

Significance
Inhibitors of cyclic nucleotide phosphodiesterase (PDE) PDE3A increase cardiac contractility in patients with heart failure, but their long-term use increases mortality. Two isoforms expressed in cardiac myocytes, PDE3A1 and PDE3A2, have amino acid sequences that are identical except for a unique N-terminal extension in PDE3A1. We found that PDE3A1 and PDE3A2 are selectively phosphorylated at alternative sites in response to the activation of PKA and PKC, respectively, resulting in differential regulation of their catalytic activity and protein interactomes. Existing PDE3 inhibitors thus target at least two functionally distinct cardiac isoforms likely with different roles in intracellular signaling. This raises the possibility that isoform-selective targeting may increase contractility in failing hearts without increasing mortality, providing a potential route for developing therapeutics.
Keywords: cAMP, protein–protein interactions
Abstract
Inhibitors of cyclic nucleotide phosphodiesterase (PDE) PDE3A have inotropic actions in human myocardium, but their long-term use increases mortality in patients with heart failure. Two isoforms in cardiac myocytes, PDE3A1 and PDE3A2, have identical amino acid sequences except for a unique N-terminal extension in PDE3A1. We expressed FLAG-tagged PDE3A1 and PDE3A2 in HEK293 cells and examined their regulation by PKA- and PKC-mediated phosphorylation. PDE3A1, which is localized to intracellular membranes, and PDE3A2, which is cytosolic, were phosphorylated at different sites within their common sequence. Exposure to isoproterenol led to phosphorylation of PDE3A1 at the 14-3-3–binding site S312, whereas exposure to PMA led to phosphorylation of PDE3A2 at an alternative 14-3-3–binding site, S428. PDE3A2 activity was stimulated by phosphorylation at S428, whereas PDE3A1 activity was not affected by phosphorylation at either site. Phosphorylation of PDE3A1 by PKA and of PDE3A2 by PKC led to shifts in elution on gel-filtration chromatography consistent with increased interactions with other proteins, and 2D electrophoresis of coimmunoprecipitated proteins revealed that the two isoforms have distinct protein interactomes. A similar pattern of differential phosphorylation of endogenous PDE3A1 and PDE3A2 at S312 and S428 is observed in human myocardium. The selective phosphorylation of PDE3A1 and PDE3A2 at alternative sites through different signaling pathways, along with the different functional consequences of phosphorylation for each isoform, suggest they are likely to have distinct roles in cyclic nucleotide-mediated signaling in human myocardium, and raise the possibility that isoform-selective inhibition may allow inotropic responses without an increase in mortality.
Cyclic nucleotide phosphodiesterases (PDEs) of the PDE3A subfamily regulate contraction and relaxation in cardiac muscle (1). Inhibitors of these enzymes are used to increase cardiac contractility in patients with heart failure by raising intracellular cAMP content and increasing the phosphorylation of PKA substrates involved in intracellular Ca2+ cycling, including phospholamban, L-type Ca2+ channels, and ryanodine-sensitive Ca2+ channels (2–5). Their long-term use is associated with increased mortality, however, possibly resulting from the phosphorylation of transcription factors by PKA and consequent proapoptotic changes in gene expression (6–9). Finding a way to obtain the inotropic benefits of PDE3 inhibition without its adverse consequences would constitute a major therapeutic advance.
Three isoforms of PDE3A, generated by transcription and translation from alternative start sites in the PDE3A gene, are expressed in cardiac myocytes (Fig. 1) (10). Their amino acid sequences are identical except for the presence of different lengths of the N-terminal sequences involved in intracellular localization, and they are indistinguishable with respect to catalytic activity and inhibitor sensitivity (11–13). PDE3A1, which has a unique N-terminal extension containing hydrophobic loops that insert into intracellular membranes, is recovered solely in microsomal fractions of cardiac muscle, whereas PDE3A2 and PDE3A3 are recovered in cytosolic as well as microsomal fractions. The three isoforms also differ with respect to the presence of phosphorylation sites involved in protein–protein interactions. Both PDE3A1 and PDE3A2 contain a PKC site, S428, through which PDE3A2 binds to 14-3-3 in HeLa cells and human platelets (14, 15), as well as a PKA site, S312, which resembles a documented 14-3-3–binding site in PDE3B (16, 17), although interactions of 14-3-3 with PDE3A1 have not yet been reported to our knowledge. The effects of phosphorylation on interactions with other proteins have not been described, and although phosphorylation of PDE3A has been shown to increase its cAMP hydrolytic activity (15, 18–20), effects on individual isoforms have not been characterized.
Fig. 1.

PDE3 isoforms. Three isoforms of PDE3A (PDE3A1, PDE3A2, and PDE3A3) were generated through transcription and translation from alternative initiation sites. Sites of phosphorylation in the N terminus, the unique N-terminal extension of PDE3A1, the common N-terminal sequence of PDE3A1 and PDE3A2, and the conserved C-terminal catalytic region (in black) are depicted. PDE3B, transcribed from a different gene, has a domain structure similar to that of PDE3A1, but with a longer N-terminal extension.
We examined the role of phosphorylation in regulating the protein–protein interactions and catalytic activity of PDE3A1 and PDE3A2. In the experiments described below, we transfected HEK293 cells with PDE3A1 and PDE3A2 and examined their modulation in response to phosphorylation by PKA and PKC. We discovered that PDE3A1 and PDE3A2 are selectively phosphorylated at alternative sites in their common N-terminal sequence; PKA activation by isoproterenol leads to phosphorylation of PDE3A1 at S312, whereas PKC activation by PMA leads to phosphorylation of PDE3A2 at S428. This differential phosphorylation of PDE3A1 and PDE3A2, which was also observed in human myocardium, was accompanied by distinct changes in their catalytic activities and protein interactomes.
Results
FLAG-Tagged PDE3A1 and PDE3A2 Localize to Separate Intracellular Compartments in Transfected HEK293 Cells.
Microsomes and cytosolic fractions were prepared from HEK293 cells transiently transfected with FLAG-tagged PDE3A1 and PDE3A2. Western blot analysis with anti-FLAG antibodies showed that PDE3A1 is recovered solely in microsomal fractions, whereas PDE3A2 is recovered solely in cytosolic fractions (Fig. S1). These findings demonstrate that the unique N-terminal extension of PDE3A1 directs the protein to intracellular membranes in transfected HEK293 cells, consistent with previous studies in other cell lines (11, 12).
PDE3A1 and PDE3A2 Bind 14-3-3 Through Phosphorylation Within Their N-Terminal Sequences.
Extracts of HEK293 cells expressing FLAG-tagged PDE3A isoforms were incubated with GST-tagged 14-3-3 in the absence and presence of PKA and ATP. Immunoprecipitation with anti-FLAG antibodies and Western blot analysis with anti-phospho PKA substrate and anti-GST antibodies showed that PDE3A1 and PDE3A2 can be phosphorylated by PKA, and that GST-14-3-3 coimmunoprecipitates only with phosphorylated forms (Fig. 2A). GST-14-3-3 does not coimmunoprecipitate with PDE3A3, which lacks the N-terminal sequence common to PDE3A1 and PDE3A2 and is not phosphorylated by PKA. These results indicate that phosphorylation sites in PDE3A1 and PDE3A2 upstream of amino acid 483, the start site of PDE3A3, regulate their interactions with 14-3-3.
Fig. 2.

Identification of the N-terminal 14-3-3–binding region of PDE3A. (A) HEK293 cells were transfected with FLAG-tagged PDE3A constructs corresponding to PDE3A1, PDE3A2, and PDE3A3. Cell lysates were incubated with GST-tagged 14-3-3 in the absence or presence of 100 nM PKA and 200 µM ATP for 20 min at 30 °C. PDE3A isoforms were immunoprecipitated using anti-FLAG antibodies. Samples were analyzed by Western blot analysis using anti-GST, anti-phospho PKA substrate, and anti-FLAG antibodies. (B) Microsomes and cytosolic fractions of human myocardium were incubated with GST-tagged 14-3-3 in the absence or presence of 100 nM PKA and 200 µM ATP for 20 min at 30 °C. GST-tagged 14-3-3 was immunoprecipitated using anti-GST antibody, and immunoprecipitated proteins were identified by Western blot analysis with anti-GST and anti-PDE3A C-terminal antibodies.
To confirm the relevance of these findings to cardiac muscle, we incubated preparations from human myocardium with GST-tagged 14-3-3 in the absence and presence of PKA and ATP. Immunoprecipitation with anti-GST antibodies and Western blot analysis with anti-PDE3A antibodies demonstrated the coimmunoprecipitation of phosphorylated PDE3A1 and PDE3A2 with 14-3-3 in microsomal fractions and of PDE3A2 in cytosolic fractions (which do not contain PDE3A1) (Fig. 2B).
Phosphorylation of PDE3A1 and PDE3A2 by PKA at S312 or S428 Results in Binding to 14-3-3.
Using peptide arrays covering the unique N-terminal sequence of PDE3A1 and the common N-terminal sequence of PDE3A1 and PDE3A2, we confirmed that 14-3-3 binds selectively to peptides with phosphoserine substitutions at S312 and S428, which lie within the common N-terminal sequence of PDE3A1 and PDE3A2 (Fig. S2A). FLAG-tagged constructs based on this sequence (i.e., PDE3A2-NT) were phosphorylated by exogenous PKA in the presence of GST-tagged 14-3-3. Immunoprecipitation of WT PDE3A2-NT resulted in the coimmunoprecipitation of 14-3-3 (Fig. S2B). Individual S312A and S428A mutations did not prevent these interactions, but binding to 14-3-3 was blocked when both sites were mutated in combination. These results indicate that the common N-terminal sequence of PDE3A1 and PDE3A2 is sufficient for binding to 14-3-3, and that phosphorylation at either S312 or S428 alone is sufficient for this interaction.
Activation of PKA by Isoproterenol in Transfected HEK293 Cells Results in Selective Phosphorylation of PDE3A1 at S312.
HEK293 cells transfected with FLAG-tagged PDE3A1 or PDE3A2 were exposed to isoproterenol to stimulate cAMP production and activate PKA. Immunoprecipitation with anti-FLAG antibodies and Western blot analysis with phosphospecific antibodies raised against PDE3A-derived peptides with phosphoserine substitutions at S312 and S428 demonstrated the phosphorylation of PDE3A1 at S312 and the coimmunoprecipitation of endogenous 14-3-3, with little phosphorylation of PDE3A2 (Fig. 3A). When PDE3A2-transfected cells were exposed to isoproterenol in the presence of 3-isobutyl-1-methylxanthine (IBMX), which converts the β-adrenergic receptor-stimulated increase in intracellular cAMP from a localized increase to a spatially uncoupled and diffuse response (21), PDE3A2 was readily phosphorylated at S312 (Fig. 3B). Furthermore, when affinity-purified FLAG-tagged PDE3A1 and PDE3A2 were phosphorylated in vitro by PKA, PDE3A2 was phosphorylated more robustly than PDE3A1 at both S312 and S428, and for both isoforms, S312 was phosphorylated more robustly than S428 (Fig. 3C). These results indicate that the selective phosphorylation of PDE3A1 at S312 in response to isoproterenol reflects the compartment-selective regulation of cAMP-mediated signaling.
Fig. 3.
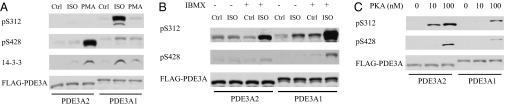
Phosphorylation of PDE3A1 and PDE3A2 in response to PKA and PKC activation. HEK293 cells were transfected with FLAG-tagged PDE3A2 and PDE3A1. (A) Before lysis and immunoprecipitation with anti-FLAG antibodies, cells were treated with 1 µM isoproterenol for 90 s or with 10 ng/mL PMA for 15 min. (B) The experiment was carried out in cells incubated in the absence and presence of 100 µM IBMX before treatment with 1 µM isoproterenol for 90 s. (C) FLAG-tagged PDE3A2 and PDE3A1, affinity-purified from transfected cells using anti-FLAG antibodies, were incubated in the absence or presence of 10 or 100 nM PKA and 200 µM ATP for 20 min at 30 °C. Phosphorylation was analyzed by Western blot analysis with phosphospecific antibodies as indicated. The coimmunoprecipitation of endogenous 14-3-3 was analyzed by immunoblotting with anti–14-3-3 antibody.
Activation of PKC by PMA in Transfected HEK293 Cells Results in Selective Phosphorylation of PDE3A2 at S428.
We compared the effects of PKC activation by PMA on the phosphorylation of FLAG-tagged PDE3A1 and PDE3A2 in transfected HEK293 cells. Exposure of PDE3A2-transfected cells to PMA led to a marked increase in phosphorylation at S428, with no increase in phosphorylation at S312, along with increased binding to endogenous 14-3-3, consistent with observations in HeLa cells (14, 15). In contrast, exposure of PDE3A1-transfected cells to PMA led to little increase in phosphorylation at S428, no increase in phosphorylation of S312, and little increase in binding to endogenous 14-3-3 (Fig. 3A).
PKA and PKC Modulate the Catalytic Activity of PDE3A1 and PDE3A2 Through Distinct Molecular Mechanisms.
In view of the reported effects of PKA and PKC on PDE3A activity (14–17), we examined the effects of phosphorylation at S312 and S428 on the cAMP hydrolytic activity of PDE3A isoforms in transfected HEK293 cells. Exposure of PDE3A2-transfected cells to PMA led to an increase in cAMP hydrolysis that was blocked by the introduction of an S428A substitution (Fig. 4A). Treatment of these cells with the nonhydrolyzable analog dibutyryl cAMP (dBcAMP), which causes a receptor-independent, noncompartmentalized activation of PKA, resulted in phosphorylation at both S312 and S428; the stimulation of catalytic activity was similar in magnitude to that seen in response to PMA, and was similarly blocked by the S428A mutation. These findings demonstrate a role for phosphorylation at S428 in regulating the catalytic activity of PDE3A2.
Fig. 4.

Stimulation of PDE3A2 and PDE3A1 activity in response to PKC and PKA activation. (A) HEK293 cells were transfected with FLAG-tagged PDE3A2, with and without alanine substitution at S428, and treated with 10 ng/mL PMA for 15 min or with 1 mM dBcAMP for 1 h. (B) HEK293 cells were transfected with FLAG-tagged PDE3A1, with and without alanine substitutions at S312 and S428, and then treated with 1 µM isoproterenol for 90 s, with 10 ng/mL PMA for 15 min, or with 1 mM dBcAMP for 1 h. PDE3 activity was assayed as described. PDE3A constructs were immunoprecipitated with anti-FLAG antibody, and phosphorylation was analyzed by immunoblotting with anti-FLAG and phosphospecific antibodies as indicated.
In contrast, exposure of PDE3A1-transfected HEK293 cells to isoproterenol did not stimulate cAMP hydrolytic activity, but exposure to dBcAMP led to a significant increase in the phosphorylation of PDE3A1 at both S312 and S428 and to a stimulation of cAMP hydrolytic activity (Fig. 4B). This stimulation was not observed when cells were treated with either the Rap guanine nucleotide exchange factor 3 (EPAC) activator 8-pCPT-2'-O-Me-cAMP or with sodium butyrate (Fig. S3). The same stimulation of catalytic activity in response to dBcAMP was observed when cells were transfected with a PDE3A1 construct into which S312A and S428A substitutions were introduced. Western blot analysis with anti-phospho PKA substrate antibodies confirmed that PDE3A1-S312A/S428A is phosphorylated at other sites in its sequence under these conditions (Fig. S4). These results indicate that neither S312A nor S428A regulates the catalytic activity of PDE3A1, the stimulation of which in response to dBcAMP is likely to result instead from the phosphorylation of other PKA sites in the PDE3A1 sequence.
Phosphorylation of PDE3A1 by PKA and of PDE3A2 by PMA Stimulates Incorporation into Distinct Interactomes.
The phosphorylation of PDE3B has been shown to lead to its incorporation into multiprotein complexes in 3T3 adipocytes, as evidenced by a shift in migration on gel-filtration chromatography (22, 23). To determine whether similar consequences of phosphorylation occur in the case of PDE3A isoforms, we analyzed protein extracts from transfected HEK293 cells, untreated or exposed to either isoproterenol or PMA, by gel-filtration chromatography, measurement of catalytic activity, and Western blot analysis. Under basal conditions, PDE3 activity and protein was recovered in a biphasic pattern in low- and high-molecular weight peaks (Fig. 5). Exposure of PDE3A1-transfected cells to isoproterenol led to a pronounced shift of activity and protein from low- to high-molecular weight peaks; exposure of PDE3A2-transfected cells to PMA had a similar effect. These results suggest that phosphorylation of PDE3A1 by PKA and of PDE3A2 by PKC promotes their incorporation into multiprotein complexes.
Fig. 5.

Superose 6 (S6) gel-filtration chromatography of HEK cell extracts. HEK293 cells transfected with FLAG-tagged PDE3A1 or PDE3A2 were treated with 1 µM isoproterenol for 90 s or with 10 ng/mL PMA for 15 min, respectively. Solubilized cell extracts (3 mg protein, 1 mL) were subjected to chromatography on S6 columns. Then 10-μL portions of eluted fractions (0.5 mL) were assayed for PDE3 activity, and 20-μL portions were used for SDS/PAGE and Western blot analysis with anti-PDE3A antibodies.
As a further test of this hypothesis, we immunoprecipitated PDE3A isoforms from transfected HEK293 cell extracts, and then analyzed coimmunoprecipitated proteins by isoelectric focusing followed by SDS/PAGE (Fig. S5). Multiple proteins were seen to coimmunoprecipitate with both PDE3A1 and PDE3A2 under basal conditions. Exposure of cells to isoproterenol increased the number of proteins that coimmunoprecipitated with PDE3A1, whereas exposure of cells to PMA increased the number of proteins that coimmunoprecipitated with PDE3A2. Different patterns of coimmunoprecipitated proteins were observed for PDE3A1 and PDE3A2 under both basal and stimulated conditions, indicating that the two isoforms have distinct protein interactomes.
PDE3A1 and PDE3A2 in Human Myocardium Are Selectively Phosphorylated at S312 and S428.
To determine whether the differential phosphorylation of PDE3A1 and PDE3A2, observed in HEK293 cells transfected with each isoform individually, occurs in cardiac myocytes, in which both PDE3A1 and PDE3A2 are expressed (10, 24), we examined the endogenous phosphorylation of PDE3A1 and PDE3A2 in microsomes and cytosolic fractions prepared from human left ventricular myocardium (Fig. 6). We found that PDE3A1, which was recovered exclusively in microsomes, is phosphorylated preferentially at S312, and that PDE3A2, which was recovered in both microsomal and cytosolic fractions, is phosphorylated only at S428. These findings demonstrate that PDE3A1 and PDE3A2 are differentially phosphorylated at separate sites in human cardiac myocytes in vivo in a pattern similar to that observed in transfected HEK293 cells.
Fig. 6.

Site-specific phosphorylation of endogenous PDE3A isoforms expressed in microsomes of human myocardium. PDE3A isoforms in microsomal and cytosolic fractions of human myocardium were immunoprecipitated using anti-PDE3A C-terminal antibody. Immunoprecipitated PDE3A isoforms were identified by immunoblotting with anti-PDE3A C-terminal antibody, and their phosphorylation was analyzed with phosphospecific antibodies as indicated.
Discussion
The presence in cardiac myocytes of three isoforms of PDE3A, transcribed and translated from alternative start sites in the PDE3A gene, has been recognized for some time (10, 24); however, apart from the role of hydrophobic domains in the unique N-terminal extension of PDE3A1 in its localization to intracellular membranes (11, 12), functional differences among these isoforms have not been identified. Our experimental approach has led to the discovery (i) that PDE3A1 and PDE3A2 are selectively phosphorylated in response to the activation of PKA- and PKC-mediated signaling pathways, respectively; (ii) that such selectivity includes their differential phosphorylation at alternative protein-binding sites within their common N-terminal sequence; (iii) that phosphorylation modulates the catalytic activity of PDE3A1 and PDE3A2 through different molecular mechanisms; and (iv) that phosphorylation promotes the association of PDE3A1 and PDE3A2 with distinct interactomes. The similar pattern of selective phosphorylation of endogenous PDE3A1 and PDE3A2 at these sites in human myocardium suggests that the functional differences between these isoforms are likely relevant in human heart, where PDE3 inhibitors exert inotropic actions that are important in the treatment of heart failure.
Considering that their amino acid sequences are otherwise identical, the functional differences between PDE3A1 and PDE3A2 must be based on the presence or absence of the unique N-terminal extension of PDE3A1. Our findings suggest that the influence of this sequence is mediated both through its effects on intracellular targeting of the enzyme and through its effects on the conformation of downstream amino acid sequences. In transfected HEK293 cells, the selective phosphorylation of PDE3A1 at S312 in response to isoproterenol clearly reflects the localization of this isoform to intracellular membranes in a manner that facilitates phosphorylation in response to a compartmentalized increase in cAMP content, as PDE3A2, which is recovered only in cytosolic fractions of these cells and is robustly phosphorylated by PKA in vitro, is phosphorylated at this site only when compartmentalized responses to β-adrenergic receptor stimulation are disrupted by IBMX.
Our finding that membrane-associated PDE3A2 in human myocardium is poorly phosphorylated at S312 suggests that the selective phosphorylation of PDE3A1 at this site in cardiac myocytes may depend on its localization to specific microdomains within intracellular membranes (to which PDE3A2 is presumably not localized), as well as on specific protein–protein interactions that may be involved, which, as our results indicate, are different for PDE3A1 and PDE3A2. Although the meager phosphorylation of PDE3A1 at S428 in response to PMA could, conversely, result from its localization and sequestration away from PKC, this explanation seems unlikely, given that exposure of HEK293 cells to PMA causes a translocation of PKC isoforms to intracellular membranes to which PDE3A1 is targeted (25), and because microsomal PDE3A2 is endogenously phosphorylated at this site in human myocardium. It is more probable that the N-terminal extension of PDE3A1 modifies the conformation surrounding S428 so as to convert it from a strong PKC site in PDE3A2 to a weak one in PDE3A1. The fact that the unique N-terminal extension has the additional effect in PDE3A1 of uncoupling phosphorylation at S428 from its allosteric stimulation of catalytic activity seen in PDE3A2 is further evidence that this sequence affects function by modifying downstream tertiary structure.
The selective phosphorylation of PDE3A isoforms may affect intracellular signaling in two ways. First, the phosphorylation of PDE3A2 by PKC has been shown to stimulate cAMP hydrolytic activity in platelets (15), and our present findings suggest that this is likely to be the case in human heart as well. The role of PKA-mediated phosphorylation in the regulation of catalytic activity appears to be more complex. With the compartmentalized activation of PKA in response to isoproterenol, there is no stimulation of either isoform, whereas with the higher, noncompartmentalized activation of PKA by dBcAMP, both PDE3A1 and PDE3A2 are stimulated. In the case of PDE3A2, this stimulation is, as with PMA, completely dependent on phosphorylation at S428. To our surprise, however, we found that the stimulation of PDE3A1 activity is independent of phosphorylation at either S428, which regulates activity in PDE3A2, or S312, which closely resembles the 14-3-3 site S318 in PDE3B through which the latter’s catalytic activity is regulated (17). The mechanism by which PKA-mediated phosphorylation influences the activity of PDE3A1 is thus fundamentally different from the mechanisms that apply to PDE3A2 and to PDE3B, and must result from phosphorylation at other PKA sites in its sequence. Whether the level and profile of PKA activation achieved by exposure to dBcAMP are relevant in cardiac myocytes is unclear, given that phosphorylation of PDE3A2 at S312 was not observed in human myocardium. Determining whether phosphorylation by PKA stimulates the catalytic activity of PDE3A1 under physiologically relevant conditions and, if so, identifying the molecular mechanisms involved are important directions for future studies.
Selective phosphorylation of PDE3A isoforms at alternative sites also may affect signaling through effects on protein–protein interactions. We examined interactions with 14-3-3, a signaling regulator that binds to a wide range of proteins to stabilize phosphorylation-induced conformational changes (26, 27). Consequences of 14-3-3 binding include enzyme activation and changes in intracellular localization (28, 29); for example, binding of 14-3-3 to PDE3B phosphorylated by PKA at S318, blocks dephosphorylation of this site and potentiates its stimulation of catalytic activity (17). What may be of greater importance is our observation that phosphorylation promotes the association of PDE3A1 and PDE3A2 with distinct protein interactomes. As noted earlier, phosphorylation leads to the recruitment of the structurally related phosphodiesterase PDE3B into multiprotein complexes in 3T3 adipocytes, and distinct insulin- and β3-adrenergic receptor agonist-induced signaling complexes have been described (22, 23). In the case of PDE3A, recent studies in animal models have demonstrated its association in signaling complexes in cardiac myocytes containing SERCA2, phospholamban, AKAP-18, PKA RII, and PP2A, through which PDE3A can modulate sarcoplasmic reticulum Ca2+ handling, and with PI3Kγ, which contributes to its regulation of β-adrenergic receptor-mediated signaling (1, 30). Identifying the proteins in human cardiac myocytes with which PDE3A1 and PDE3A2 interact, the specific phosphorylation sites through which these interactions are regulated, and the specific pathways involved in their phosphorylation are likely to provide insight into the roles of these isoforms in intracellular signaling.
The selective phosphorylation of PDE3A1 and PDE3A2, along with the distinct effects of phosphorylation on their catalytic activity and protein–protein interactions, suggest that these isoforms are likely to regulate the phosphorylation of different PKA substrates by discrete PKA subpopulations in response to the activation of separate signaling pathways. Testing this possibility directly in cardiac myocytes is challenging at present. Because PDE3A1 and PDE3A2 are generated by transcription from alternative sites in exon 1 of PDE3A (10), isoform-selective knockdown approaches are likely to be problematic. Furthermore, to our knowledge, no currently available agents selectively inhibit the catalytic activity of either isoform. The development of new agents that can selectively target these isoforms will be important in furthering this line of investigation.
The selective regulation of PDE3A isoforms may have therapeutic ramifications. As noted earlier, PDE3 inhibitors, which are used to “overcome” a reduction in receptor-mediated cAMP generation in patients with heart failure, increase contractility by increasing the phosphorylation of PKA substrates involved in intracellular Ca2+ cycling, but their long-term use is associated with an increase in mortality that may result from PKA-mediated proapoptotic changes in gene expression (2–9). Our findings indicate that existing PDE3 inhibitors are actually targeting at least two functionally distinct isoforms likely to have different roles in regulating intracellular signaling in cardiac myocytes. Although PDE3A1 and PDE3A2 are equally sensitive to these inhibitors (13), an agent capable of binding selectively to PDE3A1 or PDE3A2 and disrupting its protein–protein interactions might increase cAMP content in a particular intracellular microdomain without raising total intracellular cAMP content. By affecting the phosphorylation of a restricted set of PKA substrates in cardiac myocytes, such an agent might have inotropic effects with fewer of the adverse consequences seen with catalytic site inhibitors. The identification of agents that bind selectively to phosphorylated PDE3A1 or PDE3A2 would constitute an important step in evaluating the feasibility of this approach.
Materials and Methods
Preparation of Cytosolic and Microsomal Fractions of the Left Ventricular Myocardium.
Human myocardium was obtained from the left ventricular free walls of the hearts of explanted hearts of patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation. Cytosolic and microsomal fractions were prepared by homogenization and differential sedimentation as described previously (31). Protein was quantified by Bradford's method, with BSA fraction V as the standard (32).
Expression of Human PDE3A and 14-3-3 Constructs.
Recombinant FLAG-PDE3A and GST-14-3-3 constructs were prepared and expressed in HEK293 cells as described in SI Materials and Methods.
Measurement of cAMP Hydrolytic Activity.
cAMP hydrolytic activity was quantified at 30 °C by the two-step snake venom method with [3H]cAMP (1 µM) as a substrate (10); PDE3 activity was quantified by measuring activity in the absence and presence of cilostamide (22). The amount of protein used per assay and the incubation times were adjusted to ensure that no more than 20% of the total cyclic nucleotide was hydrolyzed during the assay.
Treatment of Cells with Pharmacologic Agents.
Cells were treated with pharmacologic agents as described in SI Materials and Methods.
Immunoprecipitation and Immunoblotting.
Immunoprecipitation and immunoblotting were performed as described in SI Materials and Methods.
In Vitro Phosphorylation of FLAG-PDE3A.
In vitro phosphorylation of FLAG-PDE3A isoforms was performed as described in SI Materials and Methods.
Synthesis of Peptides and Overlay of Peptide Arrays.
Synthesis of peptides and overlay of peptides arrays were performed as described in SI Materials and Methods.
Gel-Filtration Chromatography on Superose 6 Columns.
Solubilized HEK293 cell extracts (1.0 mL, 3 mg total protein) were applied to a Superose 6 HR 10/30 column (AKTA FPLC System; GE Healthcare) that was equilibrated and eluted with buffer containing 50 mM Hepes (pH 7.5), 1 mM EDTA, 10 mM pyrophosphate, 5 mM NaF, 150 mM NaCl, 5 mM MgCl2, 0.1 μM okadaic acid, Roche protease inhibitor mixture, and 1% Nonidet P-40. Portions of the fractions (0.5 mL) were used for assays of PDE3 activity or SDS/PAGE/Western blot analyses (using affinity-purified rabbit anti-PDE3A).
Two-Dimensional SDS/PAGE Analysis.
Two-dimensional SDS/PAGE analysis of proteins coimmunoprecipitating with PDE3A1 and PDE3A2 in HEK293 cells was performed and analyzed as described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Susan Taylor for her gift of the catalytic subunit of PKA. This work was supported by research grants from the US Department of Veterans Affairs (Merit Review CARA-029-09F and CARA-027-12S, to M.A.M.), the American Heart Association (M.A.M.), the National Institutes of Health (Grants R01 HL093183, HL088434, and P20 HL100396; a National Heart, Lung, and Blood Institute Program of Excellence in Nanotechnology Award; Contract HHSN268201000045C; Grant P50 HL112324, to R.J.H., and Grant GM089778, to J.A.W.), the UK Medical Research Council (M.D.H.), and the Fondation Leducq (06CVD02, to M.A.M. and M.D.H.; 13CVD01, to R.J.H.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305427110/-/DCSupplemental.
References
- 1.Beca S, et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res. 2013;112(2):289–297. doi: 10.1161/CIRCRESAHA.111.300003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank KF, Bölck B, Brixius K, Kranias EG, Schwinger RH. Modulation of SERCA: Implications for the failing human heart. Basic Res Cardiol. 2002;97(Suppl 1):I72–I78. doi: 10.1007/s003950200033. [DOI] [PubMed] [Google Scholar]
- 3.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87(12):1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 4.Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35(6):621–628. doi: 10.1016/j.ceca.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Marx SO, Marks AR. Regulation of the ryanodine receptor in heart failure. Basic Res Cardiol. 2002;97(Suppl 1):I49–I51. doi: 10.1007/s003950200029. [DOI] [PubMed] [Google Scholar]
- 6.Amsallem E, Kasparian C, Haddour G, Boissel JP, Nony P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev. 2005;(1):CD002230. doi: 10.1002/14651858.CD002230.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding B, et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation. 2005;111(19):2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding B, et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci USA. 2005;102(41):14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan C, Miller CL, Abe J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res. 2007;100(4):489–501. doi: 10.1161/01.RES.0000258451.44949.d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wechsler J, et al. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem. 2002;277(41):38072–38078. doi: 10.1074/jbc.M203647200. [DOI] [PubMed] [Google Scholar]
- 11.Kenan Y, Murata T, Shakur Y, Degerman E, Manganiello VC. Functions of the N-terminal region of cyclic nucleotide phosphodiesterase 3 (PDE 3) isoforms. J Biol Chem. 2000;275(16):12331–12338. doi: 10.1074/jbc.275.16.12331. [DOI] [PubMed] [Google Scholar]
- 12.Shakur Y, et al. Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3): Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J Biol Chem. 2000;275(49):38749–38761. doi: 10.1074/jbc.M001734200. [DOI] [PubMed] [Google Scholar]
- 13.Hambleton R, et al. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem. 2005;280(47):39168–39174. doi: 10.1074/jbc.M506760200. [DOI] [PubMed] [Google Scholar]
- 14.Pozuelo Rubio M, Campbell DG, Morrice NA, Mackintosh C. Phosphodiesterase 3A binds to 14-3-3 proteins in response to PMA-induced phosphorylation of Ser428. Biochem J. 2005;392(Pt 1):163–172. doi: 10.1042/BJ20051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter RW, Mackintosh C, Hers I. Protein kinase C-mediated phosphorylation and activation of PDE3A regulate cAMP levels in human platelets. J Biol Chem. 2009;284(18):12339–12348. doi: 10.1074/jbc.M807536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onuma H, et al. Identification of the insulin-regulated interaction of phosphodiesterase 3B with 14-3-3 beta protein. Diabetes. 2002;51(12):3362–3367. doi: 10.2337/diabetes.51.12.3362. [DOI] [PubMed] [Google Scholar]
- 17.Palmer D, et al. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J Biol Chem. 2007;282(13):9411–9419. doi: 10.1074/jbc.M606936200. [DOI] [PubMed] [Google Scholar]
- 18.Macphee CH, Reifsnyder DH, Moore TA, Lerea KM, Beavo JA. Phosphorylation results in activation of a cAMP phosphodiesterase in human platelets. J Biol Chem. 1988;263(21):10353–10358. [PubMed] [Google Scholar]
- 19.Grant PG, Mannarino AF, Colman RW. cAMP-mediated phosphorylation of the low-Km cAMP phosphodiesterase markedly stimulates its catalytic activity. Proc Natl Acad Sci USA. 1988;85(23):9071–9075. doi: 10.1073/pnas.85.23.9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han SJ, et al. Protein kinase B/Akt phosphorylation of PDE3A and its role in mammalian oocyte maturation. EMBO J. 2006;25(24):5716–5725. doi: 10.1038/sj.emboj.7601431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295(5560):1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad F, et al. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem J. 2007;404(2):257–268. doi: 10.1042/BJ20060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad F, et al. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the beta3-adrenergic receptor agonist CL316243: Effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem J. 2009;424(3):399–410. doi: 10.1042/BJ20090842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi YH, et al. Identification of a novel isoform of the cyclic-nucleotide phosphodiesterase PDE3A expressed in vascular smooth-muscle myocytes. Biochem J. 2001;353(Pt 1):41–50. [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Meng Q, Jing X, Xu P, Luo D. A role for protein kinase C in the regulation of membrane fluidity and Ca²(+) flux at the endoplasmic reticulum and plasma membranes of HEK293 and Jurkat cells. Cell Signal. 2011;23(2):497–505. doi: 10.1016/j.cellsig.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Mackintosh C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem J. 2004;381(Pt 2):329–342. doi: 10.1042/BJ20031332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson C, et al. Bioinformatic and experimental survey of 14-3-3–binding sites. Biochem J. 2010;427(1):69–78. doi: 10.1042/BJ20091834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saha M, et al. RSK phosphorylates SOS1 creating 14-3-3–docking sites and negatively regulating MAPK activation. Biochem J. 2012;447(1):159–166. doi: 10.1042/BJ20120938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muslin AJ, Xing H. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell Signal. 2000;12(11-12):703–709. doi: 10.1016/s0898-6568(00)00131-5. [DOI] [PubMed] [Google Scholar]
- 30.Ghigo A, et al. Phosphoinositide 3-kinase γ protects against catecholamine-induced ventricular arrhythmia through protein kinase A-mediated regulation of distinct phosphodiesterases. Circulation. 2012;126(17):2073–2083. doi: 10.1161/CIRCULATIONAHA.112.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandeput F, et al. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther. 2009;330(3):884–891. doi: 10.1124/jpet.109.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.