

Abstract
The lack of a versatile system to control gene expression in Helicobacter pylori has hampered efforts to study H. pylori physiology and pathogenesis. To overcome these limitations, we evaluated the utility of an inducible system based on the well-characterized Tet repressor (TetR) and Tet operator (tetO). As validation of this system, we introduced three copies of tetO into the promoter region upstream of the cagUT operon (encoding two virulence factors required for function of the H. pylori Cag type IV secretion system) and expressed tetR by introducing a codon-optimized gene into the chromosomal ureA locus. Introduction of the tetO copies upstream of cagUT did not disrupt promoter activity, as determined by immunoblotting for CagT. The subsequent introduction of tetR, however, did repress CagT synthesis. Production of CagT was restored when strains were cultured in the presence of the inducer, anhydrotetracycline. To demonstrate one potential application of this new tool, we analyzed the function of the Cag type IV secretion system. When the modified H. pylori strains were co-cultured with AGS cells, activity of the Cag type IV secretion system was dependent on the presence of anhydrotetracycline as evidenced by inducer-dependent induction of IL-8 secretion, CagA translocation, and appearance of type IV secretion system pili at the bacteria-host interface. These studies demonstrate the effectiveness of the tetR-tetO system to control gene expression in H. pylori and provide an improved system for studying H. pylori physiology and pathogenesis.
Keywords: Helicobacter pylori genetics/physiology, Bacterial gene expression/regulation, Bacterial genetic engineering, Genetic promoter regions
1. Introduction
Helicobacter pylori are Gram-negative bacteria that can establish persistent colonization of the human stomach (Suerbaum and Michetti 2002; Atherton and Blaser 2009; Cover and Blaser 2009). All people infected by H. pylori develop chronic superficial gastritis, and colonization of the stomach is considered a risk factor for peptic ulcer disease, gastric lymphoma, and gastric adenocarcinoma (IARC 1994; Yamada, Ahnen et al. 1994; Fox and Wang 2007; Wroblewski, Peek et al. 2010). The risk of the latter diseases is influenced by multiple factors, including host genetics, dietary or environmental factors, and strain-specific variations in H. pylori.
Most studies of H. pylori gene function have involved the study of mutant strains in which individual genes are disrupted or deleted (Copass, Grandi et al. 1997; Dailidiene, Dailide et al. 2006; Shaffer, Gaddy et al. 2011; Debowski, Gauntlett et al. 2012; Noto and Peek 2012). Such approaches have been important in characterizing a variety of virulence factors, including the H. pylori cag type IV secretion system (T4SS). This system consists of a multiprotein complex that delivers the CagA oncoprotein and peptidoglycan into epithelial cells (Backert and Selbach 2008). Upon its entry into the host cell cytoplasm, CagA can be phosphorylated by host cell kinases (Odenbreit, Puls et al. 2000; Stein, Rappuoli et al. 2000). CagA, in both phosphorylated and non-phosphorylated forms, is able to interact with host cell signaling proteins, resulting in an assortment of consequences, including cytoskeletal alterations, disruption of cellular junctions, and altered cellular adhesion and polarity (Amieva, Vogelmann et al. 2003; Hatakeyama 2004; Bourzac and Guillemin 2005). Delivery of peptidoglycan into the host cell cytoplasm by the Cag T4SS results in activation of nucleotide-binding oligomerization domain 1 (NOD1), leading to the production of proinflammatory cytokines MIP-2, β-defensin, and IL-8 (Viala, Chaput et al. 2004; Allison, Kufer et al. 2009).
Analysis of mutant strains has provided important insights into various features of the cag type IV secretion system, but knock-out or loss-of-function mutants can be blunt instruments. Such mutants do not allow one to determine whether a given gene is required throughout the infectious process or whether the gene is only required to establish or to maintain the infection (Salama, Otto et al. 2001; Liu, Gao et al. 2008; Barrozo, Cooke et al. 2013). Such mutants also are not suited for the study of essential genes (which cannot be disrupted or deleted). An easily regulated system to specifically control expression of a gene of interest could overcome these limitations, and could also provide an alternative approach, instead of comparisons of isogenic mutant and complemented mutant strains, for analyzing the functions of genes.
Previous studies have exploited inducible promoters in H. pylori, but the experimental approaches used in these studies have limitations. Multiple studies have used endogenous promoters to regulate gene expression in H. pylori (Delany, Pacheco et al. 2001; Ernst, Stoof et al. 2006; Carpenter, McDaniel et al. 2007; Muller, Pflock et al. 2007; Vannini, Agriesti et al. 2012). However, these promoters are regulated by metals (e.g., iron or nickel) known to modulate expression of multiple genes in H. pylori (Ernst, Stoof et al. 2006; Whitmire, Gancz et al. 2007; Danielli and Scarlato 2010; Pich and Merrell 2013). Thus, the addition of an inducing metal to H. pylori cultures to regulate genes expressed by these promoters complicates analysis of the gene of interest. One previous study overcame this limitation by introducing lacIq on an E. coli-H. pylori shuttle plasmid (Boneca, Ecobichon et al. 2008; Thibonnier, Thiberge et al. 2008; Thibonnier, Aubert et al. 2010). However, such shuttle plasmids can be difficult to introduce into some H. pylori strains, likely because of restriction barriers between strains.
In the present study, we deployed the well-characterized Tet-Off system, in which transcription of targeted genes is negatively regulated by the tetracycline repressor (TetR), to regulate gene expression in H. pylori (Hillen and Wissmann 1989; Gossen and Bujard 1992). In the absence of inducer (e.g., tetracycline), TetR binds to specific operator sequences (tetO) located within the promoter region of the regulated gene and represses transcription; addition of inducer disrupts TetR binding to tetO and allows transcription. We describe the introduction of a codon-optimized tetR as well as copies of the TetR binding site, tetO, into the H. pylori chromosome, and demonstrate the Tet-dependent production of CagT and multiple phenotypes associated with the Cag T4SS.
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
H. pylori strain 26695 was selected for use in this study (Table 1) (Tomb, White et al. 1997). This strain and derivatives developed herein were routinely grown at 37°C on Trypticase soy agar plates containing 5% sheep blood in ambient air containing 5% CO2. H. pylori mutant strains were selected based on resistance to chloramphenicol (2.5 μg ml−1) or resistance to kanamycin (25 μg ml−1) on Brucella agar plates (Brucella broth containing 1.35% agar and 10% fetal bovine serum). H. pylori liquid cultures were grown in Brucella broth supplemented with 10% fetal bovine serum (Atlanta Biologicals). Brucella broth and agar media were prepared without addition of sodium bisulfite (Hawrylik, Wasilko et al. 1994). When appropriate, culture media were supplemented with anhydrotetracycline (ATc, 100 ng ml−1).
Table 1.
Bacterial Strains and Plasmids
| Strain Designation | Description | Reference |
|---|---|---|
| 26695 | Sequenced reference strain | (Tomb, White et al. 1997) |
| VM120 | 26695 containing 3 copies of tetO inserted in cagUT promoter element, KmR | |
| VM122 | VM120 containing tetR expressed from the ureA locus, KmR CmR | |
| VM124 | 26695 containing tetR expressed from the ureA locus, KmR CmR | |
| Plasmid Designation | ||
| pAD1 | Vector for introducing recombinant DNA into the H. pylori ureA locus | (Ando, Israel et al. 1999; Forsyth and Cover 2000; Loh, Forsyth et al. 2004; Shaffer, Gaddy et al. 2011) |
| pMM680 | pUC57 containing the H. pylori 26695 cagU-cagV intergenic region and introduced copies of a kanamycin resistance determinant and tetO | |
| pMM682 | H. pylori codon-optimized tetR cloned into pAD1 |
2.2. Mutagenesis of cagUT Promoter and Introduction of tetR
To mutate the cagUT promoter, a synthetic DNA construct was designed that included about 500 bp of flanking sequence on either side of the identified cagUT transcription start site, derived from H. pylori strain 26695 (Sharma, Hoffmann et al. 2010; Ta, Hansen et al. 2012). The synthetic construct also included a kanamycin resistance cassette (divergently transcribed relative to cagUT) in the intergenic region between cagU and cagV, and three copies of tetO flanking the predicted cagUT promoter element (Figure S1). This synthetic construct was cloned into plasmid pUC57 and the resulting plasmid was used to transform H. pylori strain 26695, selecting for kanamycin-resistant colonies. PCR and DNA sequencing were used to confirm that homologous recombination via double crossover resulted in the introduction of the engineered cagV-cagU intergenic region into the chromosome of H. pylori.
A codon-optimized tetR gene was synthesized (Genscript; accession number KF697108). To introduce the tetR gene into H. pylori, we used a plasmid derived from pAD1, which allows genes of interest to be introduced into the H. pylori chromosomal ureA locus (Loh, Forsyth et al. 2004; Shaffer, Gaddy et al. 2011). The modified pAD1 plasmid contains a chloramphenicol resistance cassette, restriction sites to allow cloning of a gene of interest into a site downstream from the ureA promoter and a ribosomal binding site, and flanking sequences derived from the ureA locus. A plasmid (pMM682) was constructed to allow expression of a codon-optimized tetR sequence from the ureA promoter. The appropriate H. pylori strain was transformed with this plasmid, and chloramphenicol-resistant colonies were selected. PCR and DNA sequencing were used to confirm that homologous recombination via double crossover resulted in the introduction of tetR into the H. pylori chromosome.
2.3. Generation of Rabbit Polyclonal anti-CagT Serum
A fragment of CagT derived from H. pylori 26695 (amino acids 26–280) was expressed as a GST fusion protein from pGEX-5X-1 vector (GE Healthcare). CagT-GST production was induced with IPTG and the fusion protein was purified using gluthathione beads (Busler, Torres et al. 2006). Rabbits were then immunized with purified CagT-GST.
2.4. Immunoblot Analysis
To detect protein production, individual samples were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and subsequently immunoblotted using rabbit polyclonal antiserum raised against recombinant CagT, rabbit polyclonal anti-CagA (Santa Cruz), mouse monoclonal antibody to TetR (clone 9G9, Clontech), or mouse monoclonal antibody to phosphotyrosine (pY-99, Santa Cruz). Horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG was used as the second antibody. Signals were generated by enhanced chemiluminescence reaction (SuperSignal West Pico Chemiluminescent Substrate, Pierce Chemical) and detection by exposure to X-ray film.
2.5. Cell Culture Methods
AGS human gastric epithelial cells (ATCC) were grown in the presence of 5% CO2 in RPMI medium containing 10% FBS, 2 mM L-glutamine, and 10 mM HEPES buffer.
2.6. IL-8 Secretion by Gastric Cells in Contact with H. pylori
H. pylori strains were co-cultured with AGS cells at a multiplicity of infection of 100:1. IL-8 secretion into the cell culture medium was analyzed using an anti-human IL-8 sandwich ELISA (R&D Systems).
2.7. CagA Translocation Assay
Translocation of CagA into AGS cells was analyzed by co-culturing H. pylori strains with AGS cells and detecting tyrosine phosphorylation of CagA, as previously described (Busler, Torres et al. 2006; Loh, Torres et al. 2007). Briefly, H. pylori and AGS human gastric cells were co-cultured at a MOI of 100:1 for 7 h at 37°C. Cells were lysed in NP-40 lysis buffer containing Complete Mini EDTA-free Protease Inhibitor (Roche) and Phos-Stop phosphatase inhibitor (Roche), and CagA translocation was assessed by separating the soluble fraction using 7.5% SDS-PAGE and immunoblotting with an anti-phosphotyrosine antibody (α-PY99, Santa Cruz).
2.8. Electron Microscopy of H. pylori in Contact with Gastric Epithelial Cells
Imaging of H. pylori by field emission scanning electron microscopy was performed essentially as described (Shaffer, Gaddy et al. 2011). H. pylori and AGS human gastric cells were co-cultured at a MOI of 100:1 on tissue culture-treated coverslips (BD Biosciences) for 4 h at 37°C in the presence of 5% CO2. Cells were fixed with 2.0% paraformaldehyde, 2.5% glutaraldehyde in 0.05 M sodium cacodylate buffer for 1 h at 37°C. Coverslips were washed with sodium cacodylate buffer and secondary fixation was performed with 1% osmium tetroxide at room temperature for 30 minutes. Coverslips were washed with sodium cacodylate buffer and dehydrated with sequential washes of increasing concentrations of ethanol. Samples were then dried at the critical point, mounted onto sample stubs, grounded with a thin strip of silver paint at the sample edge, and sputter-coated with gold-palladium before viewing with a FEI Quanta 250 FEG scanning electron microscope.
3. Results and Discussion
3.1. Introduction of tetO Sites Upstream of cagUT
We selected the cagUT operon as a potential target to regulate and thereby evaluate the effectiveness of the Tet system in H. pylori. This operon has a promoter sequence containing a predicted -10 element upstream of a previously identified transcription start site, appears to be a simple operon with a well-defined start site and does not appear to be complicated by alternate start sites or antisense transcripts (Sharma, Hoffmann et al. 2010; Ta, Hansen et al. 2012). One of the products (CagT) can be monitored by immunoblotting, and the gene products are of interest in the H. pylori research community because of their roles in the Cag T4SS (Fischer, Puls et al. 2001; Kutter, Buhrdorf et al. 2008). To regulate expression from the cagUT promoter, we first introduced three copies of the tetO sequence flanking the cagUT promoter into H. pylori strain 26695, resulting in strain VM120 (Table 1 and Figures 1 and S1). To determine whether CagT production was disrupted by these alterations in the cagUT promoter region, we analyzed CagT levels in strain VM120 and the parental strain 26695 by immunoblotting. Results indicated that the promoter alterations did not disrupt CagT production (Figure 2A).
Figure 1. Introduction of tetO upstream of cagUT.
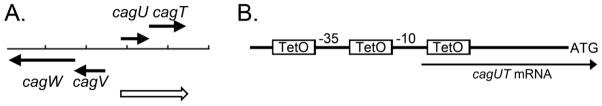
A. The genetic organization of cagUT and flanking genes is depicted. Filled arrows represent individual open reading frames. H. pylori cagU and cagT are co-transcribed as part of an operon illustrated by the open arrow (Sharma et al. 2010; Ta et al. 2012). B. Three copies of tetO were introduced into the predicted promoter region upstream of cagU: upstream from the predicted -35 region, between the predicted -35 and -10 regions, and downstream from the identified transcription start site (Sharma et al. Nature 2010). The DNA sequence of the modified promoter region is shown in Figure S1.
Figure 2. TetR-dependent regulation of CagT.
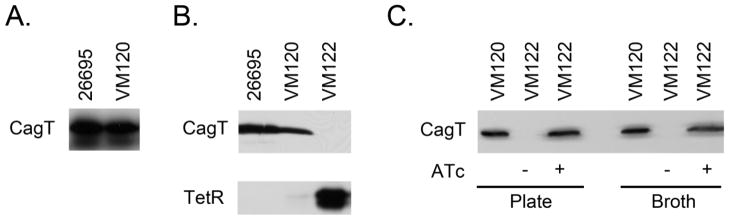
A. Three copies of tetO were inserted upstream of cagUT in H. pylori strain 26695 to yield strain VM120. Samples were standardized by protein concentration and an immunoblot using anti-CagT polyclonal antiserum was used to monitor CagT production. Results indicated that the presence of the tetO copies upstream of cagUT did not disrupt CagT production. B. A codon-optimized copy of tetR was introduced into the ureA locus of strain VM120 to produce strain VM122. Samples were standardized by protein concentration and immunoblots using anti-CagT polyclonal antiserum or anti-TetR monoclonal antibody were used to monitor CagT and TetR production, respectively. Results indicated that CagT production was disrupted by the presence of tetR in strain VM122. C. H. pylori strains were cultured on agar medium or in broth culture for 16 hours in the absence or presence of ATc (100 ng ml−1). Samples were standardized by protein concentration and an immunoblot using anti-CagT polyclonal antiserum was used to monitor CagT production. Results indicated that CagT production in strain VM122 could be induced by the addition of ATc.
3.2. TetR-Mediated Repression of Gene Expression
We next introduced a codon-optimized copy of the Tn10 tetR gene into the ureA locus of strain VM120, generating strain VM122. An immunoblot to detect TetR production and PCR of the chromosomal ureA locus were used to confirm construction of the desired strain (Figure 2B and data not shown). The introduction of tetR resulted in repression of the cagUT operon as evidenced by reduced CagT levels in strain VM122 compared to strains VM120 and VM124 (Figure 2B and data not shown).
3.3. Induction of Gene Expression Using ATc
We next cultured strain VM122 in the absence or presence of the TetR inducer ATc, a derivative of tetracycline that binds the Tet repressor with greater affinity than tetracycline and exhibits markedly reduced antibiotic activity (Degenkolb, Takahashi et al. 1991; Rasmussen, Noller et al. 1991; Oliva, Gordon et al. 1992; Lederer, Kintrup et al. 1996). Cultures then were analyzed for CagT production by immunoblotting. Results indicated that the addition of ATc to the culture medium relieved TetR-mediated repression of the cagUT promoter (Figure 2C).
3.4. Inducible Cag T4SS Activity
CagU and CagT are essential to the function of the Cag T4SS, the multiprotein complex that delivers the CagA oncoprotein to gastric epithelial cells (Fischer, Puls et al. 2001; Kutter, Buhrdorf et al. 2008). Although the function of CagU is unknown, CagT is purported to be a part of the “core complex” that spans the bacterial inner and outer membranes and is thus believed to be an important structural component of the secretion apparatus. Using strain VM122, we analyzed Cag T4SS activity and pilus assembly.
As an initial experiment, strain VM122 grown in the absence of inducer was added to AGS cell monolayers in the absence or presence of ATc. One means of assessing activity of the Cag T4SS was by measuring IL-8 secretion by AGS cells. These results indicated that IL-8 secretion induced by strain VM122 was ATc dependent (Figure 3A). A second means of assessing activity of the Cag T4SS was by immunoblotting to detect phosphorylated CagA in H. pylori-AGS co-cultures. Detection of phosphorylated CagA provides an assessment of CagA entry into cells, since only CagA that enters the host cytoplasm is phosphorylated. Phosphorylated CagA was not detected among AGS cells cultured in the absence of H. pylori or among AGS cells cultured with H. pylori strain VM122 in the absence of ATc (Figure 3B). In contrast, phosphorylated CagA was detected among AGS cells cultured with H. pylori strain VM122 in the presence of ATc (Figure 3B). These results indicated that CagA translocation by strain VM122 also was ATc dependent. Together, these results demonstrated that the tetR-tetO system of gene regulation worked as predicted in the H. pylori–AGS co-culture system.
Figure 3. Inducible H. pylori cag type IV system function.
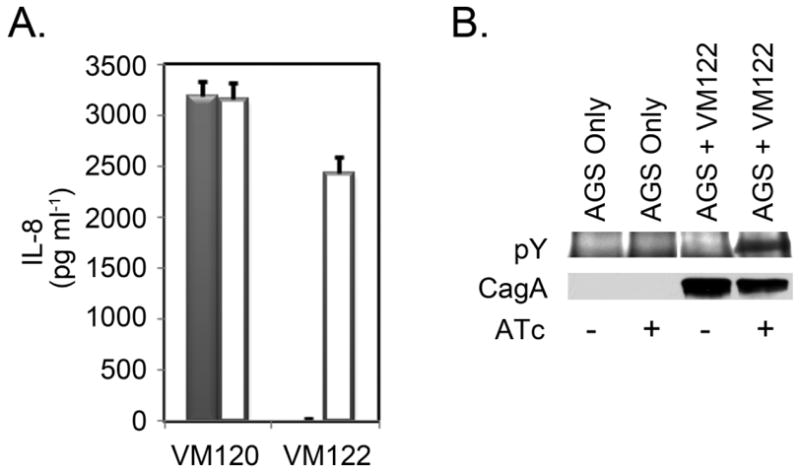
H. pylori strains were cultured in broth for 16 hours in the absence of ATc. Bacteria then were added to AGS cell monolayers at an MOI of 100:1 in the absence or presence of ATc (200 ng ml−1) and co-cultured for 7 hours. A. Tissue culture supernatants were collected, separated from detached cells and bacteria by centrifugation, and analysed for IL-8 content by ELISA. Results indicated that the ability of H. pylori strain VM122 to induce IL-8 production by AGS cells was dependent on the presence of ATc (Filled columns: co-cultures in the absence of ATc. Open columns: co-cultures in the presence of ATc) Results represent the mean and standard deviation of triplicate samples. B. CagA translocation was monitored by immunoblotting cell lysates using anti-phosphotyrosine (pY) antibodies and total CagA was detected using anti-CagA antibodies. Results indicated that the ability of H. pylori strain VM122 to translocate CagA into AGS cells was dependent on the presence of ATc.
Finally, we monitored the production of Cag T4SS pili at the bacterial-host interface. Strain VM122 was grown in the absence of inducer before being added to AGS cell monolayers in the absence or presence of ATc. Analyses by scanning electron microscopy revealed no detectable pili at bacterial-host interfaces in co-cultures lacking ATc (Figure 4A). In contrast, numerous pili were detected in co-cultures grown in the presence of ATc (Figure 4B and C).
Figure 4. Inducible formation of Cag T4SS pili in an H. pylori-AGS cell co-culture.
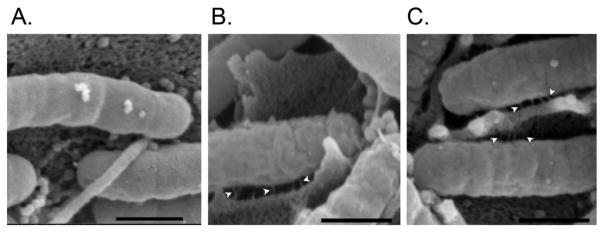
H. pylori strain VM122 was cultured in broth for 16 hours in the absence of ATc. Bacteria then were added to AGS cell monolayers at an MOI of 100:1 in the absence or presence of ATc (200 ng ml−1) and co-cultured for 2 or 4 hours. Monolayers then were fixed and analyzed by SEM. Results indicated that the ability of H. pylori strain VM122 to form Cag T4SS pili at the bacterial-host interface was dependent on the presence of ATc. Representative images are shown (A: 4 hours, no ATc; B: 2 hours with ATc; C: 4 hours with ATc). Arrowheads indicate T4SS pilus structures. The bar in each image denotes 500 nm.
4. Conclusions
This study is the first to report on the use of a tetracycline-regulated promoter in H. pylori. Using this system, we demonstrated that CagU and/or CagT are not only required for functioning of the H. pylori Cag T4SS, but also that one or both of these proteins is required for assembly of T4SS pili at the microbial-host interface. Additional studies beyond the scope of the current study are required to differentiate between CagU- and CagT-specific effects. The assembly of the T4SS pili occurs rapidly upon induction of cagUT expression, with pili detected as early as 2 hours after addition of the inducer, ATc. This result is in agreement with previous reports investigating the time course of H. pylori-induced IL-8 induction (Sharma, Tummuru et al. 1995; Eftang, Esbensen et al. 2012). In addition to being useful in time-course studies, this method provides an alternative approach (instead of knock-out and complementation) for studying gene function, and may be applied to the study of essential genes in H. pylori. Furthermore, with minor modifications, the application of the method could be expanded. For example, by introducing mutations into tetR, the method could be modified such that regulated genes are expressed in the absence of drug and repressed by the addition of drug (i.e., Tet-on) (Urlinger, Baron et al. 2000). Alternatively, by restoring expression of ureA (which is required for H. pylori colonization of animals) in TetR-producing strains, the method could be applied to regulating H. pylori gene expression in animal models of disease.
Supplementary Material
Highlights.
The tet repressor-operator system can be used to control gene expression in H. pylori
agU and/or CagT are required for assembly of the type IV secretion system pili at the H. pylori-epithelial interface
The assembly of the type IV secretion system pili occurs rapidly, with pili detected as early as 2 hours after induction of the cagUT operon
Acknowledgments
We thank Carrie Shaffer for helpful discussions. This study was supported by NIH AI039657, CA116087, AI068009, and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development. Experiments in the Vanderbilt Cell Imaging Shared Resource were supported by the Vanderbilt University Digestive Disease Research Center (NIH grant P30DK058404) and the Vanderbilt University Ingram Cancer Center (NIH grant P30 CA068485).. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviation
- ATc
anhydrotetracycline
- T4SS
type IV secretion system
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mark S. McClain, Email: mark.s.mcclain@vanderbilt.edu.
Stacy S. Duncan, Email: stacy.s.duncan@gmail.com.
Jennifer A. Gaddy, Email: jennifer.a.gaddy@vanderbilt.edu.
Timothy L. Cover, Email: timothy.l.cover@vanderbilt.edu.
References
- Allison CC, Kufer TA, et al. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol. 2009;183(12):8099–109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, et al. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300(5624):1430–4. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando T, Israel DA, et al. HP0333, a member of the dprA family, is involved in natural transformation in Helicobacter pylori. J Bacteriol. 1999;181(18):5572–80. doi: 10.1128/jb.181.18.5572-5580.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119(9):2475–87. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10(8):1573–81. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- Barrozo RM, Cooke CL, et al. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog. 2013;9(2):e1003189. doi: 10.1371/journal.ppat.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boneca IG, Ecobichon C, et al. Development of inducible systems to engineer conditional mutants of essential genes of Helicobacter pylori. Appl Environ Microbiol. 2008;74(7):2095–102. doi: 10.1128/AEM.01348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourzac KM, Guillemin K. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 2005;7(7):911–9. doi: 10.1111/j.1462-5822.2005.00541.x. [DOI] [PubMed] [Google Scholar]
- Busler VJ, V, Torres J, et al. Protein-protein interactions among Helicobacter pylori Cag proteins. J Bacteriol. 2006;188(13):4787–800. doi: 10.1128/JB.00066-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter BM, McDaniel TK, et al. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl Environ Microbiol. 2007;73(23):7506–14. doi: 10.1128/AEM.01084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copass M, Grandi G, et al. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect Immun. 1997;65(5):1949–52. doi: 10.1128/iai.65.5.1949-1952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–73. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailidiene D, Dailide G, et al. Contraselectable streptomycin susceptibility determinant for genetic manipulation and analysis of Helicobacter pylori. Appl Environ Microbiol. 2006;72(9):5908–14. doi: 10.1128/AEM.01135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielli A, Scarlato V. Regulatory circuits in Helicobacter pylori: network motifs and regulators involved in metal-dependent responses. FEMS Microbiol Rev. 2010;34(5):738–52. doi: 10.1111/j.1574-6976.2010.00233.x. [DOI] [PubMed] [Google Scholar]
- Debowski AW, Gauntlett JC, et al. Xer-cise in Helicobacter pylori: one-step transformation for the construction of markerless gene deletions. Helicobacter. 2012;17(6):435–43. doi: 10.1111/j.1523-5378.2012.00969.x. [DOI] [PubMed] [Google Scholar]
- Degenkolb J, Takahashi M, et al. Structural requirements of tetracycline-Tet repressor interaction: determination of equilibrium binding constants for tetracycline analogs with the Tet repressor. Antimicrob Agents Chemother. 1991;35(8):1591–5. doi: 10.1128/aac.35.8.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delany I, Pacheco AB, et al. Iron-dependent transcription of the frpB gene of Helicobacter pylori is controlled by the Fur repressor protein. J Bacteriol. 2001;183(16):4932–7. doi: 10.1128/JB.183.16.4932-4937.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftang LL, Esbensen Y, et al. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 2012;12:9. doi: 10.1186/1471-2180-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst FD, Stoof J, et al. NikR mediates nickel-responsive transcriptional repression of the Helicobacter pylori outer membrane proteins FecA3 (HP1400) and FrpB4 (HP1512) Infect Immun. 2006;74(12):6821–8. doi: 10.1128/IAI.01196-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Puls J, et al. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42(5):1337–48. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- Forsyth MH, Cover TL. Intercellular communication in Helicobacter pylori: luxS is essential for the production of an extracellular signaling molecule. Infect Immun. 2000;68(6):3193–9. doi: 10.1128/iai.68.6.3193-3199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117(1):60–9. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547–51. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4(9):688–94. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- Hawrylik SJ, Wasilko DJ, et al. Bisulfite or sulfite inhibits growth of Helicobacter pylori. J Clin Microbiol. 1994;32(3):790–2. doi: 10.1128/jcm.32.3.790-792.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen W, Wissmann A. Tet repressor-tet operator interaction. In: Saenger W, Heinemann U, editors. Protein-Nulceic Acid Interaction, Topics in Molecular and Structural Biology. Vol. 10. London: Macmillan; 1989. pp. 143–162. [Google Scholar]
- IARC. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 61. World Health Organization International Agency for Research on Cancer; Lyon, France: 1994. Schistosomes, liver flukes and Helicobacter pylori. [PMC free article] [PubMed] [Google Scholar]
- Kutter S, Buhrdorf R, et al. Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J Bacteriol. 2008;190(6):2161–71. doi: 10.1128/JB.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer T, Kintrup M, et al. Tetracycline Analogs Affecting Binding to Tn10-Encoded Tet Repressor Trigger the Same Mechanism of Induction. Biochemistry. 1996;35(23):7439–46. doi: 10.1021/bi952683e. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao P, et al. An in vivo gene deletion system for determining temporal requirement of bacterial virulence factors. Proc Natl Acad Sci U S A. 2008;105(27):9385–90. doi: 10.1073/pnas.0801055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, Forsyth MH, et al. Growth phase regulation of flaA expression in Helicobacter pylori is luxS dependent. Infect Immun. 2004;72(9):5506–10. doi: 10.1128/IAI.72.9.5506-5510.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh JT, V, Torres J, et al. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res. 2007;67(10):4709–15. doi: 10.1158/0008-5472.CAN-06-4746. [DOI] [PubMed] [Google Scholar]
- Muller S, Pflock M, et al. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol Res. 2007;162(1):1–14. doi: 10.1016/j.micres.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Noto JM, Peek RM., Jr Genetic manipulation of a naturally competent bacterium, Helicobacter pylori. Methods Mol Biol. 2012;921:51–9. doi: 10.1007/978-1-62703-005-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287(5457):1497–500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Oliva B, Gordon G, et al. Evidence that tetracycline analogs whose primary target is not the bacterial ribosome cause lysis of Escherichia coli. Antimicrob Agents Chemother. 1992;36(5):913–9. doi: 10.1128/aac.36.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pich OQ, Merrell DS. The ferric uptake regulator of Helicobacter pylori: a critical player in the battle for iron and colonization of the stomach. Future Microbiol. 2013;8(6):725–38. doi: 10.2217/fmb.13.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen B, Noller HF, et al. Molecular basis of tetracycline action: identification of analogs whose primary target is not the bacterial ribosome. Antimicrob Agents Chemother. 1991;35(11):2306–11. doi: 10.1128/aac.35.11.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Otto G, et al. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect Immun. 2001;69:730–736. doi: 10.1128/IAI.69.2.730-736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer CL, Gaddy JA, et al. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog. 2011;7(9):e1002237. doi: 10.1371/journal.ppat.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma CM, Hoffmann S, et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. 2010;464(7286):250–5. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- Sharma SA, Tummuru MK, et al. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63(5):1681–87. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Rappuoli R, et al. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A. 2000;97(3):1263–8. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347(15):1175–86. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- Ta LH, Hansen LM, et al. Conserved transcriptional unit organization of the Cag pathogenicity island among Helicobacter pylori strains. Front Cell Infect Microbiol. 2012;2:46. doi: 10.3389/fcimb.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibonnier M, Aubert S, et al. Study of the functionality of the Helicobacter pylori trans-translation components SmpB and SsrA in an heterologous system. BMC Microbiol. 2010;10:91. doi: 10.1186/1471-2180-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibonnier M, Thiberge JM, et al. Trans-translation in Helicobacter pylori: essentiality of ribosome rescue and requirement of protein tagging for stress resistance and competence. PLoS One. 2008;3(11):e3810. doi: 10.1371/journal.pone.0003810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb JF, White O, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388(6642):539–47. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Urlinger S, Baron U, et al. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci U S A. 2000;97(14):7963–8. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Agriesti F, et al. A convenient and robust in vivo reporter system to monitor gene expression in the human pathogen Helicobacter pylori. Appl Environ Microbiol. 2012;78(18):6524–33. doi: 10.1128/AEM.01252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala J, Chaput C, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5(11):1166–74. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- Whitmire JM, Gancz H, et al. Balancing the double-edged sword: metal ion homeostasis and the ulcer bug. Curr Med Chem. 2007;14(4):469–78. doi: 10.2174/092986707779941069. [DOI] [PubMed] [Google Scholar]
- Wroblewski LE, Peek RM, Jr, et al. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–39. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Ahnen D, et al. NIH Consensus Conference. Helicobacter pylori in peptic ulcer disease. NIH Consensus Development Panel on Helicobacter pylori in Peptic Ulcer Disease. Jama. 1994;272(1):65–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.