

Abstract
BACKGROUND
CFF practice guidelines recommend patients ≥ age 6 use dornasealfa and hypertonic saline daily, and those ≥ age 6 colonized with P. aeruginosa use inhaled tobramycin and oral azithromycin to improve lung function and reduce pulmonary exacerbations. A decline in FEV1 was noted in our 2008 CF Center Report. We hypothesized that increasing adherence to prescribing guidelines for these pulmonary medications would improve mean FEV1.
METHODS
This was a quality improvement project completed at a US CF center. CFF practice guidelines were reviewed with the center physicians. Patients were identified that were eligible to receive recommended therapies and it was determined whether they were prescribed the therapies. Baseline FEV1 data was collected. Adherence rates and FEV1 were followed quarterly for 1 year. Providers received a quarterly report card with adherence rates, mean FEV1 compared to colleagues, and a list of eligible patients that were not prescribed recommended therapies.
RESULTS
92 patients were included. At baseline, the overall adherence rate was 59%. Overall adherence increased quarterly (p=<0.001). Each quarter there was improvement in adherence to prescribing for each medication (p<0.001). Except in quarter 1, FEV1 increased quarterly (p=0.092). There was moderate correlation (r=0.533) between improved adherence and improved FEV1.
CONCLUSIONS
Educating clinicians about guidelines, providing feedback on adherence to guidelines, and monitoring prescribing patterns improves prescribing adherence. FEV1 showed improvement after months of sustained adherence, trending towards significance. Longer follow-up is necessary to determine if improved prescribing adherence translates into improved FEV1 or slows rate of decline in FEV1.
Keywords: cystic fibrosis, dornasealfa, hypertonic saline, tobramycin, azithromycin, guidelines, adherence
Introduction
Cystic Fibrosis (CF) is an inherited disease that impacts multiple organs. Abnormalities in the cystic fibrosis transmembrane conductance regulator (CFTR) cause abnormal sodium absorption and chloride secretion. These abnormalities result in the formation of thick mucus plugs that clog the airways, decrease mucociliary clearance, and provide a nidus for bacterial infection. Chronic infection results in a neutrophil-dominated inflammatory response.1,2 Over time the inflammation remodels the airways and causes progressive destruction, decline in lung function, and respiratory failure.2
Over the last 20 years there has been a considerable increase in life expectancy for patients with CF.3 This increase has been attributed to many contributing factors, including: antibiotics, anti-inflammatories, mucolytics, and consistent care at a CF Center.4–18 Respiratory complications are the largest source of morbidity and mortality for CF patients, so many therapies are aimed at maintaining and improving lung function in patients with CF. The Cystic Fibrosis Foundation (CFF) pulmonary guidelines recommend that all patients ≥ age 6 should use dornasealfa and hypertonic saline on a daily basis, and those ≥ age 6 colonized with Pseudomonas aeruginosa should use inhaled tobramycin (TOBI) and oral azithromycin to improve lung function and reduce pulmonary exacerbations.19
The CFF provides an annual report to each CF center summarizing how each center compares on a national level for specific measures. Measures and outcomes are outlined on an annual basis by the CFF. Centers enter site-specific data in the CF Patient Registry, which is then analyzed and compared with the other CF accredited centers. This comparative data set is reported back to the center. Forced expiratory volume in one second (FEV1) data is reported in several age groupings: 6–12 years, 13–17 years, 6–17 years, 18–29 years, and >30 years. FEV1 is monitored closely in patients with CF as it is associated with morbidity and mortality. Preservation of FEV1 is one of the goals of the pulmonary therapies used to treat CF. A decline was noted in mean FEV1 in the 6–17 and 13–17 year age groups in the 2008 CF Center Patient Registry Report for the CF Center at Children’s Hospitals and Clinics of Minnesota. The mean FEV1 decreased from 99.7 to 97.3 in the 6–17 age group, and 100.2 to 92.5 in the 13–17 age group. Although this is a small decrease, there was concern about progression of the decline and potential long-term impact on morbidity and mortality for our patients. Concomitantly it was recognized that physician adherence to the pulmonary medication guidelines, although in some cases higher than the national average, had room for improvement (only 85.7% of eligible patients were on azithromycin, 82.3% on TOBI, 78.2% on dornasealfa, and 29.5% on hypertonic saline; compared to the national averages of 65.9%, 68.1%, 78.3%, and 42.7%, respectively).20,21 The objective of this study was to use a simple intervention to increase provider prescribing adherence to the CFF pulmonary medication guidelines. We hypothesized that increasing clinician adherence to the guidelines for these four pulmonary medications would improve the mean FEV1 in the 13–17 year olds and would stabilize FEV1, in hopes of preventing further decline in the 6–17 year olds.
Materials and Methods
This was a quality improvement project which used the Plan, Do, Study, Act process.22,23,24 The study was approved by the Institutional Review Board at Children’s Hospitals and Clinics of Minnesota (CHCM) and was completed at CHCM (University of Minnesota CF affiliate).
At the start of the study, CFF pulmonary guidelines were reviewed with the CF Center physicians.19 Each physician received a copy of the guidelines along with a one-page summary of the recommendations for dornasealfa, hypertonic saline, TOBI, and azithromycin. A CF Action Plan was created that included the four targeted pulmonary medications, airway clearance strategies, nutrition/dietary recommendations, and other CF medications (Figure 1). The CF Action Plan is completed by physicians at each visit and is given to patients and their families. Our goal, by listing all the medications on one document, was to prompt providers to remember to prescribe the recommended therapies.
Figure 1.

CF Action Plan – Provided to all CF patients. Included targeted therapies to remind physicians of pulmonary medication guideline recommendations.
The CF Patient Registry was used to identify patients who, based on CFF pulmonary guidelines, were eligible to receive dornasealfa, hypertonic saline, TOBI, and azithromycin, and to collect baseline FEV1 data. Patients’ charts were reviewed to determine whether they were prescribed the recommended therapies. Prescribing adherence rates and FEV1 were followed quarterly for one year. Providers received a quarterly report card with prescribing adherence rates and mean FEV1 compared to their colleagues, as well as a list of eligible patients that were not on recommended therapies (Figure 2).
Figure 2.

Example of the quarterly report card that each provider received. Compares prescribing adherence rates between providers and identifies patients not on recommended therapies.
Statistical analysis
Chi-square compared prescribing adherence rates between physicians and quarters. Two-sample t-test compared mean FEV1 between patients who were prescribed medications and patients who were not. ANOVA compared mean FEV1 by quarter. Pearson correlation coefficient described the relationship between overall adherence rate and FEV1. All statistical analyses were conducted using the Statistical Program for Social Sciences version 15.0 software (SPSS).
Results
127 patients with CF were seen at CHCM in 2008. 92 patients were included who were eligible to receive dornasealfa, hypertonic saline, TOBI, and/or azithromycin. According to the 2008 CF Patient Registry report (baseline), the overall clinician prescribing adherence rate to CFF guidelines was 59%. Each quarter there was improvement in adherence to prescribing for all medications (p<0.001, p<0.001, p =0.001, and p=0.003 for dornasealfa, hypertonic saline, TOBI and azithromycin, respectively) (Figure 3). The overall clinician adherence rate increased quarterly (Figure 4) (p<0.001). For the 6–17 year olds, mean FEV1was 97% at baseline. For the 13–17 year olds, mean FEV1 was 93% at baseline. Each quarter following the intervention, FEV1 increased (p=0.092) (Figure 4). There was a moderate correlation (r=0.533) between improved adherence and improved mean FEV1 (Figure 4). Patients who were prescribed hypertonic saline and TOBI had a significantly higher mean FEV1 (p=0.018 and p=0.011, respectively).
Figure 3.
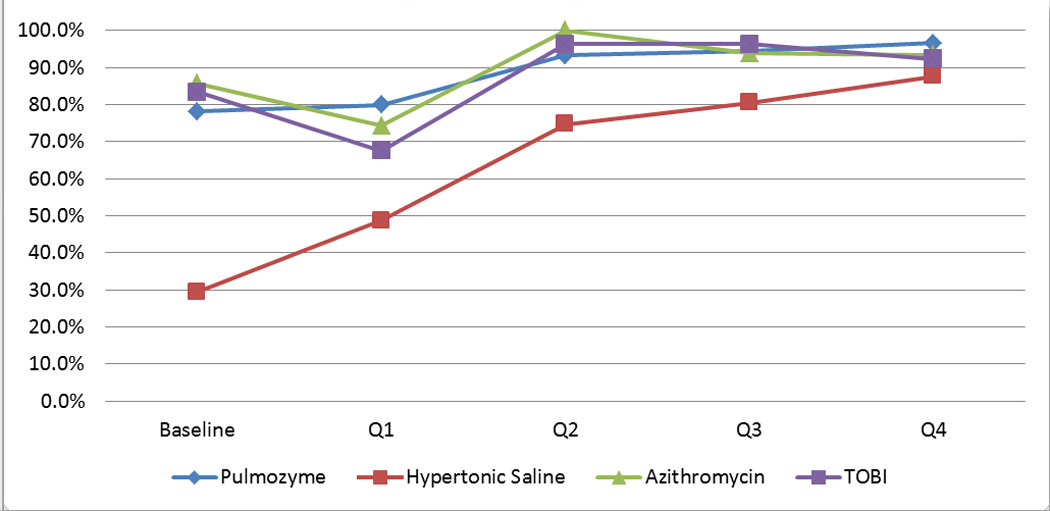
Change in prescribing adherence rates for pulmonary medications
Figure 4.
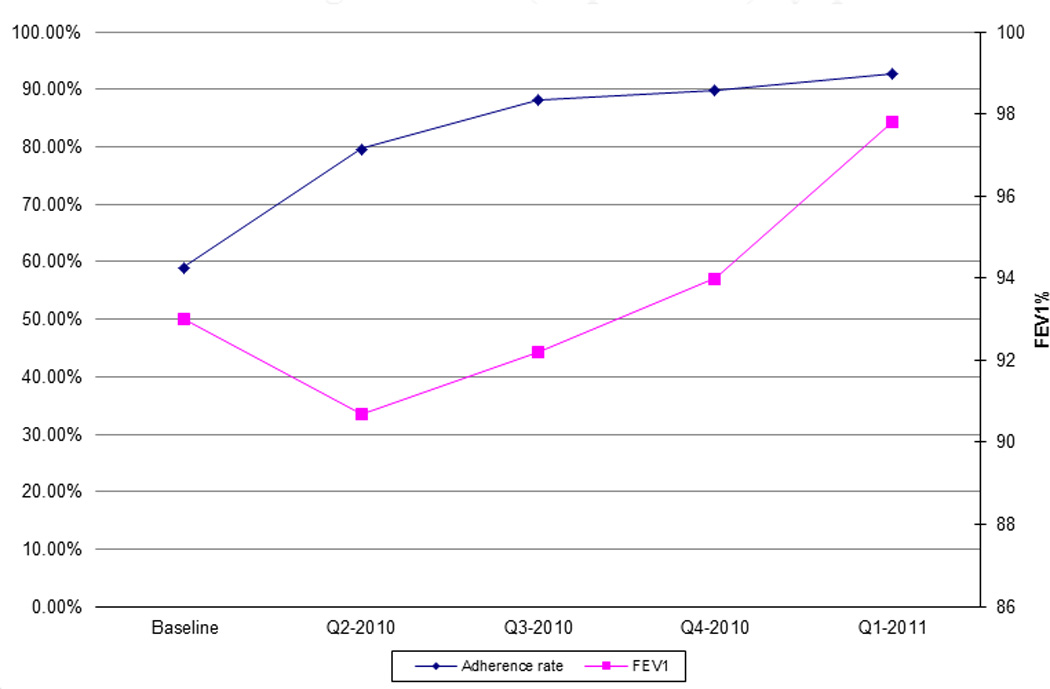
Change in prescribing adherence rate versus change in FEV1 (% predicted) by quarter
Discussion
This study followed the steps outlined by Garber, et al., for improving guideline adherence. Baseline practices were measured, guidelines were disseminated, staff and patients were educated, implementation of practices was monitored, and patient outcomes were measured.25 The results are similar to those of McPhail, et al., who found that educating physicians about prescribing guidelines, providing feedback on prescribing adherence failures, and monitoring prescribing patterns over time improves prescribing adherence.26 As the authors advocate, prescribing guidelines alone do not improve patient outcomes. Physicians must be aware of the guidelines, accept the recommendations, and subsequently implement the guidelines. The results of Garber and McPhail are echoed in the work by Glauser, et al., who note that one time dissemination of guidelines is not sufficient for changing clinician behavior. System changes are necessary for providers to adapt to guidelines.27
A simple intervention, demonstrated by this study, increased provider adherence to guidelines and, unlike the work of McPhail and colleagues, demonstrated a trend for improvement in patient outcomes. It showed that increased provider adherence to prescribing guidelines moderately correlates with improved FEV1.26 Improvement in FEV1 is the expected goal for use of the four pulmonary medications: dornasealfa, hypertonic saline, TOBI, and oral azithromycin. Dornasealfa and hypertonic saline are aimed at decreasing the viscosity of airway secretions, thus improving airway secretion clearance.10,28 TOBI is used to eradicate and suppress chronic infection/colonization of Pseudomonas aeruginosa which is associated with a more rapid decline in lung function.29 Azithromycin, both by its anti-inflammatory effect and anti-microbial effect has been shown to improve FEV1 and decrease frequency of pulmonary exacerbations of CF.6,8, 31
This study shows a trend of improvement in FEV1 when compared to baseline but did not reach statistical significance. This may be due to several reasons: (1) our baseline adherence rate to prescribing guidelines was already above the national average. There was room for improvement in all areas, but for three of the four medications we were already above or near 80%, (2) our baseline FEV1 was 93% predicted, a decline compared to the previous years, but still within normal and above the national average of 89%, and (3) pulmonary medications may maintain lung function and/or prevent pulmonary exacerbations, rather than actually improve lung function, therefore it may take longer than a year (the length of this study) to see a statistically significant improvement.
Prior to this intervention, it was custom to rely on the physicians to educate themselves about practice guidelines. There was not a system in place to disseminate new information. This intervention has demonstrated that the practice needs to take an active role in disseminating new information to providers, and clarifying expectations as to how they relate to new guidelines.
This study has several limitations. It was performed at a single CF center with eight CF providers and one CF nurse. Presumably it is easier to implement this type of intervention in a smaller practice setting. Consequently, the results are similar to those of McPhail, et al.26 This study relied heavily on the data entered in the CF Patient Registry. If data was missing or contradictory, every attempt was made to obtain accurate prescribing information. There may have been times when the database did not reflect the most up-to-date information.
Conclusions
Educating clinicians about prescribing guidelines, providing feedback on adherence to guidelines, and monitoring prescribing patterns over time improve prescribing adherence. Mean FEV1 shows improvement after several months of sustained adherence and trended towards statistical significance. Longer follow-up is necessary to determine if improved prescribing adherence translates into improved mean FEV1or a slower rate of decline in FEV1.
Abbreviations
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CFF
Cystic Fibrosis Foundation
- TOBI
inhaled tobramycin
- FEV1
forced expiratory volume in 1 second
- CHCM
Children’s Hospitals and Clinics of Minnesota
- ANOVA
Analysis of variance
References
- 1.Boucher RC. Pathogenesis of cystic fibrosis airways disease. Trans Am ClinClimatol Assoc. 2001;112:99–107. [PMC free article] [PubMed] [Google Scholar]
- 2.Welsh MJ. 23rd Goldman: Cecil Medicine, Chapter 89 – CYSTIC FIBROSIS. [Google Scholar]
- 3.Cystic Fibrosis Foundation. [accessed June 22, 2012];Patient Registry Report. 2010 Available from: http://www.cff.org/LivingWithCF/QualityImprovement/PatientRegistryReport.
- 4.Cohen-Cymberknoh M, Shoseyov D, Kerem E. Managing Cystic Fibrosis Strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;183:1463–1471. doi: 10.1164/rccm.201009-1478CI. [DOI] [PubMed] [Google Scholar]
- 5.Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand. 1983;72:651–657. doi: 10.1111/j.1651-2227.1983.tb09789.x. [DOI] [PubMed] [Google Scholar]
- 6.Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters incystic fibrosis: a randomised trial. Thorax. 2002;57:212–216. doi: 10.1136/thorax.57.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yousef AA, Jaffe A. The role of azithromycin in patients with cystic fibrosis. Paediatr Respir. 2010;11:108–114. doi: 10.1016/j.prrv.2009.12.003. Rev. [DOI] [PubMed] [Google Scholar]
- 8.Saiman L, Marshall BC, Mayer-Hamblett N, Burns JL, Quittner AL, Cibene DA, Coquillette S, Fieberg AY, Accurso FJ, Campbell PW., III Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2003;290:1749–1756. doi: 10.1001/jama.290.13.1749. [DOI] [PubMed] [Google Scholar]
- 9.Konstan MW, Schluchter MD, Xue W, Davis PB. Clinical use of ibuprofen is associated with slower FEV1 decline in children with cystic fibrosis. Am J Respir Crit Care Med. 2007;176:1084–1089. doi: 10.1164/rccm.200702-181OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331:637–642. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- 11.Hodson ME, Shah PL. RhDNase trials in cystic fibrosis. Eur Respir J. 1995;8:1786–1791.12. doi: 10.1183/09031936.95.08101786. [DOI] [PubMed] [Google Scholar]
- 12.Quan JM, Tiddens HA, Sy JP, McKenzie SG, Montgomery MD, Robinson PJ, Wohl ME, Konstan MW. Pulmozyme Early Intervention Trial Study Group: a two-year randomized, placebo-controlled trial of dornasealfa in young patients with cystic fibrosis with mild lung function abnormalities. J Pediatr. 2001;139:813–820. doi: 10.1067/mpd.2001.118570. [DOI] [PubMed] [Google Scholar]
- 13.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye PT. A controlled trial of longterm inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]
- 14.Wark P, McDonald VM. Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev. 2009;2:CD001506. doi: 10.1002/14651858.CD001506.pub3. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen OH, Thomsen BL, Green A, Andersen PK, Hauge M, Schiotz PO. Cystic fibrosis in Denmark 1945 to 1985. An analysis of incidence, mortality and influence of centralized treatment on survival. Acta Paediatr Scand. 1988;77:836–841. doi: 10.1111/j.1651-2227.1988.tb10765.x. [DOI] [PubMed] [Google Scholar]
- 16.Mahadeva R, Webb K, Westerbeek RC, Carroll NR, Dodd ME, Bilton D, Lomas DA. Clinical outcome in relation to care in centres specialising in cystic fibrosis: cross sectional study. BMJ. 1998;316:1771–1775. doi: 10.1136/bmj.316.7147.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebecque P, Leonard A, De Boeck K, De Baets F, Malfroot A, Casimir G, Desager K, Godding V, Leal T. Early referral to cystic fibrosis specialist centre impacts on respiratory outcome. J Cyst Fibros. 2009;8:26–30. doi: 10.1016/j.jcf.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Johnson C, Butler SM, Konstan MW, Morgan W, Wohl ME. Factors influencing outcomes in cystic fibrosis: a center-based analysis. Chest. 2003;123:20–27. doi: 10.1378/chest.123.1.20. [DOI] [PubMed] [Google Scholar]
- 19.Flume PA, O’Sullivan BP, Robinson KA, et al. Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2007;176(10):957–969. doi: 10.1164/rccm.200705-664OC. [DOI] [PubMed] [Google Scholar]
- 20.2007 Patient Registry Report
- 21.2008 Patient Registry Report
- 22.Berwick DM. Developing and testing changes in delivery of care. Ann Intern Med. 1998;128:651–656. doi: 10.7326/0003-4819-128-8-199804150-00009. [DOI] [PubMed] [Google Scholar]
- 23.Langley GJ, Nolan KM, Nolan TW, Norman CL, Provost LP. The Improvement Guide: A Practical Approach to Enhancing Organizational Performance. San Francisco, Calif: Jossey-Bass; 2009. [Google Scholar]
- 24.Varkey P, Reller MK, Resar RK. Basics of Quality Improvement in Health Care. Mayo Clin Proc. 2007;82(6):735–739. doi: 10.4065/82.6.735. [DOI] [PubMed] [Google Scholar]
- 25.Garber E, Deseia M, Zhou J, Alba L, Angst D, Cabana M, Saiman L. CF Infection Control Study Consortium. Barriers to adherence to cystic fibrosis infection control guidelines. Pediatr Pulmonol. 2008;43:900–907. doi: 10.1002/ppul.20876. [DOI] [PubMed] [Google Scholar]
- 26.McPhail GL, Weiland J, Acton JD, Ednick M, Chima A, VanDyke R, Fenchel MC, Amin RS, Seid M. Improving Evidence-Based Care in Cystic Fibrosis Through Quality Improvement. Arch Pediatr Adolesc Med. 2010;164(10):957–960. doi: 10.1001/archpediatrics.2010.178. [DOI] [PubMed] [Google Scholar]
- 27.Glauser TA, Nevins PH, Williamson JC, Abdolrasulnia M, Salinas GD, Zhang J, Debonnett L, Riekert KA. Adherence to the 2007 Cystic Fibrosis Pulmonary Guidelines: A National Survey of CF Care Centers. Pediatr Pulmonol. 2012;47:434–440. doi: 10.1002/ppul.21573. [DOI] [PubMed] [Google Scholar]
- 28.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–250. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 29.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34:91–100. doi: 10.1002/ppul.10127. [DOI] [PubMed] [Google Scholar]
- 30.Equi A, Balfour-Lynn IM, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet. 2002;360:978–984. doi: 10.1016/s0140-6736(02)11081-6. [DOI] [PubMed] [Google Scholar]