

Abstract
The encapsulation of viable tissues via layer-by-layer polymer assembly provides a versatile platform for cell surface engineering, with nanoscale control over capsule properties. Herein, we report the development of a hyperbranched polymer-based, ultrathin capsule architecture expressing bioorthogonal functionality and tailored physiochemical properties. Random carbodiimide-based condensation of 3,5-dicarboxyphenyl glycineamide on alginate yielded a highly branched polysaccharide with multiple, spatially restricted, and readily functionalizable terminal carboxylate moieties. Poly(ethylene glycol) (PEG) was utilized to link azido end groups to the structured alginate. Together with phosphine functionalized poly(amido amine) (PAMAM) dendrimer, nanoscale layer-by-layer coatings, covalently stabilized via Staudinger ligation, were assembled onto solid surfaces and pancreatic islets. The effects of electrostatic and/or bioorthogonal covalent interlayer interactions on the resulting coating efficiency and stability, as well as pancreatic islet viability and function, were studied. These hyperbranched polymers provide a flexible platform for the formation of covalently stabilized ultrathin coatings on viable cells and tissues. In addition, the hyperbranched nature of the polymers presents a highly functionalized surface capable of bioorthogonal conjugation of additional bioactive or labeling motifs.
Keywords: Layer-by-layer, Staudinger ligation, dendritic polymer, conformal coating, cell encapsulation
Introduction
Pancreatic islet transplantation is a promising treatment for Type 1 diabetes mellitus. Current clinical protocols, however, require the use of systemic immunosuppressive drugs to prevent allograft islet rejection, imposing a significant burden on the patient and limiting widespread applicability.1 Alternatively, islet encapsulation within semipermeable membranes can dampen host immune reactivity to the transplanted tissue by blocking direct alloantigen recognition.2, 3 Further, inflammatory pathways may be dampened via masking of inflammatory proteins and agents with benign polymers.
Approaches in the encapsulation of pancreatic islets vary from simple PEGylation to more elaborate semi-permeable protective membranes.4 Typically, islets are encapsulated within microscale hydrogel-based beads, on the order of 600–1000 μm;3, 5 however, imposing large barriers between the cell surface and its environment commonly leads to impairment of cellular function and responsiveness, while also substantially increasing the overall volume of the implant.6, 7 Cellular encapsulation via layer-by-layer (LbL) film deposition is a highly desirable technique, wherein the chemical and physical properties of the coating can be precisely tailored at the nanometer scale, resulting in ultrathin, conformal coatings. A variety of intermolecular interactions can be employed to facilitate layer formation, such as electrostatic,8, 9 covalent,10, 11 hydrogen bonding,12 and molecular recognition.13 The most common type of coating strategy, electrostatic complexation, exhibits major drawbacks when applied to encapsulating viable cells, including cytotoxicity of cationic polymers and long-term instability of resulting coatings. Modulating the net positive charge of cationic polymers, e.g. PEG polymer grafting, can minimize cell damage,9 although long term stability still presents a challenge.
The stability and homogeneity of LbL coatings can be enhanced through the incorporation of interpolymer covalent bonding, which further permits the use of moderately charged or even neutral polymers. While covalent crosslinking within polymeric layers has been achieved through multiple ligation strategies,10, 11, 14 the use of bioorthogonal reactions, i.e. orthogonal reactions that can proceed in an aqueous environment, under defined environmental conditions (e.g. temperature 25–37 °C, pH 7–7.5, osmolarity ~300 mOsM), and involve non-native chemical handles and nontoxic catalysts and/or reaction by-products,15, 16 are highly desirable for viable cell encapsulation. Staudinger ligation, a reaction between an azide and phosphine to form an amide bond, meets these criteria.17 Not only is the reaction scheme bioorthogonal, the resulting covalent bond is resistant to hydrolysis, oxidation, and reduction.18 We have recently demonstrated the applicability of this ligation scheme for enhancing the stability of hydrogels for micro-scale cellular encapsulation.19, 20
The use of hyperbranched polymers and dendrimers in LbL assembly offers a facile platform, with a high degree of end groups for ease in functionalization and efficiency in covalent crosslinking, modulation of chemical property (e.g. biodegradability and stimuli responsive), and spatially restrictive functional end groups. The latter is important for efficient LbL assembly of polymers containing hydrophobic functional end groups, such as phosphine. Hyperbranched polymers have been utilized for a variety of biomedical applications, such as drug delivery carriers,21 biosensors,22, 23 transfection,24 protective coatings,15 and self-assembly of multimolecular structures.25
Considering that alternate deposition of dendrimer and hyperbranched polymers on surfaces may result in coating materials with tunable properties, this study utilizes these highly branched polymers to assemble an innovative capsule architecture expressing bioorthogonal functionality and tailored physiochemical properties for cellular encapsulation and cell surface engineering. Specifically, we have developed hyperbranched alginate and poly(amido amine) (PAMAM) dendrimers functionalized with the complementary Staudinger ligation reactive groups, azide and methyl-2-diphenylphosphino-terephthalate (MDT), respectively (Scheme 1). Alginate was selected as the polymeric backbone for hypergrafting due to its known biocompatibility. The effects of polymer functionalization and electrostatic interactions on the efficiency of coating and resulting film stability were evaluated. Subsequently, selected polymers were used to generate ultrathin coatings on viable cell spheroids, specifically pancreatic rat islets, (Scheme 2) whereby resulting cytotoxicity and cellular function were evaluated. The implications of this novel bioorthogonal, covalent, layer-by-layer platform for cell surface engineering, as well as post-functionalization of terminal end groups for chemoselective conjugation of additional bioactive motifs, are discussed.
Scheme 1.

Synthetic steps for generation of hyperbranched Alg-N3 and MDT/GA PAMAM, specifically: (1) 3,5-dicarboxyphenyl glycineamide; (2) PAMAM functionalized with MDT and GA; and (3) hyperbranched alginate azide. Key: (a) Chloroacetyl chloride, NaOH; (b) NH3; (c) EDC, NHS; (d) H2N-PEG-N3, EDC, NHS; and (e) 2-(diphenylphosphino)terephthalic acid 1-methyl-4-pentafluorophenyl diester, triethylamine, and glutaric anhydride.
Scheme 2. Schematic of ultrathin coating assembly.
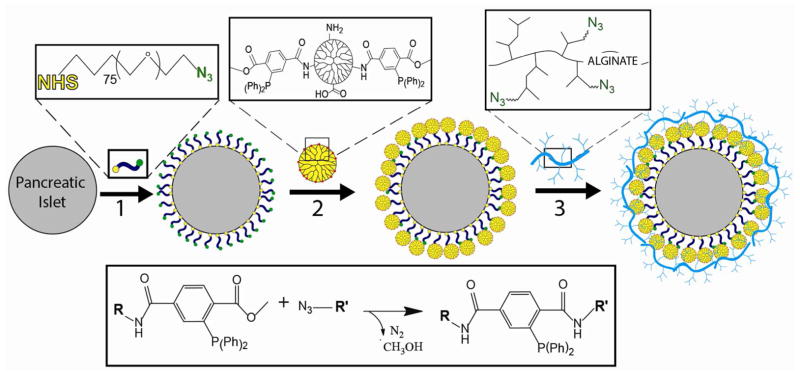
For electrostatic assembly, MDT functionalized PAMAM (2) is deposited directly onto the islet surface via electrostatic interactions. For covalent assembly, N3-PEG-NHS is first covalently bound to free amines on the islet surface (1), followed by covalent linkage of MDT functionalized PAMAM (2). Subsequently, hyperbranched alginate azide is covalently linked to the exposed MDT functionalized PAMAM coating (3). Interlayer covalent bonds are formed between complementary azide and MDT groups via Staudinger ligation (inset reaction scheme). Additional layers can be built via step-wise incubation of MDT functionalized PAMAM (2) and hyperbranched alginate azide (3), until desired number of layers is achieved.
Material and Methods
Materials
Sodium alginate (PRONOVA UP VLVG, Mw 29 kDa, G/M ratio ≥ 1.5) was purchased from NovaMatrix. Fluorescein-5-thiosemicarbazide was purchased from Marker Gene Technologies. Thermo Scientific SnakeSkin regenerated cellulose membrane of 22 mm and 10 kDa molecular weight cut-off was used for dialysis. 11-Bromoundecyltrichlorosilane was purchased from Gelest. Silicon wafers (single side polished, no dopant, 2 inches diameter x 0.5 mm thick), TSKgel GMPWxl (1-8000 kDa MW range) column, poly(amido amine) (PAMAM, 5% wt/v in methanol, ethylenediamine core, generation 5, MW 28,826 g/mol, 5.4 nm diameter, 128 primary amino end groups), 4-pentafluorophenyl ester of 1-methyl-2-diphenylphosphino-terephthalic acid (MDT-pfp), organic solvents, and other high purity reagents were purchased from Sigma-Aldrich. Culture buffers and CMRL media were purchased from Cellgro and Gibco, respectively. Cell viability stains and picogreen DNA determination assays were purchased from Invitrogen. Insulin quantification ELISA assay was purchased from Mercodia.
Polymer Characterization
PerkinElmer Spectrum 100 spectrometer was utilized to obtain attenuated total reflectance Fourier-transform infrared (ATR-FT-IR) spectra for all samples. Spectra were an average of 4 scans at 4 cm−1 resolution. The spectrometer was equipped with a KBr beam splitter, electronically and temperature-stabilized fast recovery deuterated triglycine sulfate detector, and 1 bounce universal ATR sampling accessory. Proton nuclear magnetic resonance spectra for all samples were obtained using a Bruker 400 MHz NMR with autotuning multinuclear probe, housed at the Chemistry Department of the University of Miami. Dynamic light scattering measurements were obtained on a WYATT Technology DynaPro Titan DLS. Ten reads were performed for each sample, with at least 3 independent assessments for each tested polymer and polymer mixtures. Dextran of varying molecular weights (from 10 – 500 kDa) were used to correlate z-average radius to its respective molecular weight. For z-average radii above 10 nm, a logarithmic fit was used, based the collected dextran calibration curve (R2 = 0.994). For z-average radii between 1 and 10 nm, a linear fit was used, again based on the collected dextran calibration curve within that range (R2 = 0.969). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometric (MALDI-TOF-MS, Bruker Bi-Flex IV) measurements with α-cyano-4-hydroxy-cinnamic acid as the matrix were used for molecular weight assessment of N3-PEG-NHS (Chemistry Department, University of Miami). MALDI was attempted for hyperbranched alginate, but was unsuccessful. A Hitachi LaChrom Elite HPLC, TSKgel GMPWxl (1-8000 kDa MW range) column, and a flow rate of 0.6 mL/min were utilized for size exclusion chromatography (SEC). Hyperbranched alginate azide (0.2 mg/mL), labeled with FITC, and FITC-dextran calibrations of 10, 70, or 500 kDa (0.01 mg/mL) were tested. A fluorescence detector recorded emission at 520 nm upon excitation at 480 nm wavelength. FITC was used to enhance measurement sensitivity. PBS (1x, pH 7.4, 135 mM NaCl) was used as the elution and solvent buffer. To fully and rapidly solubilize the Hyp-Alg-N3, the pH of the solution was adjusted to 12 with 5 M NaOH and mixed well, followed by deliberate pH neutralization to 7.2 via 5 M HCl.
Synthesis of 3,5-dicarboxyphenyl glycineamide
The synthesis protocol for 3,5-dicarboxyphenyl glycineamide was adapted from the literature.26 First, 2 mL of chloroacetyl chloride was added at a rate of 500 μL every 10 min to a stirring solution containing 5 g of 5-aminoisophthalic acid, dissolved in 60 mL of 4 M NaOH, while cooling the flask over an ice-water bath. After 20 min, the pH of the solution was adjusted to 1.5 with 10 M HCl. A precipitate formed and was collected by filtration, rinsed with cold water, and dried under reduced pressure. Yield was 5.9 g of a light, brown solid with selected characteristic ATR-FT-IR bands at 3336, 3222, 2901, 2636, 1715, 1689, 1631, 1562, 790, 767, 750, and 700 cm−1.
Next, 5 g of the above product (3,5-dicarboxyphenyl chloroacetamide) was reacted overnight with 70 mL of 29.5% aqueous ammonia. Ammonia was removed under reduced pressure. The resulting solution was cooled in ice-water bath and the pH was adjusted to 1 with 10 M HCl. The resulting precipitate was collected via filtration, rinsed with cold water, and dried under reduced pressure. Yield was 2.1 g of a light, brown solid. Kaiser’s test was positive. Selected characteristic ATR-FT-IR bands were found at 3088, 2611, 1917, 1688, 1618, 1550, 1386, 1211, 899, and 759 cm−1. 1H NMR (D2O with 0.05% TSP and NaOH) peaks: δ 3.89 (2H), 8.04 (2H), and 8.12 (1H) ppm.
Synthesis of hyperbranched alginate
A mixture of 200 mg alginate, 16 mg of N-hydroxysuccinimide (NHS), 240 mg of 2-(N-morpholino) ethanesulfonic acid (MES), and 20 mL purified water was stirred until fully dissolved. 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (800 mg) was added in small incremental portions. Subsequently, 200 mg of 3,5-dicarboxyphenyl glycineamide, dissolved in 3.8 mL of 0.42 M NaOH solution, was added at a rate of 267 μL/min under agitation. After 25 min, 240 μL of 5 M NaOH was added at a rate of 2.67 μL/min. The product was precipitated with 40 mL acetone, collected by filtration, rinsed twice with 4 mL acetone, dissolved in 4 mL 50 mM NaCl, precipitated with 8 mL acetone, rinsed twice with 33% (v/v) water in acetone, rinsed twice with 8 mL acetone, and dried under reduced pressure. Yield was 350 mg of a white, crystalline-like solid with selected characteristic ATR-FT-IR bands at 3271, 3087, 2934, 1694, 1607, 1565, 1396, 1361, 1088, and 1024 cm−1.
Azido-functionalization of hyperbranched alginate
The amount of 50 mg of hyperbranched alginate, 5 mg of NHS, 30 mg of MES, 50 mg of H2N-PEG-N3 (Mw of 372 g/mol, synthesized as previously reported 19), and either 0 or 2 mg fluorescein-5-thiosemicarbazide were dissolved in 5 mL purified water. In small portions, 125 mg of EDC was added while stirring. After 20 min, 30 μL of 5 M NaOH was added at a rate of 1 μL/min. The product was precipitated with 20 mL acetone, collected by centrifugation (2643×g for 2 min), dissolved twice in 3 mL of 50 mM NaCl, precipitated with 15 mL acetone, rinsed twice with 15 mL acetone, and dried under reduced pressure. The resulting dried precipitate was dissolved in 4 mL dH2O water and purified via dialysis against 500 mL water (replaced every 20 min for 2 h). For the first hour, 10 μL of 5 M NaOH and 200 μL of 1 M NaCl were introduced to the solution within the dialysis bag every 20 min. The pure solution was filter-sterilized (0.2 μm pore size filter) and freeze-dried. Yield was 54 mg of a white or orange solid. Kaiser’s test was negative. Selected ATR-FT-IR bands: 3278, 2877, 2110, 1645, 1600, and 1540 cm−1. 1H NMR (D2O with 0.05% TSP) peaks: δ 2.24, 2.84, 3.28–3.82, and 7.52–8.48 ppm. Molecular weight: 1H NMR = 76 kDa average MW, based on end-group analysis and the average MW of base alginate of 29 kDa (for details on calculation of degree of end group functionalization, see Results); SEC = 65–850 kDa, based on dextran calibration curve; and DLS = 70–250 kDa, based on z-average hydrodynamic radius and dextran calibration curve. Of note, a broad MW distribution was expected for the hyperbranched alginate due to the nature of hypergrafting and alginate monodispersity.27, 28 In addition, SEC and DLS methods are affected by polymer aggregation in solution, with may contribute to the wider MW range detected by these techniques.29–31
MDT-Functionalization of PAMAM
A solution of 14, 28, or 37 mg MDT-pfp in 500 μL anhydrous dichloromethane (DCM) was drop-wise injected into 1 mL of 5% w/v PAMAM in methanol under Ar atmosphere and stirred for 30 min. For selected PAMAM-MDT where GA was added, 25 μL of triethylamine in 500 μL DCM was first drop-wise injected, followed by the slow injection of 0–10 mg glutaric anhydride (GA) in 500 μL DCM. After stirring for additional 30 min, 20 μL of glacial acetic acid was added. The product was precipitated with 10 mL diethyl ether and collected by centrifugation (200×g for 1 min). The resulting precipitate was dissolved or suspended in 1 mL 50% v/v DCM in methanol with vortex under Ar atmosphere, precipitated with 10 mL diethyl ether, collected by centrifugation, and dried under reduced pressure. The product was dissolved in 2 mL purified water containing 20 μL glacial acetic acid, otherwise pH was increased with NaOH until dissolved. The solution was purified by dialysis for 2 h against 500 mL purified water (replaced every 20 min) under constant Ar bubbling. During the first hour, 100 μL of 1 M NaCl was added every 20 min. The resulting solution was filter-sterilized and freeze-dried. Successful functionalization and chemical moieties were confirmed by proton NMR and ATR-FT-IR spectroscopy. Yield was 52–57 mg. Selected ATR-FT-IR bands: 1717, 1637, 1539, 746, and 697 cm−1. 1H NMR (D2O with 0.05% TSP) peaks: d 1.83, 2.07–3.66 and 6.5–8.2 ppm. Additional characteristics, including MW, listed in Table 1.
Table 1.
Properties of selected PAMAM derivatives with varying MDT and GA functionalization.
| PAMAM (ID) | MDT (%) | GA (%) | Net Charge | MW (g/mol) |
|---|---|---|---|---|
| 30/0 | 29 | 0 | + 91 | 41,670 |
| 15/0 | 14 | 0 | +110 | 35,026 |
| 15/20 | 14 | 23 | + 51 | 38,412 |
| 15/30 | 14 | 35 | + 21 | 40,178 |
| 15/40 | 14 | 44 | −3 | 41,503 |
| 15/50 | 14 | 58 | −38 | 43,564 |
Synthesis of N3-PEG-NHS
First, 34 mg GA in 500 μL DCM was injected at a rate of 8 μL/min into 1 g H2N-PEG-NH2 (Mw of 3513 g/mol, synthesized as previously reported19) dissolved in 5 mL DCM with rapid stirring and reacted for 2 h under Ar atmosphere. DCM was removed under reduced pressure. The product was dissolved in 7 mL purified water and passed through a 20 mL column of QAE-Sephadex beads using water as solvent. The collected eluted samples containing PEG were combined and freeze-dried. PEG was detected by precipitation test upon mixing same volumes of samples with 1% polyacrylic acid in 1 N HCl solution. The above procedure was repeated, using SP-Sephadex beads instead. The resulting solid was dissolved in 5 mL anhydrous DCM, filtered through a polypropylene filter (0.45 μm pore size), and dried under reduced pressure. Yield was 460 mg of a white solid powder. Kaiser’s test was positive. Selected characteristic ATR-FT-IR bands: 2883, 1652, 1552, and 1099 cm−1. Proton NMR (CDCl3 with 0.03% TMS): δ 1.92–2.03 (m, 2H), 2.28–2.45 (dt, 4H, J = 6.06–7.07 Hz), 3.65 (s, 354H), 6.46 (s, 1H), and 7.91 (s, 2H) ppm.
Second, 200 mg of product from step 1 (H2N-PEG-COOH) was reacted with 49 mg of N3-PEG-NHS (Mw of 914 g/mol, synthesized as previously reported32), and 31 μL of triethylamine in 700 μL anhydrous N,N-dimethylformamide (DMF) for 25 min under Ar atmosphere. The product was precipitated with 7 mL cold (ice-water bath) diethyl ether, collected by centrifugation (2643×g for 5 min at 0 °C), dissolved in 7 mL 200 proof ethanol at 37 °C, passed through a silica gel pad (5×15mm in a Pasteur pipet), precipitated by cooling in ice-water bath with frequent vortex, collected by centrifugation, rinsed with 7 mL cold diethyl ether, and dried under reduced pressure. Yield was 0.17 g of a white solid. Kaiser’s test was negative. Selected characteristic ATR-FT-IR bands: 2884, 2105, 1732, 1660, 1544, and 1102 cm−1.
Finally, 140 mg of product from step 2 (N3-PEG-COOH) was reacted with 11 mg of NHS, and 30 μL of N,N′-diisopropylcarbodiimide in 500 μL anhydrous DMF for 2 h under Ar atmosphere. The product was precipitated with 5 mL cold diethyl ether, collected by centrifugation (2643xg for 5 min at 0 °C), dissolved in 5 mL 200 proof ethanol at 37 °C, precipitated by cooling in ice-water bath with frequent vortex, collected by centrifugation, rinsed with 5 mL cold diethyl ether, and dried under reduced pressure. Yield was 120 mg of a white solid powder. Selected characteristic ATR-FT-IR bands: 2884, 2105, 1812, 1782, 1739, 1668, 1543, 1207, and 1100 cm−1. 1H NMR (CDCl3 with 0.03% TMS): δ 1.92–2.42 (11H), 2.63–2.98 (5H), and 3.13–3.87 (498H) ppm. Mw by MALDI was 4023 g/mol.
Azido-functionalization of Si wafers
A silicon wafer was cut to approximately 11×13 mm. It was treated with 2 mL of 0.1 M NaOH for 3 min at 80 °C, rinsed three times with 2 mL pure water, once with 2 mL 0.1 M HCl, three times with 2 mL water, three times with 2 mL ethanol, and blow-dried with Ar. The wafer was then treated with 6 μL of 11-bromoundecyltrichlorosilane in 2 mL toluene for 30 min at 80 °C under Ar atmosphere and frequent swirling. The Si wafer was rinsed three times with 2 mL toluene, three times with 2 mL 5% (v/v) water in DMF, three times with 2 mL ethanol, blow-dried with Ar, and cured at 110 °C for 10 min under Ar atmosphere. Finally, the Si wafer was treated with 10 mg NaN3 in 2 mL anhydrous DMF under Ar atmosphere for 1 h at 80 °C and frequent swirling. The Si wafer was rinsed three times with 2 mL of DMF, six times with 2 mL of pure water, three times with 2 mL of 50% (v/v) water in ethanol, three times with 2 mL of ethanol, blow-dried with Ar, and stored under Ar at 4 °C in the dark.
Coating and characterization of azido-functionalized Si wafers
For each layer deposited, a Si wafer was placed in a petri dish with water reservoir and covered to prevent dehydration. The azido-functionalized surface was covered with the specific polymeric solution at 3 mg/mL in PBS (1x, pH 7.4) and incubated for 10 min at 37 °C. The solution was mixed 2–3 times during the incubation via a pipette. A large test tube and pipette was utilized to rinse the wafer three times with PBS, dH2O, and blow-dried with Ar. Film thickness on Si wafer surfaces was measured with J.A. Woollam Co. alpha-SETM spectroscopic ellipsometer at a 70° angle in standard mode. Data was fitted to the “Si with transparent film” model. Samples for elemental composition of surface films on Si wafers were submitted to the NanoScience Technology Center of the University of Central Florida for X-ray photoelectron spectroscopy (XPS) (Physical Electronics 5400 ESCA XPS).
Coating of pancreatic islets
Lewis rats islets were isolated as described previously.33 Islets were coated after 48 h post-isolation of culture. Islets were transferred into a 15 mL polypropylene centrifuge tube and pelleted by centrifugation (682 rpm for 14 s). For electrostatic assembly of layers, islets were suspended in 3 mg/mL polymeric solution (in supplemented CMRL1066 culture media) for 30 s at room temperature. The CMRL medium was supplemented with 8.7% (v/v) fetal bovine serum (HyClone from Thermo Scientific), 330 mg/L of L-glutamine, 96 U/mL of Penicillin, 96 mg/L of Streptomycin, and 22 ml/L of 1 M HEPES buffer. All reagents were purchased from Cellgro.
For covalent assembly of each layer, islets were first reacted with N3-PEG-NHS at 20 mg/mL in PBS (1x, supplemented with 0.3 mg/mL of D-glucose, pH 7.8) for 20 min at room temperature. To deposit additional PAMAM or hyperbranched alginate layers, islets were incubated in 3 mg/mL polymeric solution (in supplemented CMRL1066 culture media) for 10 min at 37 °C.
Coating uniformity and stability was assessed 24 and 120 hrs following layer-by-layer assembly via detection of fluorescently labeled hyperbranched alginate. Images were collected using a Leica SP5 spectral confocal inverted microscope. Multi-slice images (4–8 μm thickness; 50–110 slices per image; 1024×1024; 20x objective) were collected and compiled using 3-D projection function. Islets were assessed via MTT metabolic activity, live/dead imaging, and glucose stimulated insulin secretion, as previously described.34 For MTT, triplicates of 300 IEQ were evaluated. MTT results were normalized to control values and represented as fold change from control absorbance. For insulin evaluation, triplicates of 100 IEQ were assessed. Rat ELISA kit (Mercordia) and a Molecular Devices SpectraMax M5 Microplate/cuvette reader was utilized to quantify insulin levels. Coated islets were also assessed via transmission electron microscopy (TEM) 24 h after coating. Islets were pelleted and immersed in 2% glutaraldehyde. The fixed islets were then embedded in Epon/Araldite resin, sectioned, mounted on grids, and stained with 4% uranyl acetate (in 100% methanol) for TEM imaging. TEM images were obtained using a Jeol Jem-1400 transmission electron microscope operating at 80 kV.
For bioorthogonal labeling of the ultrathin coating, islets were coated non-fluorescently labeled polymers. Following assembly of layers, control and coated islets were incubated with 3 mg/mL of FITC-PEG5000-MDT or FITC-PEG5000-CH3 (control) for 30 mins. Confocal imaging of resulting coatings was performed as described above.
Statistical Analysis
Data are expressed as the mean ± standard deviation, with n ≥ 3 for each experimental group. A minimum of three independent experiments were made for each assay, with graphs summarizing results from a representative experiment. For islet viability and insulin secretion experiments, comparisons between groups were made only using the same islet preparation, to minimizing variations due to islet isolation procedures. Statistical analysis was performed on all samples using one-way ANOVA, and Tukey multiple comparison test to evaluate significant difference between groups with P < 0.05.
Results and Discussion
Characterization of dendritic polymers
Synthetic steps and illustrations of hyperbranched polymers are summarized in Scheme 1. A convenient one-pot carbodiimide-based coupling protocol was utilized to hypergraft branched polymeric chains of 3,5-dicarboxyphenyl-glycineamide onto the alginate backbone. Subsequently, hyperbranched alginate was functionalized with linear, heterobifunctional poly(ethylene glycol) (PEG, Mw 350 g/mol), expressing a primary amino and an azido end group. The resulting condensation reaction formed stable amide bonds. ATR-FT-IR spectroscopy exhibited characteristic vibrational bands of hyperbranched alginate azide (Alg-N3), including the alginate O-H stretch (3278 cm−1), alginate CO2− asymmetric stretch (1600 cm−1), amide C=O stretch (1645 cm−1), amide N-H bending (1540 cm−1), PEG C-H symmetric stretch (2877 cm−1), and azide asymmetric stretch (2110 cm−1) (Figure 1A). Degree of functionalization was derived from proton NMR spectroscopy and peak integration (Figure 1B). A calibration curve of PEG (δ 3.28–3.82 ppm) at different concentrations against the constant TSP standard was also used to determine the degree of PEG on the sample. The integration ratio between PEG and aromatic ring protons (δ 7.52–8.48 ppm) was used to quantify the amount of 3,5-dicarboxyphenylglycineamide. Subtracting the derived weights of PEG and 3,5-dicarboxyphenylglycineamide from that of the sample in solution provided the amount of alginate. Data indicated that 1 mole of alginate had 106 moles of 3,5-dicarboxyphenylglycineamide and 70 moles of H2N-PEG-N3, exhibiting a functionalization increase of greater than 350% over our previous, non-branched, alginate functionalization method.19
Figure 1. Characterization of hyperbranched alginate.

(A) ATR-FT-IR spectra of (I) alginate, (II) 3,5-dicarboxyphenylglycineamide, (III) Hyperbranched alginate, (IV) hyperbranched alginate azide, and (V) fluorescent hyperbranched alginate azide. (B) Proton NMR of hyperbranched alginate azide.
For fabrication of MDT functionalized polyamido amine (PAMAM) dendrimer, a 15, 30, or 40% functionalization of primary amino end groups on PAMAM with MDT was evaluated. All resulting PAMAMs were soluble in PBS; however, solutions of 40% functionalized PAMAM, turned cloudy upon warming to 37 °C and were thus excluded from further study. The resulting PAMAM was highly cationic (Table 1). A decreased positive or net neutral polyelectrolyte composition may be more desirable for cellular encapsulation, given that reduction of polymer cationic strength trends to increased biocompatibility.35 As such, to manipulate the net surface charge of the dendrimer, a subset of the remaining primary amino groups was subsequently reacted with glutaric anhydride (GA). Functionalization of 30% MDT PAMAM with 15–30% GA resulted in aggregation at 37 °C. Thus, MDT functionalization was decreased to 15% to permit soluble PAMAMs with GA functionalization. ATR-FT-IR spectra of PAMAM MDT/GA dendrimers revealed characteristic bands of the MDT group (1719, 746, and 697 cm−1), as well as amide bonding (1637 and 1539 cm−1) (Figure 2A). Degree of functionalization was determined via NMR peak integration ratio between aromatic (δ 6.5–8.2 ppm) or GA (δ 1.83 ppm) against PAMAM (δ 2.07–3.66 ppm) protons (Figure 2B). Characterization data of selected MDT and GA functionalized PAMAM dendrimers are summarized in Table 1. Of note, the surface net charge listed for the specific dendrimer assumes every primary amino or carboxylate group is charged, thus the true net charge depends on pH of the solution. Through the manipulation of MDT and GA functionalization, variation in the degree of covalent linkage points, as well as the overall cationic charge of the polymer, can be modulated. This degree of control permits the evaluation of the effects of electrostatic and/or covalent interactions on film formation, thickness, and stability.
Figure 2. Characterization of PAMAM dendrimers.

(A) ATR-FT-IR spectra of (I) PAMAM 15/0, (II) PAMAM 30/0, (III) PAMAM 15/20, and (IV) PAMAM 15/40. (B) Proton NMR of PAMAM 30/0.
Polymers were further assessed via dynamic light scattering, which provides evaluation of the z-average hydrodynamic radius (r) of the polymers. As shown in Figure 3, PAMAM 15/0 peaked at r = 3.2 nm, while hyperbranched Alg-N3 exhibited a broad distribution centered at r = 15.0 nm. Random grafting and branching of the polysaccharide backbone may be a contributing factor for the lack of z-average hydrodynamic radius homogeneity for the hyperbranched alginate. Heterogeneity of polymer size was further supported by size exclusion chromatography (see Supplemental Figure S-1). Of interest, the addition of a small amount (0.15 mg/mL) of the hyperbranched polymer to the functionalized PAMAM solution reproducibly resulted in instant shifting of the z-average hydrodynamic radius to r = 292 nm (Figure 3).
Figure 3.
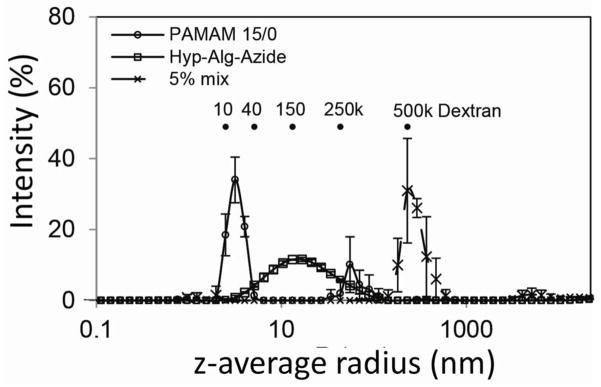
Assessment of particle size via dynamic light scattering (DLS) for PAMAM 15/0 (3 mg/mL), Hyp-Alg-Azide (3 mg/mL), and a mix of PAMAM 15/0 (3 mg/mL) with Hyp-Alg-Azide (0.15 mg/mL or 5%). Error=SD; N=3.
Coating of Si wafer
The ability of these complementary polymers to form stable, covalently linked coatings in a layer-by-layer manner was first evaluated using idealized silicon planar substrates functionalized with azide groups. Azido-functionalization of Si wafers was confirmed via selective binding of mPEG-MDT (but not mPEG-N3) on the Si surface (Figure 4A). The resulting polymer coating was found to be stable upon repeated rinsing with PBS and 4 M NaCl (Figure 4A). Varying combinations of MDT/GA functionalized PAMAM and hyperbranched Alg-N3 polymers were screened for their capacity for layer-by-layer (LbL) assembly, specifically: (1) PAMAM 30/0 with hyperbranched Alg-N3; (2) PAMAM 15/0 with hyperbranched Alg-N3; and (3) PAMAM 15/40 with hyperbranched Alg-N3. Efficient and complete layer-by-layer polymer deposition was achieved for all experimental groups (Figure 4B), with film thickness linearly increasing (R2 ≥ 0.99) after deposition of the third layer. Control groups consisted of (4) PAMAM 0/0 with hyperbranched alginate, (5) PAMAM 15/40 with hyperbranched alginate, and (6) PAMAM 15/40 with Alg-N3 (not hyperbranched). For highly cationic dendrimers (e.g. PAMAM 0/0, 15/0, and 30/0), covalent interactions were not required for LbL assembly (e.g. Figure 4B, #4); however, interpolymer covalent bonding led to thicker individual polymeric layers. For example, increasing MDT functionalization from 0 to 15% to 30% (i.e. PAMAM 0/0 to 15/0 to 30/0) resulted in increased film thickness (Figure 4B, #4 to #2 to #1). A comparison of the films formed using cationic (PAMAM 15/0) and neutral (PAMAM 15/40) dendrimers of identical MDT functionalization (Figure 4B, #2 & #3), found that increased cationic charge resulted in a moderate increase in overall film thickness; highlighting the benefits, but not the requirement, of electrostatic attraction to efficient film deposition. For neutral dendrimers (PAMAM 15/40), covalent linkages were critical for LbL assembly, as illustrated by comparing films formed using hyperbranched alginate with or without azide functionalization (Figure 4B, # 3 vs #5); emphasizing the importance of covalent linkages in film deposition when electrostatic interactions were absent. Negatively charged PAMAM dendrimers (e.g. PAMAM 15/50) did not exhibit LbL deposition with hyperbranched Alg-N3, likely due to electrostatic repulsion between the negatively charged PAMAM and alginate polymers (see Supplemental Figure S-2).
Figure 4. Layer-by-layer assembly of functionalized hyperbranched alginate and PAMAM polymers on planar substrates.
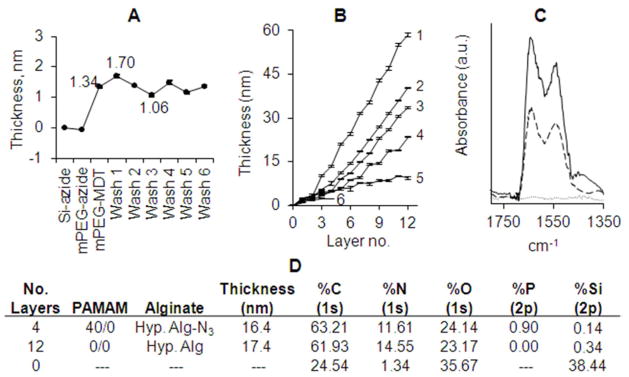
(A) Selectivity and stability of azide-functionalized Si wafer illustrated via incubation of silicone azide surface with mPEG-N3 (control), followed by incubation with mPEG-MDT (bioorthogonal conjugation). Wafer was subsequently washed with PBS (Wash 1–3), followed by 4 M NaCl (Wash 4–6). (B) Ellipsometry film thickness measurements on planar azide-functionalized Si surfaces after deposition of 12 alternating polymeric layers of either: (1) PAMAM 30/0 with hyperbranched Alg-N3; (2) PAMAM 15/0 with hyperbranched Alg-N3; and (3) PAMAM 15/40 with hyperbranched Alg-N3. Controls consisted of (4) PAMAM 0/0 with hyperbranched alginate, (5) PAMAM 15/40 with hyperbranched alginate, and (6) PAMAM 15/40 with Alg-N3 (not hyperbranched). Error = standard deviation. (C) ATR-FT-IR spectra of the deposited films (2) (solid line) and (3) (dashed line). (D) Elemental composition of surfaces of Si wafers by X-ray photoelectron spectroscopy.
Covalently linked films were found to be highly stable over time in both PBS buffer and 4 M NaCl solutions at 37 °C (<1% decrease in film thickness), when compared to films generated solely via electrostatic interactions (8.5 and 20% loss in film thickness following PBS and NaCl washes, respectively). Overall, while film assembly was successful either through covalent and/or electrostatic interactions, the combination of covalent and electrostatic interactions resulted in the largest overall film thickness and greatest stability.
Of note, layer-by-layer film deposition was not achieved when a non-hyperbranched Alg-N3 was used with PAMAM 15/40 (Figure 4B, #6). Hence, the hyperbranches are responsible, at least in part, for the observed layer formation. It has been reported in the literature that LbL film assemblies of poly(allylamine hydrochloride) incorporating poly(styrenesulfonate) (PSS), which contains similar negative (sulfonate) groups on benzene rings, exhibit strong colloidal stability due to charge density and pKa.36 Thus, the resulting strong polyelectrolyte nature of the hyperbranched alginate could be the contributing factor; however, other non-specific interactions such as π-π stacking, π-cation interaction, and H-bonding could contribute to the observed assembly and stability of films.
ATR-FT-IR spectra of coated Si wafers confirmed successful film deposition by the appearance of two IR bands centered at 1647 and 1549 cm−1, principally corresponding to the amide I and II bonds, respectively, on the assembled films (Figure 4C). ATR-FT-IR spectra revealed higher IR absorbance for the polymer combination resulting in both electrostatic and covalent interactions (2), in agreement with ellipsometric measurements. X-ray photoelectron spectroscopy (XPS) characterization of selected uncoated versus coated films further confirmed ellipsometric and ATR-FT-IR results, whereby multi-layer films containing MDT-functionalized PAMAM had detectable phosphorus levels (Figure 4D).
Coating of primary pancreatic islets
The ability of complementary PAMAM and alginate polymers to form nanothin, covalently stabilized, polymeric films on cell clusters was evaluated using primary pancreatic rat islets (Scheme 2). Initial evaluation of islet coating and resulting cytotoxicity was performed via deposition of three polymer layers: PAMAM dendrimer, hyperbranched Alg-N3 (labeled with fluorescein for imaging), and PAMAM dendrimer. In this manner, the first layer assembled onto the islet via electrostatic or, possibly hydrogen bonding, only. For this preliminary three-layer assessment, PAMAM derivatives 15/0, 15/20, 15/30, and 15/40 were screened. Ultrathin, conformal coatings were observed on islet spheroids; however, a trend of decreasing fluorescent intensity and coating uniformity with decreasing PAMAM cationic charge was observed, with minimal detection of fluorescence for the net neutral PAMAM 15/40 (Figure 5, #1–4). These results indicate final film assembly using this approach was dependent on electrostatic interactions at the primary layer. LbL uniformity using neutral PAMAM polymer was enhanced via addition of a primary PEG-N3 layer onto the islet surface through incubation of islets with NHS-PEG-N3. PEG coating was achieved via NHS ester conjugation with primary amino groups on the islet’s surface. Incubation of PEG-N3 coated islets with PAMAM 15/40 resulted in a significant increase in overall LbL assembly (Figure 5, #4 vs #5). These results illustrate the ability to form LbL coatings on islets using neutral PAMAM dendrimers, once the islet surface was terminated in azido groups.
Figure 5. Coating uniformity and cell viability of primary rat pancreatic islets following 3-layer encapsulation via PAMAM and alginate.
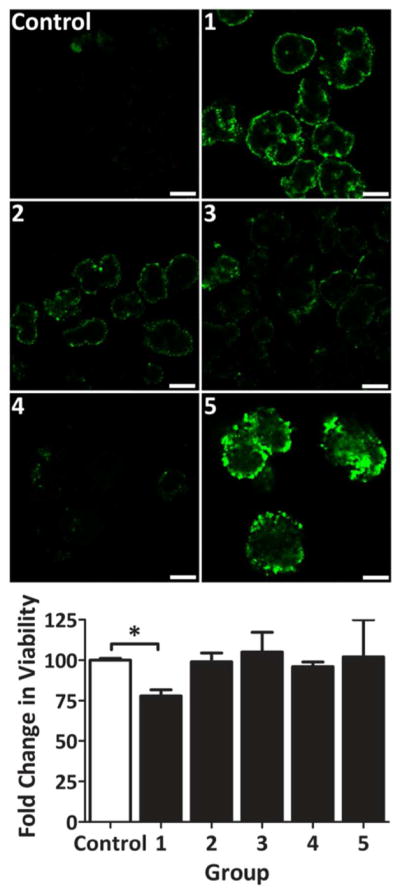
Single plane, confocal fluorescent images of rat islets cross-section 24 h post-encapsulation with 3-layer coatings (Images). Islet viability, via MTT metabolic assay, normalized to control (Graph). Groups: uncoated (Control); 3-layer coating using either PAMAM 15/0 (1), 15/20 (2), 15/30 (3), or 15/40 (4), interlayered with fluorescently-labeled hyperbranched Alg-N3; and NHS-PEG-N3, followed by 3-layer coating using PAMAM 15/40 interlayered with fluorescently-labeled hyperbranched Alg-N3. Error = standard deviation. Scale bar = 200 μm. * P < 0.05
Assessment of cellular viability following the 3-layer coating procedure via MTT metabolic assay (Figure 5) revealed that highly cationic PAMAM dendrimers, e.g. PAMAM 15/0, were cytotoxic, resulting in decreased overall metabolic activity. Cytotoxicity was concentration and time dependent (e.g. metabolic activity decreased from 97 to 27% when PAMAM 30/0 concentration and incubation time was increased from 0.1 mg/mL / 1 min to 3.5 mg/mL / 30 min). This trend in cytotoxicity was expected, as the toxicity of highly cationic polymers, such as poly-L-lysine (PLL) and poly(allylamine hydrochloride) (PAH), for numerous cell types has long been documented.13, 35, 37 Charge neutralization via glutaric anhydride by 20% or higher (e.g. PAMAM 15/20, 15/30, and 15/40) decreased cytotoxicity; consistent with published reports on the neutralization of polymers, such as PEG grafting of PLL.37 After three days of culture, resulting 3-layer coatings revealed instability, as coating fragmentation was observed.
Following 3-layer screening of polymer combinations, evaluation of the most promising dendrimers, i.e. PAMAM 15/0, 15/20, and 15/40, was performed via layer-by-layer assembly of 6 total layers. PAMAM 15/0 was layered via electrostatic assembly and directly applied onto the islet surface. For PAMAM 15/20 or PAMAM 15/40, PAMAM was layered onto the cell spheroid following islet PEGylation via NHS-PEG-N3. For all coatings, fluorescently labeled, hyperbranched Alg-N3 was utilized as the alternating layer. Following the deposition of 6 layers of PAMAM and hyperbranched Alg-N3, nanothin coatings on the islet surface were visualized via confocal microscopy (Figure 6). Strong coverage was observed for coatings containing PAMAM 15/0 or PAMAM 15/20 dendrimers. A trend of decreased coating coverage was observed as the GA functionalization on PAMAM dendrimer increased. Single plane images highlight the localization of the coatings to the periphery of the cell spheroid (Figure 6, bottom panel). The presence of ultrathin PAMAM/alginate coatings was further confirmed via transmission electron microscopy (TEM). As illustrated in Figure 7, the polymeric coatings were relegated to the islet periphery. The coating thickness, per TEM analysis, ranged from 70 – 100 nm. The consistency of the coating was further verified to be high for coating schemes utilizing PAMAM 15/0 and 15/20, while the coating scheme employing PAMAM 15/40 was found to be irregular.
Figure 6. Six-layer encapsulation of primary rat pancreatic islets via layer-by-layer assembly of alginate and PAMAM.

Evaluation of capsule formation via fluorescein labeled alginate and confocal z-stack projection (A–C) or single plane (a–c) imaging of rat pancreatic islets 24 hr after coating. Groups: (1) electrostatic assembly via three bilayers of PAMAM 15/0 and fluorescently-labeled hyperbranched Alg-N3; (2) primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/20 and fluorescentlylabeled hyperbranched Alg-N3; and (3)primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/40 and fluorescently-labeled hyperbranched Alg-N3. Scale bar = 50 μm
Figure 7. TEM images of primary rat pancreatic islets coated with 6 layers via layer-by-layer assembly of alginate and PAMAM.

Groups: (1) electrostatic assembly via three bilayers of PAMAM 15/0 and fluorescently-labeled hyperbranched Alg-N3; (2) primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/20 and fluorescently-labeled hyperbranched Alg-N3; and (3) primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/40 and fluorescently-labeled hyperbranched Alg-N3. Scale bar = 1 μm
Confocal images collected 5 days after coating found highly stable capsules when PAMAM 15/0 and 15/20 were used, while a moderate decrease in coating uniformity was detected for PAMAM 15/40 coatings (see Supplemental Figures, S-3). Results illustrate the enhanced stability of resulting covalently stabilized LbL coatings, even for dynamic, viable tissue surfaces, when 6 polymeric layers were utilized.
The inhomogeneity of the coatings built on a primary NHS-PEG-N3 layer may be attributed to incomplete saturation of the primary PEG coating on the islet surface. Published reports found that a single treatment with activated ester molecules such as NHS-PEG may not result in complete cell coverage.38 Thus, future efforts will focus on improving the homogeneity of this PEG base layer via multiple incubations or doping the islet surface with amine-rich proteins, while preserving islet viability.
Characterization of islet viability and function following encapsulation with PAMAM and alginate polymers was conducted via live/dead imaging, MTT, and glucose stimulated insulin secretion. Metabolic assessment of islets 48 h post-encapsulation via MTT revealed a significant decrease in viability for coated islets containing PAMAM 15/0, when compared to controls (Figure 8A). On the contrary, capsules containing PAMAM 15/20 or neutral PAMAM 15/40 resulted in no significant decrease in overall cell viability, when compared to control islets. Tracking islet viability 5 d post-encapsulation found no detectable difference in viability from controls, as the viability of the control islet significantly decreased from day 2 to day 5 (P < 0.001). This decrease in viability for control islets was expected, given the trend of decreased viability for islets following long culture times.39 Of interest, no significant decline in islet viability from day 2 to day 5 was observed for coated islets, regardless of the polymer combination used.
Figure 8.
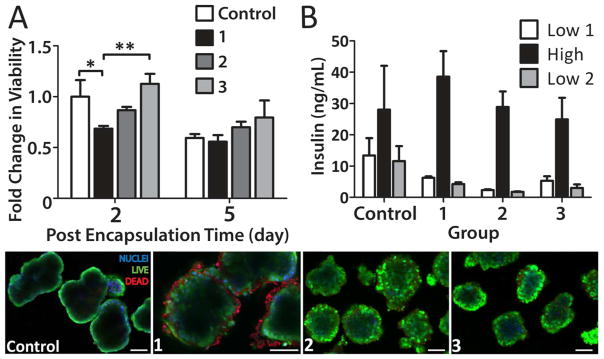
Assessment of islet viability and function after 6-layer coating via MTT metabolic assay (A), glucose-stimulated insulin release (B), and live/dead confocal microscopy imaging (bottom panel; blue, nuclei; live, green; dead, red). Groups: (1) electrostatic assembly via three bilayers of PAMAM 15/0 and fluorescently-labeled hyperbranched Alg-N3; (2) primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/20 and fluorescently-labeled hyperbranched Alg-N3; and (3)primary layer of NHS-PEG-N3, followed by three bilayers of PAMAM 15/40 and fluorescently-labeled hyperbranched Alg-N3. Note: nonspecific binding of the nuclei staining (ethidium homodimer-1, red fluorescence) was observed for (1). *P < 0.05 **P < 0.01 Scale bar = 50 μm
Islet function and the ability of insulin to diffuse out of the coating was evaluated via glucose stimulated insulin release, whereby islets were exposed to 1 h intervals of low, high, and low glucose challenges. All groups exhibited comparable function and responsive insulin secretion when challenged with high glucose (Figure 8B). Live/dead imaging of the coated islets support metabolic activity results, with moderate peripheral cell death observed for islets treated with PAMAM 15/0 (Figure 8, bottom panel). Of note, binding of the red dead cell stain (ethidium homodimer-1) to the highly cationic PAMAM 15/0 was detected, complicating imaging analysis. While this probe was designed to not fluoresce prior to interaction with DNA, co-incubation of this probe with functionalized dendritic polymers exhibited red fluorescence, (see Supplemental Figure S-4). Counterstaining of cells with hoechst nuclei stain permitted delineation between non-specific binding and nuclei binding of dead cells.
With establishment of the benign nature of the layer-by-layer scheme using PAMAM and hyperbranched alginate polymers, future studies will seek to evaluate the capacity of these coatings to mask incendiary cells markers, as well as impart immunoprotective effects. These studies include extensive evaluation of coating permselectivity, detailed characterization of coating uniformity, assessment of the coating’s capacity to mask inflammatory proteins and antigens, as well as in vivo testing using allograft and xenograft transplant models.
Bioorthogonal labeling of ultrathin coating
While encapsulation with biocompatible polymers is highly desirable for masking the cell surface, engineering the surface to express bioactive motifs could provide additional benefits via modulating the local implant environment to assist engraftment, vascularization, decrease inflammation, and/or direct immune responses. Through the use of highly branched and rigid polymers, the resulting coating surface can express a high degree of either phosphine or azide groups, whereby additional agents of interest, such as labels, particles, or proteins, can be easily tethered, in a bioorthogonal manner, via Staudinger ligation chemistry. We have demonstrated this ability, and its subsequent specificity, using fluorescently labeled PEGs expressing a complementary Staudinger ligation group (Figure 9). Thus, these coatings represent a versatile platform for further surface functionalization via highly chemoselective ligation, which can be desirable for cell and tissue engineering applications.
Figure 9. Bioorthogonal tethering of FITC-PEG-MDT to islets following layer-by-layer encapsulation.
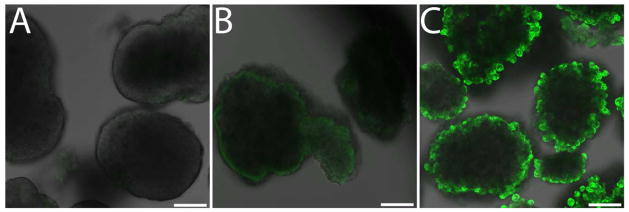
Confocal planar slice image of uncoated control islets incubated in FITC-PEG5000-MDT (A); islet coated with 6 layers of PAMAM 15/20 and hyperbranched alginate-N3, followed by incubation with FITC-PEG5000-CH3 (B); and islet coated with 6 layers of PAMAM 15/20 and hyperbranched alginate-N3, followed by incubation with FITC-PEG5000-MDT (C). Scale bar = 50 μm.
Conclusions
We report the development of functionalized hyperbranched alginate and dendritic polymers that provide a facile platform for the generation of tailored, covalently stabilized, ultrathin coatings on inert and viable surfaces. Hyperbranched structures were found to facilitate efficient, uniform, and stable film deposition. Due to the cytocompatibility of the bioorthogonal ligation strategy, coated pancreatic islets exhibited unimpaired viability and function, when dendrimer cationic strength was mitigated. With the high stability of resulting coatings and film deposition efficiency, these conformal coating systems may find potential applications in the immunoprotection of islets for the treatment of diabetes. Furthermore, the flexibility of this platform to bioorthogonally tether supplemental agents to the surface provides an additional tool to modulate the implant microenvironment.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health through the Type 1 Diabetes Pathfinder Award Program (1DP2 DK08309601) and the Diabetes Research Institute Foundation. We thank the DRI Preclinical and Translational Models Core for providing the rodent islets used for this study and the DRI Analytical Imaging Core for use of their facilities. We thank the University of Central Florida Advanced Materials Processing and Analysis Center, in particular Kirk Scammon, for XPS measurements and analysis. We thank the University of Miami EM Core, particularly Peggy Bates, Vania Almeida, and Yelena Pressman for their highly efficient TEM processing and imaging. We especially thank Dr. Camillo Ricordi, Director of the Diabetes Research Institute, for his continual guidance, support, and consult.
References
- 1.Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, Oberholzer J, Odorico JS, Garfinkel MR, Levy M, Pattou F, Berney T, Secchi A, Messinger S, Senior PA, Maffi P, Posselt A, Stock PG, Kaufman DB, Luo X, Kandeel F, Cagliero E, Turgeon NA, Witkowski P, Naji A, O’Connell PJ, Greenbaum C, Kudva YC, Brayman KL, Aull MJ, Larsen C, Kay TW, Fernandez LA, Vantyghem MC, Bellin M, Shapiro AM. Diabetes Care. 2012;35:1436–45. doi: 10.2337/dc12-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giraldo JA, Weaver JD, Stabler CL. Journal of Diabetes Science & Technology. 2010;4:1238–47. doi: 10.1177/193229681000400525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teramura Y, Iwata H. Adv Drug Del Rev. 2010;62:827–40. doi: 10.1016/j.addr.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Scharp DW, Marchetti P. Adv Drug Del Rev. 2013 doi: 10.1016/j.addr.2013.07.018. in press. [DOI] [PubMed] [Google Scholar]
- 5.Lim F, Sun A. Science. 1980;210:908–10. doi: 10.1126/science.6776628. [DOI] [PubMed] [Google Scholar]
- 6.Trivedi N, Keegan M, Steil GM, Hollister-Lock J, Hasenkamp WM, Colton CK, Bonner-Weir S, Weir GC. Transplantation. 2001;71:203–11. doi: 10.1097/00007890-200101270-00006. [DOI] [PubMed] [Google Scholar]
- 7.van Schilfgaarde R, de Vos P. J Mol Med. 1999;77:199–205. doi: 10.1007/s001090050336. [DOI] [PubMed] [Google Scholar]
- 8.Zhi ZL, Kerby A, King AJ, Jones PM, Pickup JC. Diabetologia. 2012;55:1081–90. doi: 10.1007/s00125-011-2431-y. [DOI] [PubMed] [Google Scholar]
- 9.Wilson JT, Cui W, Kozlovskaya V, Kharlampieva E, Pan D, Qu Z, Krishnamurthy VR, Mets J, Kumar V, Wen J, Song Y, Tsukruk VV, Chaikof EL. J Am Chem Soc. 2011;133:7054–64. doi: 10.1021/ja110926s. [DOI] [PubMed] [Google Scholar]
- 10.El Haitami AE, Thomann J-Sb, Jierry Lc, Parat A, Voegel J-C, Schaaf P, Senger B, Boulmedais F, Frisch Bt. Langmuir. 2010;26:12351–12357. doi: 10.1021/la101670g. [DOI] [PubMed] [Google Scholar]
- 11.Buck ME, Lynn DM. Adv Eng Mater. 2011;13:B343–B352. doi: 10.1002/adem.201080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozlovskaya V, Zavgorodnya O, Chen Y, Ellis K, Tse HM, Cui X, Thompson JA, Kharlampieva E. Adv Funct Mater. 2012;22:3389–3398. doi: 10.1002/adfm.201200138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson JT, Cui W, Chaikof EL. Nano Lett. 2008;8:1940–8. doi: 10.1021/nl080694q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Such GK, Quinn JF, Quinn A, Tjipto E, Caruso F. J Am Chem Soc. 2006;128:9318–9319. doi: 10.1021/ja063043+. [DOI] [PubMed] [Google Scholar]
- 15.Paez JI, Brunetti V, Strumia MC, Becherer T, Solomun T, Miguel J, Hermanns CF, Calderon M, Haag R. J Mater Chem. 2012;22:19488–19497. [Google Scholar]
- 16.Best MD. Biochemistry (Mosc) 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 17.Saxon E, Bertozzi CR. Science. 2000;287:2007–10. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 18.Saxon E, Armstrong J, Bertozzi C. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- 19.Gattas-Asfura KM, Stabler CL. Biomacromolecules. 2009;10:3122–9. doi: 10.1021/bm900789a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall KK, Gattas-Asfura KM, Stabler CL. Acta Biomater. 2011;7:614–24. doi: 10.1016/j.actbio.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaminskas LM, McLeod VM, Porter CJH, Boyd BJ. Mol Pharm. 2012;9:355–373. doi: 10.1021/mp2005966. [DOI] [PubMed] [Google Scholar]
- 22.Qu Y, Sun Q, Xiao F, Shi G, Jin L. Bioelectrochemistry. 2010;77:139–144. doi: 10.1016/j.bioelechem.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Vieira NCS, Figueiredo A, de Queiroz AAA, Zucolotto V, Guimaraes FEG. Sensors. 2011;11:9442–9449. doi: 10.3390/s111009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin G-w, Koo H, Nam K, Kim H, Lee S, Park J-S, Lee Y. Polymer. 2011;52:339–346. [Google Scholar]
- 25.Zhou Y, Huang W, Liu J, Zhu X, Yan D. Adv Mater. 2010;22:4567–4590. doi: 10.1002/adma.201000369. [DOI] [PubMed] [Google Scholar]
- 26.Vinogradov SA. Org Lett. 2005;7:1761–1764. doi: 10.1021/ol050341n. [DOI] [PubMed] [Google Scholar]
- 27.Gao C, Yan D. Prog Polym Sci. 2004;29:183–275. [Google Scholar]
- 28.Schull C, Frey H. ACS macro letters. 2012;1:461–464. doi: 10.1021/mz200250s. [DOI] [PubMed] [Google Scholar]
- 29.Ahrer K, Buchacher A, Iberer G, Josic D, Jungbauer A. J Chromatogr A. 2003;1009:89–96. doi: 10.1016/s0021-9673(03)00433-3. [DOI] [PubMed] [Google Scholar]
- 30.Nobbmann U, Connah M, Fish B, Varley P, Gee C, Mulot S, Chen J, Zhou L, Lu Y, Shen F, Yi J, Harding SE. Biotechnol Genet Eng Rev. 2007;24:117–28. doi: 10.1080/02648725.2007.10648095. [DOI] [PubMed] [Google Scholar]
- 31.Dollinger G, Cunico B, Kunitani M, Johnson DL, Jones R. J Chromatogr A. 1992;592:215–228. [Google Scholar]
- 32.Gattas-Asfura KM, Fraker CA, Stabler C. J Biomed Mater Res A. 2011;99:47–57. doi: 10.1002/jbm.a.33162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pileggi A, Molano RD, Ricordi C, Zahr E, Collins J, Valdes R, Inverardi L. Transplantation. 2006;81:1318–24. doi: 10.1097/01.tp.0000203858.41105.88. [DOI] [PubMed] [Google Scholar]
- 34.Pedraza E, Coronel MM, Fraker CA, Ricordi C, Stabler CL. Proc Natl Acad Sci U S A. 2012;109:4245–50. doi: 10.1073/pnas.1113560109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. Biomaterials. 2003;24:1121–1131. doi: 10.1016/s0142-9612(02)00445-3. [DOI] [PubMed] [Google Scholar]
- 36.Mak WC, Cheung KY, Trau D. Chem Mater. 2008;20:5475–5484. [Google Scholar]
- 37.Wilson JT, Krishnamurthy VR, Cui W, Qu Z, Chaikof EL. J Am Chem Soc. 2009;131:18228–9. doi: 10.1021/ja908887v. [DOI] [PubMed] [Google Scholar]
- 38.Lee DY, Yang K, Lee S, Chae SY, Kim KW, Lee MK, Han DJ, Byun Y. J Biomed Mater Res. 2002;62:372–7. doi: 10.1002/jbm.10246. [DOI] [PubMed] [Google Scholar]
- 39.Kin T, Senior P, O’Gorman D, Richer B, Salam A, Shapiro AMJ. Transpl Int. 2008;21:1029–1035. doi: 10.1111/j.1432-2277.2008.00719.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.