-loaded saposins promote lipid transfer onto CD1d molecules (9), it remains unclear whether they also accelerate the rate of dissociation of lipids already bound to CD1d molecules. To address this question, we developed a surface plasmon resonance assay (SPR or BIAcore) based on the binding of soluble iNKT TCR to CD1d molecules coated onto BIAcore chips in the presence or absence of recombinant saposin molecules.
In initial experiments using a combination of cellular and plate-bound assays, we compared all four recombinant saposins for their ability to load iNKT cell agonists onto CD1d molecules. In agreement with previously published reports (8, 32), we showed a dominant role of saposin B in accelerating and overall enhancing loading of soluble lipids onto CD1d molecules (Fig. S6). Based on these results we decided to use recombinant saposin B for the cell-free studies. To prove the ability of the recombinant saposin B to bind synthetic iNKT cell agonists, we synthesized radiolabeled ThrCer (14C-ThrCer). We demonstrated that saposin B binds to 14C-ThrCer at a range of concentrations and, as expected, with higher affinity at pH 5 (Kd = 1.8 μM) than at pH 7 (Kd = 4.5 μM) (Fig. 3A).
Fig. 3.

Cell-free assays to study saposin-mediated lipid transfer onto CD1d molecules. (A) Binding curve of Saposin B to 14C-ThrCer at pH5 or 7 using in vitro scintillation proximity assay. (Inset) The Scatchard plots of the same data. (B–D) Real-time saposin B-dependent lipid-loading of CD1d molecules immobilized on a streptavidin BIAcore chip. Lipid, recombinant saposin B or premixed lipid–saposin B complexes were passed on the immobilized CD1d, and serial dilutions of recombinant iNKT TCR were passed to assess the loading of the CD1d molecules. [(B) α-GalCer, (C) ThrCer, (D) C20:2]. Saturation plots show binding at equilibrium of increasing concentrations of iNKT TCR to loaded CD1d molecules. Binding affinities (Kd) measured at equilibrium are also shown in each panel. (Insets) The Scatchard plots of the same data. (E) Real-time exchange of CD1d-bound lipids by saposin B. Immobilized CD1d molecules were loaded to saturation with ThrCer, followed by the injection of SapB premixed with GalGalCer (black bars), or SapB alone (white bars) to displace ThrCer. CD1d loading was detected with the soluble iNKT TCR (which recognizes ThrCer–CD1d complexes but not GalGalCer–CD1d, Left) or the 9B antibody (which recognizes the galactose head group and not ThrCer–CD1d, (Right). Plotted are the Bmax values, representing saturating amounts of TCR bound to CD1d. (F) Half-life of CD1d–ThrCer complexes in the presence or absence of lipid-loaded saposin B. Bacterially expressed human CD1d molecules, refolded with ThrCer, were immobilized on three flow cells of a BIAcore chip. One flow cell was left untreated to assess the spontaneous off-rate of the ThrCer–CD1d complex. SapB alone or premixed with GalGalCer was injected onto the second and third flow cells, respectively, for 5 min after 60, 120, and 240 min (time is indicated on the x axis). ThrCer–CD1d complexes were quantified passing serial dilution of the iNKT TCR and the response units at saturation are plotted on the y axis.
We next measured saposin B-mediated lipid-loading onto CD1d molecules in a BIAcore assay. Lipids (α-GalCer or ThrCer), recombinant saposin B, or a premix of saposin B-lipid were injected, each onto one flow-cell of a BIAcore chip where the same amount of CD1d was immobilized (Fig. 3 B and C). The level of CD1d loading with the antigenic lipids was assessed with the iNKT TCR. In all cases, loading of lipids alone for the short time of this assay did not result in detectable iNKT TCR recognition (Fig. 3 B and C). However, we detected a significant increase in iNKT TCR binding to CD1d molecules loaded with α-GalCer or ThrCer in the presence of saposin B (Fig. 3 B and C). Notably, the α-GalCer analog C20:2 could be loaded on CD1d molecules even in the absence of saposin B (Fig. 3D). However, saposin B markedly enhanced loading of C20:2 (Fig. 3D), as also observed in a cell-free plate-bound assay. This result is consistent with the observation that the diunsaturated C20:2 analog has less stringent requirements for loading (21) and can be presented equally well by prosaposin-deficient THP-1 (Fig. S2A). No difference in the affinity of binding of the iNKT TCR for CD1d-C20:2 complexes was observed in the presence or absence of saposin B, demonstrating that saposin B mainly accelerates the loading process without altering the position of the lipid in the antigen binding groove.
Having established that recombinant saposin B can bind to exogenous lipids and enhances their loading onto CD1d molecules, we set up an assay to assess whether lipid-loaded saposin B is also capable of displacing lipids already bound in the CD1d groove. To specifically test the lipid exchange activity of saposin B by BIAcore assay, we used, in addition to the soluble iNKT TCR, the phage display-derived recombinant antibody 9B, which recognizes CD1d molecules loaded with lipids linked to α-anomeric galactose head groups (33, 34). Because the iNKT TCR and the 9B antibody have different specificity, they can be used to distinguish between CD1d molecules loaded with GalGalCer (which are specifically seen by the 9B antibody, but not by the iNKT TCR) and CD1d molecules loaded with ThrCer (which are specifically seen by the iNKT TCR, but not by the 9B antibody). As expected, after loading to saturation CD1d molecules immobilized on the BIAcore chip with saposin B and ThrCer, these complexes could only be detected by the iNKT TCR (Fig. 3E, Left, first and third columns), but not by the 9B antibody (Fig. 3E, Right, first column). We then injected in the ThrCer/CD1d-saturated flow cells either saposin B loaded with GalGalCer complexes or saposin B alone, and compared the binding of iNKT TCR and the 9B antibody to CD1d molecules. We demonstrated that injection of saposin B alone in the ThrCer/CD1d-saturated flow cell was not sufficient to reduce the iNKT TCR signal (Fig. 3E, Left, fourth column, white), but injection of saposin B–GalGalCer complexes led to a decrease of the iNKT TCR signal (Fig. 3E, Left, second column) and an increase in the 9B signal (Fig. 3E, Right, second column).
Finally, confirming the ability of saposin B to displace CD1d-bound lipids, we observed that the off-rate of ThrCer bound to CD1d molecules was enhanced by saposin B–GalGalCer complexes, but not by saposin B alone (Fig. 3F).
These results indicate that only lipid-loaded saposin B is able to increase the off-rate of CD1d-bound lipids, thus facilitating the loading of its lipid cargo onto the CD1d groove. Because it is likely that lysosomal saposin B will be loaded with a spectrum of lipids, we speculate that the efficiency of its lipid-exchange ability may vary depending on the molecular identity of the lipids bound to both saposin B and CD1d molecules, underscoring the fundamental role for saposins in editing the profile of lipids bound to CD1 molecules.
Mathematical Model of Saposin-Mediated Lipid Exchange.
To illustrate the observed effect of saposin B on lipid-loading onto CD1d, we constructed a mathematical model that includes the essential features of these molecular interactions (Fig. 4). The model includes CD1d molecules that are either loaded with irrelevant (C) or relevant (C*) lipids that exchange based on two possible mechanisms (Fig. 4A): a constitutive saposin-independent exchange at a low basal rate and a saposin-dependent exchange at a higher rate. Importantly, we did not include any binding preference of the relevant or irrelevant lipid with saposin or CD1d (i.e., CD1d and saposin interacted with both L and L* with identical reaction parameters). Calculations of the steady-state concentration of CD1d bound to relevant lipids (C*) increased as the concentration of the relevant lipid (L*) was increased, but saposin had no effect on lipid-loading (Fig. 4B). In agreement with this, when we measured the stability of CD1d–lipid complexes immunoprecipitated from saposin-competent or -deficient cells, we observed no differences (Fig. 4C). However, when we examined the kinetic effects of saposin B, we observed that it decreased the timescale of lipid exchange (Fig. 4D), allowing the newly added relevant lipids to be quickly loaded onto CD1d molecules previously occupied with irrelevant lipids, in agreement with the experimental data shown in Fig. 3. Of note, the present model is similar—and is expected to give qualitatively similar results in the steady-state limit—to a recently published model of the role of Tapasin in mediating peptide-exchange from MHC molecules (35).
Fig. 4.

A mathematical model of saposin-mediated lipid exchange. (A) Simplified schematic showing the reactions included in the mathematical model. (B) The steady-state normalized concentration of CD1d bound to relevant lipid (C*/(C* + C) (y axis) was determined for increasing concentrations of relevant lipids at a fixed concentration of irrelevant lipids (L*, x axis) for the indicated concentrations of saposin B. (C) Immunoprecipitation of CD1d/β2M complexes, as indicated by the presence of β2m in precipitates with anti CD1d42.1 antibody. Lysates of THP-1 cells transduced with a control or prosaposin shRNA were incubated on ice for 2 h (lanes 1, 5, 9, 13) or at 37 °C for 45 min, 2 h, or 2 h in the presence of α-GalCer (10 µg/mL) (Upper) or 4.5 h with and without α-GalCer (Lower). A conformation-specific anti-CD1d antibody was used to precipitate intact CD1d/β2M complexes that had not fallen apart during the incubation period. (D) Kinetics of lipid exchange at the indicated concentration of saposin B. The normalized concentration of CD1d with relevant lipids (C*/(C* + C) (y axis) is plotted as a function of time following the addition of 1 μM of relevant lipids. Increasing the saposin concentration decreases the timescale to reach the maximum concentration of C*. Note that the maximum reached after a long time (steady state) is identical at all saposin concentrations, as expected based on B. The model equations and parameters used to generate B and D of this figure are described in SI Text.
Discussion
In this study, using a soluble iNKT TCR and iNKT cell lines, we have demonstrated an important role of prosaposin in loading human CD1d molecules with iNKT cell agonists that are up-regulated following bacterial infection of APCs. We have also provided evidence for a previously unappreciated role for lipid-loaded saposin B in increasing the off-rate of CD1d-bound lipids, thus promoting lipid exchange.
To investigate the role of saposins in modulating CD1d-dependent human iNKT cells autoreactivity to myeloid cells upon microbial recognition, we set up an in vitro system using THP-1 cells in which prosaposin expression was silenced by lentiviral shRNA. Using a soluble iNKT TCR, we have demonstrated that prosaposin-deficient THP-1 cells pulsed with synthetic iNKT cell agonists displayed fewer CD1d–lipid complexes at the cell surface, and therefore elicited reduced iNKT cell activation. This defect could be corrected either reexpressing prosaposin in the prosaposin-deficient THP-1 cells or adding recombinant saposins (as also seen by ref. 8). In agreement with previous results obtained by Kang and Cresswell with saposin-deficient murine fibroblasts expressing human CD1d (10), we did not detect any significant difference in iNKT basal autoreactivity between saposin-competent and -deficient cells. However, upon exposure to a range of different bacteria or incubation with TLR agonists, prosaposin-deficient cells displayed fewer CD1d–lipid complexes at the cell surface that could be recognized by a soluble tetrameric iNKT TCR. Although we have no biochemical evidence of the lipids recognized upon bacterial infection of THP-1, silencing of UGCG, the enzyme responsible for β-GlcCer biosynthesis, prevented detection of CD1d–lipid complexes at the cell surface upon bacterial infection of prosaposin-competent cells, consistent with the role of the self-glycosphingolipid β-GlcCer (25) as a dominant self-antigen. Furthermore, we have shown that presentation of synthetic β-GlcCer is also saposin-dependent.
The cellular compartment in which β-GlcCer or other self-lipids responsible for iNKT cell autoreactivity could be loaded onto CD1d molecules is currently unknown, and it might be anywhere between the luminal side of the secretory compartment and the saposin-positive endolysosomal compartment. It is known that β-GlcCer is synthesized on the cytoplasmic face of the Golgi apparatus from the precursor ceramide that is transported from the ER to the Golgi via a ceramide transport protein-dependent mechanism (36). β-GlcCer is then flipped to the luminal Golgi side [although the molecular identity of the flippase is still controversial (37)] and transported by the lipid transfer protein FAPP2 (38) toward the distal parts of the Golgi for biosynthesis of more complex glycosphingolipids. By vesicular and nonvesicular transport through the cell, β-GlcCer and other self-lipids might then access the endolysosomal compartment, where exposure to acidic hydrolases and lipid transfer proteins, such as GM2 activator (GM2a) and saposins, could orchestrate their loading onto recycling CD1d molecules. Cellular lipids bound to human CD1d molecules isolated from different cellular compartments have been determined by high-resolution mass spectrometry (39–41). Neo-synthesized CD1d molecules have been shown to acquire phosphatidylcholine in the ER, which is then exchanged for sphingomyelin when CD1d traffics through the Golgi (41). The identity of the lipid transfer protein facilitating exchange of CD1d-bound lipids in the Golgi is currently unknown; however, prosaposin itself might be involved, as it possesses functional lipid transfer activity and binds gangliosides (42, 43). Indeed, in preliminary experiments we have observed that prosaposin enhances loading of β-GlcCer on plate-bound CD1d molecules at pH 7. Whether β-GlcCer could be identified as a major species associated with secreted or recycling CD1d molecules remains an open question, as the previous analysis was performed on resting Epstein-Barr virus-B cells (39, 41) or in myeloid cells after digestion with ceramide glycanase, which does not cleave monohexoses from the ceramide backbone (40).
To further dissect the role of saposins in lipid-loading in the secretory versus the endolysosomal compartments, we expressed in prosaposin-sufficient and -deficient THP-1 cells CD1d molecules lacking the cytoplasmic motif known to be important for its internalization and endo-lysosomal targeting (TD-CD1d) (30). By confocal microscopy we observed that tail-deleted CD1d molecules could still reach the lysosomal Lamp-1+ compartment (Fig. S7), although less efficiently (likely because of association with invariant chain), which is highly expressed in myeloid cells (30, 44, 45). Consequently, presentation of exogenous synthetic iNKT cell agonists was only moderately affected by tail-deleted CD1d molecules, yet it remained dependent on prosaposin expression. However, following bacterial infection, fewer CD1d-self lipid complexes were displayed at the cell surface of THP-1–expressing TD-CD1d molecules, and no complexes were detected in the absence of prosaposin. The observation that basal but not inflammatory autoreactivity is maintained in the absence of prosaposin expression suggests that a qualitative and quantitative difference in the lipid profile exists between uninfected and infected THP-1. For example, basal autoreactivity to phospholipids, such as phosphatidylinositol, has been demonstrated for murine iNKT cells (46), and this may not be prosaposin-dependent despite the ability of saposin B to bind and transfer phospholipids in vitro at acidic pH (47). Furthermore, the saposin-dependence of peroxisome-derived lipids (48) or of lysophospholipids (49), which stimulate thymic and peripheral iNKT cells, remains to be determined. It is also possible that in myeloid cells association of the TD-CD1d molecules with the invariant chain in the ER (30, 44, 45) may alter the kinetics of trafficking to the lysosomes, rendering these CD1d molecules more sensitive to the presence or absence of prosaposin. Our results underscore the important role for prosaposin and CD1d trafficking through the endolysosomal compartment for CD1d-restricted presentation of endogenous lipids by infected human APCs. Ultimately, a high-resolution analysis of the lipid repertoire associated with secreted and recycling CD1d molecules will unambiguously reveal the identity of the lipid profile of immature versus stimulated myeloid cells and the changes in the presence or absence of prosaposin.
Prosaposin deficiency leads to altered cellular sphingolipid metabolism and storage of multiple sphingolipids (50). To investigate the role of saposins as lipid transfer proteins independently of any storage phenotype, we designed cell-free assays and measured binding of Escherichia coli-purified recombinant saposins to synthetic lipids and subsequent loading of CD1d molecules. Although there is evidence that glycosylation influences the ability of recombinant saposins to mobilize lipids from membranes and liposomes (51, 52), nonglycosylated proteins have been shown to retain comparable lipid binding and hydrolase-activating capacity. Despite high homology, the four saposins have different lipid specificities, as demonstrated by the phenotype of patients with selective deficiency of individual molecules (53). We limited our study to the role of saposin B, because evidence from our own works and from the literature (8, 11, 32) suggested its dominant role in loading soluble lipids onto CD1d molecules. We determined the binding affinity of saposin B to a radiolabeled version of ThrCer at pH5 and pH7 (1.8 μM and 4.5 μM, respectively), and observed that it reached a plateau when saposin B was present in molar excess. Furthermore, competing lipids or detergent molecules easily displaced binding of ThrCer, consistent with the ability of saposin B to bind a variety of lipids. Our results are in agreement with published data on saposin B–lipid interactions, obtained through different experimental methods: disk gel electrophoresis, pH shift gel electrophoresis, gel filtration, lipid solubilization assays, ion exchange assays (50), or competition assays to measure relative binding affinities to a variety of lipids (54). The majority of investigators have reported the formation of equimolar complexes (1 mol per lipid per mole of saposin B dimer).
The main effect of recombinant saposins was to enhance and accelerate the loading of CD1d molecules with iNKT cell agonists, both in a plate-bound assay and in a real-time SPR assay. Notably, a soluble iNKT TCR bound with similar affinity to CD1d molecules loaded with C20:2 in the presence or absence of saposin B, hence formally demonstrating that saposin-assisted loading of iNKT cell agonists onto CD1d molecules does not alter the binding affinity of the iNKT TCR.
The recombinant human CD1d molecules used in our cell-free assays are purified from HEK293T cells and are loaded with a spectrum of cell lipids derived from the ER, Golgi, and secretory compartment (55), some of which are reported to be antigenic for human iNKT cells (56). Therefore, the capacity of lipid-bound saposin B to load these recombinant CD1d molecules also implied its ability to promote lipid exchange. Indeed, using a combination of reagents that selectively recognize CD1d bound to ThrCer or GalGalCer, we have been able to demonstrate specific lipid exchange occurring in the SPR assay. In agreement with results shown by Zhou et al. (9) and Yuan et al. (8), saposin B alone (not precomplexed with an iNKT cell agonist) could not remove lipids already bound to CD1d molecules, unlike what has been shown for CD1e (57). However, we have shown that lipid-loaded saposin B specifically increased the off-rate of lipids bound to CD1d molecules. Importantly, our experimental data are consistent with a mechanistic mathematical model whereby lipid exchange can take place by both a saposin-independent (low exchange rate) and a saposin-dependent (high exchange rate) mechanism. This conclusion suggests a role for saposins similar to that played by tapasin in peptide exchange on MHC class I molecules, which has been recently modeled (35). Together, these results highlight the important role of saposins as lipid-editors for CD1d antigen presentation, in addition to GM2a (9) and Niemann Pick type C2 protein (NPC2) (58). In contrast to DM molecules, which edit the repertoire of peptides bound to MHC class II molecules (59), saposins do not have an enzymatic mechanism of action and we and others have not been able to demonstrate physical association with human CD1d molecules (8), unlike what has been shown for murine CD1d and saposin A (9) or human CD1b and CD1c and saposin C (7, 11). Despite extensive in vivo and in vitro studies, the ultimate mechanism of hydrolase activation by saposins is unclear and that of lipid transfer onto CD1d molecules remains unknown. The two main proposed modes of action of saposin (11, 60, 61) are the “detergent-like or solubilizer” (saposin B and GM2a), in which saposins extract lipids from bilayers and present them to hydrolases as a soluble protein–lipid complex, and the “liftase model” (saposin C), whereby saposins remodel the bilayer surface facilitating binding of the hydrolases to the relative substrates. We speculate that saposin B molecules, dimeric at neutral and low pH and with a large hydrophobic cavity (31), may readily solubilize lipids and offer them to recycling CD1d molecules throughout the endocytic compartment, actively promoting lipid exchange in the groove of CD1d molecules by increasing the off-rate of already bound ligands. Lipid transfer proteins might be required to facilitate exchange of complex lipids bound to CD1d molecules, but the acidic pH in the presence of competing lipids (naturally present in the lysosome) already induces rapid, spontaneous dissociation of iNKT cell agonists with shorter acyl or phytosphingosine chains (62). In conclusion, in concert with their ability to promote glycosphingolipid degradation, the four members of the saposin family may use different strategies to facilitate access of a variety of endogenous and microbial lipid antigens to the CD1d groove, thus shaping the repertoire of agonists available for iNKT cell recognition.
Experimental Procedures
Medium and Reagents.
The complete medium (CM) used throughout was RPMI 1640 (Gibco) for THP-1, IMDM (Gibco) for iNKT cells, and DMEM (Gibco) for fibroblasts. CM was supplemented with 2 mM l-glutamine, 1% nonessential amino acids, 1% sodium pyruvate, 1% pen/strep, 5 × 10−5 2 ME (all from Gibco), and serum: 10% (vol/vol) FCS (Sigma) for THP-1; 5% (vol/vol) Human AB Serum (Sigma) for iNKT cells, and 20% (vol/vol) FCS for fibroblasts. Recombinant human IL-2 was produced in our laboratory, as previously described (63).
Lipids: C24:1 Glucosyl(β)Ceramide [D18:1/24:1 (15Z)] was purchased from Avanti Polar Lipids. α-GalCer, C20:2, Gal(α1→2)GalCer, and Threitol Ceramide were synthesized by a strategy described previously (64–66) and their structures were confirmed by mass spectrometry. The dried lipids were dissolved at 10 mg/mL in a solution of chloroform:methanol:water [10:10:3 (vol/vol/vol)], followed by dilution in 150 mM NaCl, 0.5% Tween 20 (vehicle solution) at 100–200 μg/mL stock solution (depending on solubility). The solution was heated at 80 °C for 5 min followed by sonication for 5 min in an ultrasonic water bath. 14C-ThrCer was synthesized as previously described (67).
cDNAs encoding full-length prosaposin (10) or mutants deleted of each individual saposin (8) were cloned in the retroviral expression vector pBMN and retroviruses produced according to published protocols (www.stanford.edu/group/nolan/protocols/pro_helper_dep.html).
Recombinant hexahistidine-tagged human saposins were cloned and purified from E. coli as described previously (68), and their identity confirmed with saposin-specific antibodies (69). His6-tagged full-length prosaposin was expressed HEK293T cells using the pHLSec vector and purified as described previously (70).
Recombinant human CD1d molecules were cloned and purified from HEK293T cells as described previously (71). The α-GalCer-CD1d specific antibody 9B was generated and purified as described (33). For the experiment described in Fig. 3F, bacterially purified human CD1d was refolded with ThrCer, as previously described (22).
Generation of iNKT Cells.
Blood was purchased from the U.K. National Blood Service. Human iNKT cells were isolated by cell sorting with CD1d–α-GalCer tetramers or Vα24 and Vβ11 antibodies (Immunotech) directly from peripheral blood mononuclear cells or after expansion with autologous DCs pulsed with α-GalCer, as described previously (72). iNKT cells were grown in CM [containing 5% (vol/vol) human AB serum instead of FCS] supplemented with 1,000 U/mL IL-2 and periodically restimulated.
Generation of THP-1 Cells Overexpressing Human CD1d Constructs.
Full-length human CD1d was cloned in the pHR-SIN lentiviral vector. Lentivirus particles were made as previously described (19) and used to infect THP-1 cells (ATCC). Tail-deleted (TD) human CD1d, lacking the last 10 amino acids, was cloned in the pHR-SIN lentiviral vector using a strategy previously described (30).
Stimulation Assays.
THP-1 and THP-1-CD1d cells were plated at 50,000 per well in U bottom 96-well plates and used to stimulate iNKT cells (20,000–30,000 per well in duplicate or triplicate) in the presence of lipids at the indicated concentration.
THP-1 and iNKT cell activation was assessed by ELISA (IFN-γ, IL-4, IL-12 p40, and IL-8, all from BD Pharmingen; GM-CSF from eBiosciences) on supernatants harvested after 36 h.
Bacterial Infection.
L. monocytogenes (a mutant with deletion of actA and inlB) was grown in BHI medium. E. faecalis and K. aerogenes were grown in LB. An overnight culture of the bacteria was reinoculated at a dilution of 1:100 and expanded for further 4 h before harvesting and extensive washing with PBS. OD600 was measured to determine bacterial numbers and bacteria were UV killed. Bacteria were applied to THP-1 at the indicated multiplicity of infection (MOI) for 3 h. Cells were washed and incubated overnight before analysis with the soluble iNTK TCR. Alternatively, after washing, infected THP-1 were incubated with iNKT cell for 36 h and supernatants collected for ELISA. In some experiments, the anti-CD1d blocking antibody CD1d-42 (Pharmingen) was added at 25 μg/mL 1 h before addition of iNKT cells.
Preparation of Soluble Heterodimeric TCRs and iNKT Tetramer Staining.
Soluble iNKT TCR heterodimers were generated as previously described (19). The biotinylated TCR heterodimers were tetramerized by mixing at a 4:1 molar ratio with Streptavidin PE (Molecular Probes) or PE-Cy7 (eBiosciences).
THP-1–CD1d cells were pulsed overnight with different concentrations of lipids or infected with bacteria. Cells were washed and incubated with 0.5–1 μg iNKT-tetramer or equivalent amounts of streptavidin or control tetramer biotin at 4° C for 1 h. Cells were washed and samples were analyzed on a Cyan flow cytometer (Dako). Data were processed using Flowjo software (TreeStar).
Scintillation Proximity Assay.
Different concentrations of His6-tagged SapB were preincubated with different concentrations of 14C-ThrCer in a total volume of 100 μL in phosphate-citrate buffer (pH5 or 7) for 1 h at room temperature, to reach equilibrium. The complex was added to 110 μL prewashed Ni-NTA agarose beads (Qiagen) and continuously mixed at 4 °C for 45 min. The beads were washed three times, the supernatant discarded, and the radioactive signal was quantified by scintillation.
SPR Assay.
SPR experiments were performed on a BIAcore 3000. Approximately 1,000 response units (RU) biotinylated hCD1d was immobilized on streptavidin-coated sensor chips (GE Healthcare). Various concentrations of lipids alone or premixed with SapB were passed on the captured CD1d molecules, followed by serial dilutions of iNKT TCR to assess the level of loading of CD1d molecules. All SPR experiments were performed at 25° C in the standard BIAcore running buffer HBS-EP and data are shown after subtraction from a channel loaded with empty CD1d molecules, which remained untreated throughout the experiment.
Data analysis to obtain the binding curves shown was performed using BIAevaluation 3.1 (BIAcore) and Prism 5.0 (GraphPad) by globally fitting the data to a simple 1:1 Langmuir ligand-binding model defined by the following formula:
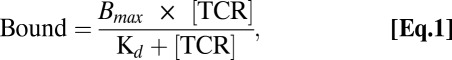 |
where “Bound” is measured in response units, Bmax is the maximum response (in RU), and Kd is the equilibrium dissociation constant.
Exchange Assays.
Biotinylated hCD1d molecules were loaded onto streptavidin-coated BIAcore chip (∼1,000 RU) and were saturated with ThrCer. For the exchange, an equilibrium mix of 225 μg SapB ± 3 μg GalGalCer was injected onto the chip; serial dilutions of iNKT TCR or the α-GalCer-CD1d–specific 9B antibody were used to assess the unloading of ThrCer and loading of GalGalCer, respectively.
Plate-Bound Assay.
Biotinylated human CD1d molecules purified from HEK 293T cells were bound to streptavidin-coated 96-well plates in citrate-phosphate buffer pH5, as described previously (73), and loaded with lipids in the presence or absence or recombinant saposins for the indicated times (Fig. S6 B and C). iNKT cells were added and their activation was measured by IFN-γ ELISA on supernatants harvested after 36 h.
Immunoprecipitation.
The protocol for immunoprecipitation was adapted from ref. 74. For this protocol, 107 THP-1 cells expressing wild-type or mutant CD1d were lysed for 30 min on ice in 2 mL 0.5% Triton X 100 lysis buffer containing protease inhibitors mixture (Roche). Cell debris was removed by 10 min at 15,000 × g 4 °C centrifugation and supernatant was split into several tubes. One tube was left on ice and the others were incubated at 37 °C for 45 min, 2 h, or 4.5 h with or without α-GalCer (10 µg/mL). Samples were shaken throughout. After incubation all samples were transferred to ice and precleared for 2 h using protein G Plus-agarose (Santa Cruz), which was then pelleted and removed. Anti-CD1d42.1 antibody (Pharmingen) was added to supernatant (10 µg/mL) and left on ice for 1 h, protein G Plus-agarose was added and tubes rotated at 4 °C for 1 h. Agarose was washed four times in lysis buffer, resuspended in loading buffer containing SDS and DTT (New England Biolabs), and heated to 80 °C while shaking for 5 min. Proteins in the samples were separated using SDS/PAGE and transferred to PVDF membranes (Bio-Rad), which were then blocked with 5% (vol/vol) skimmed milk in 0.5% Tween 20 in PBS for 2 h at room temperature. The membranes were then probed using antihuman β-2 microglobulin specific antibody (BBM.1) and HRP conjugated goat antimouse secondary antibody. HRP reaction was developed using Supersignal West Pico kit (Thermo Scientific).
Statistical Analysis.
Descriptive statistics are expressed as the mean ± SD values. Comparisons between groups were performed using two-tailed t test and a P value of <0.05 was considered significant. See Tables S1 and S2 for phenotypes of the iNKT cell lines and parameter values used in this paper.
Supplementary Material
Acknowledgments
We thank Prof. Alain R. Townsend (University of Oxford) for the gift of 293T cells secreting biotin-tagged human CD1d molecules. This work was supported by The Wellcome Trust Grants 084923/B/08/Z (to G.S.B.) and 084923Z/08/Z (to V.C.), Cancer Research UK Programme Grant C399/A2291 (to V.C.), Advanced Immunization Technologies Grant 280873 (to V.C.), and The Medical Research Council. G.S.B. acknowledges support in the form of a Personal Research Chair from Mr. James Bardrick.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.K. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1310050110/-/DCSupplemental.
References
- 1.Barral DC, Brenner MB. CD1 antigen presentation: How it works. Nat Rev Immunol. 2007;7(12):929–941. doi: 10.1038/nri2191. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: What’s in a name? Nat Rev Immunol. 2004;4(3):231–237. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- 3.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 4.Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9(1):28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 5.Dougan SK, et al. Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen-presenting cells. J Exp Med. 2005;202(4):529–539. doi: 10.1084/jem.20050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeissig S, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med. 2012;18(7):1060–1068. doi: 10.1038/nm.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winau F, et al. Saposin C is required for lipid presentation by human CD1b. Nat Immunol. 2004;5(2):169–174. doi: 10.1038/ni1035. [DOI] [PubMed] [Google Scholar]
- 8.Yuan W, et al. Saposin B is the dominant saposin that facilitates lipid binding to human CD1d molecules. Proc Natl Acad Sci USA. 2007;104(13):5551–5556. doi: 10.1073/pnas.0700617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou D, et al. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 2004;303(5657):523–527. doi: 10.1126/science.1092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang SJ, Cresswell P. Saposins facilitate CD1d-restricted presentation of an exogenous lipid antigen to T cells. Nat Immunol. 2004;5(2):175–181. doi: 10.1038/ni1034. [DOI] [PubMed] [Google Scholar]
- 11.León L, et al. Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc Natl Acad Sci USA. 2012;109(12):4357–4364. doi: 10.1073/pnas.1200764109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishimoto Y, Hiraiwa M, O’Brien JS. Saposins: Structure, function, distribution, and molecular genetics. J Lipid Res. 1992;33(9):1255–1267. [PubMed] [Google Scholar]
- 13.Hiraiwa M, et al. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): Its mechanism and inhibition by ganglioside. Arch Biochem Biophys. 1997;341(1):17–24. doi: 10.1006/abbi.1997.9958. [DOI] [PubMed] [Google Scholar]
- 14.O’Brien JS, Kishimoto Y. Saposin proteins: Structure, function, and role in human lysosomal storage disorders. FASEB J. 1991;5(3):301–308. doi: 10.1096/fasebj.5.3.2001789. [DOI] [PubMed] [Google Scholar]
- 15.Darmoise A, Maschmeyer P, Winau F. The immunological functions of saposins. Adv Immunol. 2010;105:25–62. doi: 10.1016/S0065-2776(10)05002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendelac A, Bonneville M, Kearney JF. Autoreactivity by design: Innate B and T lymphocytes. Nat Rev Immunol. 2001;1(3):177–186. doi: 10.1038/35105052. [DOI] [PubMed] [Google Scholar]
- 17.Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003;4(12):1230–1237. doi: 10.1038/ni1002. [DOI] [PubMed] [Google Scholar]
- 18.Paget C, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity. 2007;27(4):597–609. doi: 10.1016/j.immuni.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 19.Salio M, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci USA. 2007;104(51):20490–20495. doi: 10.1073/pnas.0710145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brigl M, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011;208(6):1163–1177. doi: 10.1084/jem.20102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu KO, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci USA. 2005;102(9):3383–3388. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silk JD, et al. Cutting edge: nonglycosidic CD1d lipid ligands activate human and murine invariant NKT cells. J Immunol. 2008;180(10):6452–6456. doi: 10.4049/jimmunol.180.10.6452. [DOI] [PubMed] [Google Scholar]
- 23.Prigozy TI, et al. Glycolipid antigen processing for presentation by CD1d molecules. Science. 2001;291(5504):664–667. doi: 10.1126/science.291.5504.664. [DOI] [PubMed] [Google Scholar]
- 24.Arrunategui-Correa V, Kim HS. The role of CD1d in the immune response against Listeria infection. Cell Immunol. 2004;227(2):109–120. doi: 10.1016/j.cellimm.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Brennan PJ, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol. 2011;12(12):1202–1211. doi: 10.1038/ni.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cernadas M, et al. Lysosomal localization of murine CD1d mediated by AP-3 is necessary for NK T cell development. J Immunol. 2003;171(8):4149–4155. doi: 10.4049/jimmunol.171.8.4149. [DOI] [PubMed] [Google Scholar]
- 27.Chiu YH, et al. Distinct subsets of CD1d-restricted T cells recognize self-antigens loaded in different cellular compartments. J Exp Med. 1999;189(1):103–110. doi: 10.1084/jem.189.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiu YH, et al. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat Immunol. 2002;3(1):55–60. doi: 10.1038/ni740. [DOI] [PubMed] [Google Scholar]
- 29.Elewaut D, et al. The adaptor protein AP-3 is required for CD1d-mediated antigen presentation of glycosphingolipids and development of Valpha14i NKT cells. J Exp Med. 2003;198(8):1133–1146. doi: 10.1084/jem.20030143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, et al. Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J Immunol. 2007;178(10):6181–6190. doi: 10.4049/jimmunol.178.10.6181. [DOI] [PubMed] [Google Scholar]
- 31.Ahn VE, Faull KF, Whitelegge JP, Fluharty AL, Privé GG. Crystal structure of saposin B reveals a dimeric shell for lipid binding. Proc Natl Acad Sci USA. 2003;100(1):38–43. doi: 10.1073/pnas.0136947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Im JS, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity. 2009;30(6):888–898. doi: 10.1016/j.immuni.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denkberg G, et al. Phage display-derived recombinant antibodies with TCR-like specificity against alpha-galactosylceramide and its analogues in complex with human CD1d molecules. Eur J Immunol. 2008;38(3):829–840. doi: 10.1002/eji.200737518. [DOI] [PubMed] [Google Scholar]
- 34.McCarthy C, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med. 2007;204(5):1131–1144. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dalchau N, et al. A peptide filtering relation quantifies MHC class I peptide optimization. PLOS Comput Biol. 2011;7(10):e1002144. doi: 10.1371/journal.pcbi.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanada K, et al. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426(6968):803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
- 37.Futerman AH, Riezman H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005;15(6):312–318. doi: 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 38.D’Angelo G, et al. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature. 2007;449(7158):62–67. doi: 10.1038/nature06097. [DOI] [PubMed] [Google Scholar]
- 39.Cox D, et al. Determination of cellular lipids bound to human CD1d molecules. PLoS ONE. 2009;4(5):e5325. doi: 10.1371/journal.pone.0005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muindi K, et al. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d. Proc Natl Acad Sci USA. 2010;107(7):3052–3057. doi: 10.1073/pnas.0915056107. and correction (2010) 107(13):6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan W, Kang SJ, Evans JE, Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol. 2009;182(8):4784–4791. doi: 10.4049/jimmunol.0803981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiraiwa M, et al. Isolation, characterization, and proteolysis of human prosaposin, the precursor of saposins (sphingolipid activator proteins) Arch Biochem Biophys. 1993;304(1):110–116. doi: 10.1006/abbi.1993.1328. [DOI] [PubMed] [Google Scholar]
- 43.Hiraiwa M, Soeda S, Kishimoto Y, O’Brien JS. Binding and transport of gangliosides by prosaposin. Proc Natl Acad Sci USA. 1992;89(23):11254–11258. doi: 10.1073/pnas.89.23.11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang SJ, Cresswell P. Regulation of intracellular trafficking of human CD1d by association with MHC class II molecules. EMBO J. 2002;21(7):1650–1660. doi: 10.1093/emboj/21.7.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jayawardena-Wolf J, Benlagha K, Chiu YH, Mehr R, Bendelac A. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity. 2001;15(6):897–908. doi: 10.1016/s1074-7613(01)00240-0. [DOI] [PubMed] [Google Scholar]
- 46.Gumperz JE, et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity. 2000;12(2):211–221. doi: 10.1016/s1074-7613(00)80174-0. [DOI] [PubMed] [Google Scholar]
- 47.Ciaffoni F, et al. Saposin B binds and transfers phospholipids. J Lipid Res. 2006;47(5):1045–1053. doi: 10.1194/jlr.M500547-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Facciotti F, et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat Immunol. 2012;13(5):474–480. doi: 10.1038/ni.2245. [DOI] [PubMed] [Google Scholar]
- 49.Paduraru C, et al. Role for lysosomal phospholipase A2 in iNKT cell-mediated CD1d recognition. Proc Natl Acad Sci USA. 2013;110(13):5097–5102. doi: 10.1073/pnas.1302923110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: Stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 51.Remmel N, Locatelli-Hoops S, Breiden B, Schwarzmann G, Sandhoff K. Saposin B mobilizes lipids from cholesterol-poor and bis(monoacylglycero)phosphate-rich membranes at acidic pH. Unglycosylated patient variant saposin B lacks lipid-extraction capacity. FEBS J. 2007;274(13):3405–3420. doi: 10.1111/j.1742-4658.2007.05873.x. [DOI] [PubMed] [Google Scholar]
- 52.Locatelli-Hoops S, et al. Saposin A mobilizes lipids from low cholesterol and high bis(monoacylglycerol)phosphate-containing membranes: Patient variant Saposin A lacks lipid extraction capacity. J Biol Chem. 2006;281(43):32451–32460. doi: 10.1074/jbc.M607281200. [DOI] [PubMed] [Google Scholar]
- 53.Schulze H, Kolter T, Sandhoff K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim Biophys Acta. 2009;1793(4):674–683. doi: 10.1016/j.bbamcr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 54.Fluharty CB, Johnson J, Whitelegge J, Faull KF, Fluharty AL. Comparative lipid binding study on the cerebroside sulfate activator (saposin B) J Neurosci Res. 2001;63(1):82–89. doi: 10.1002/1097-4547(20010101)63:1<82::AID-JNR10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 55.Haig NA, et al. Identification of self-lipids presented by CD1c and CD1d proteins. J Biol Chem. 2011;286(43):37692–37701. doi: 10.1074/jbc.M111.267948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fox LM, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PLoS Biol. 2009;7(10):e1000228. doi: 10.1371/journal.pbio.1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Facciotti F, et al. Fine tuning by human CD1e of lipid-specific immune responses. Proc Natl Acad Sci USA. 2011;108(34):14228–14233. doi: 10.1073/pnas.1108809108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schrantz N, et al. The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J Exp Med. 2007;204(4):841–852. doi: 10.1084/jem.20061562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denzin LK, Cresswell P. HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell. 1995;82(1):155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 60.Alattia JR, Shaw JE, Yip CM, Privé GG. Molecular imaging of membrane interfaces reveals mode of beta-glucosidase activation by saposin C. Proc Natl Acad Sci USA. 2007;104(44):17394–17399. doi: 10.1073/pnas.0704998104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alattia JR, Shaw JE, Yip CM, Privé GG. Direct visualization of saposin remodelling of lipid bilayers. J Mol Biol. 2006;362(5):943–953. doi: 10.1016/j.jmb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 62.Bai L, et al. Lysosomal recycling terminates CD1d-mediated presentation of short and polyunsaturated variants of the NKT cell lipid antigen alphaGalCer. Proc Natl Acad Sci USA. 2009;106(25):10254–10259. doi: 10.1073/pnas.0901228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Traunecker A, Oliveri F, Karjalainen K. Myeloma based expression system for production of large mammalian proteins. Trends Biotechnol. 1991;9(4):109–113. doi: 10.1016/0167-7799(91)90038-j. [DOI] [PubMed] [Google Scholar]
- 64.Jervis PJ, et al. Synthesis and biological activity of alpha-glucosyl C24:0 and C20:2 ceramides. Bioorg Med Chem Lett. 2010;20(12):3475–3478. doi: 10.1016/j.bmcl.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Veerapen N, et al. Synthesis and biological activity of alpha-galactosyl ceramide KRN7000 and galactosyl (alpha1—>2) galactosyl ceramide. Bioorg Med Chem Lett. 2009;19(15):4288–4291. doi: 10.1016/j.bmcl.2009.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wojno J, et al. Amide analogues of CD1d agonists modulate iNKT-cell-mediated cytokine production. ACS Chem Biol. 2012;7(5):847–855. doi: 10.1021/cb2005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia Diaz YR, Wojno J, Cox LR, Besra GS. Synthesis of threitol ceramide and [14C]threitol ceramide, non-glycosidic analogues of the potent CD1d antigen a-galactosyl ceramide. Tetrahedron Asymmetry. 2009;20(6–8):747–753. [Google Scholar]
- 68.Qi X, Leonova T, Grabowski GA. Functional human saposins expressed in Escherichia coli. Evidence for binding and activation properties of saposins C with acid beta-glucosidase. J Biol Chem. 1994;269(24):16746–16753. [PubMed] [Google Scholar]
- 69.Chu Z, Witte DP, Qi X. Saposin C-LBPA interaction in late-endosomes/lysosomes. Exp Cell Res. 2005;303(2):300–307. doi: 10.1016/j.yexcr.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 70.Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 10):1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- 71.Schimanski LM, et al. In vitro binding of HFE to the cation-independent mannose-6 phosphate receptor. Blood Cells Mol Dis. 2009;43(2):180–193. doi: 10.1016/j.bcmd.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 72.Gadola SD, Dulphy N, Salio M, Cerundolo V. Valpha24-JalphaQ-independent, CD1d-restricted recognition of alpha-galactosylceramide by human CD4(+) and CD8alphabeta(+) T lymphocytes. J Immunol. 2002;168(11):5514–5520. doi: 10.4049/jimmunol.168.11.5514. [DOI] [PubMed] [Google Scholar]
- 73.Naidenko OV, et al. Binding and antigen presentation of ceramide-containing glycolipids by soluble mouse and human CD1d molecules. J Exp Med. 1999;190(8):1069–1080. doi: 10.1084/jem.190.8.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Odyniec AN, et al. Regulation of CD1 antigen-presenting complex stability. J Biol Chem. 2010;285(16):11937–11947. doi: 10.1074/jbc.M109.077933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.