

Abstract
Ruxolitinib, a JAK1/JAK2 inhibitor, is currently the only pharmacological agent approved for the treatment of myelofibrosis. Approval was based on findings from two phase 3 trials comparing ruxolitinib with placebo (COMFORT-I) and with best available therapy (COMFORT-II) for the treatment of primary or secondary myelofibrosis. In those pivotal trials, ruxolitinib rapidly improved splenomegaly, disease-related symptoms, and quality of life and prolonged survival compared with both placebo and conventional treatments. However, for reasons that are currently unclear, there were only modest histomorphological changes in the bone marrow, and only a subset of patients had significant reductions in JAK2 V617F clonal burden. Here we describe a patient with post-polycythemia vera myelofibrosis who received ruxolitinib at our institution (Guy’s and St. Thomas’ NHS Foundation Trust, London, United Kingdom) as part of the COMFORT-II study. While on treatment, the patient had dramatic improvements in splenomegaly and symptoms shortly after starting ruxolitinib. With longer treatment, the patient had marked reductions in JAK2 V617F allele burden, and fibrosis of the bone marrow resolved after approximately 3 years of ruxolitinib treatment. To our knowledge, this is the first detailed case report of resolution of fibrosis with a JAK1/JAK2 inhibitor. Trial registration: ClinicalTrials.gov Identifier: NCT00934544.
Introduction
Primary myelofibrosis is a myeloproliferative neoplasm that is characterized clinically by bone marrow fibrosis, progressive anemia, and extramedullary hematopoiesis, which often manifests as splenomegaly.1,2 Myelofibrosis may also develop as a manifestation of disease progression in other myeloproliferative neoplasms, particularly polycythemia vera (PV) and essential thrombocythemia (ET). At the stage of myelofibrosis in all of these conditions, symptoms arise largely from fibrotic bone marrow replacement. Primary symptoms include those resulting from anemia or splenomegaly (e.g., early satiety, abdominal pain) and constitutional symptoms (e.g., fever, weight loss, night sweats).3,4 Alleviation of these symptoms is a near-term goal of treatment, mainly because conventional therapies have been unable to affect the biology of the disease meaningfully.2
Allogeneic hematopoietic stem cell transplantation is currently the only treatment option that has curative potential in myelofibrosis.5 However, allogeneic transplantation is associated with significant morbidities, and most patients are not suitable candidates. Additionally, with the exception of allogeneic hematopoietic stem cell transplantation, no therapy has consistently been associated with stabilization or resolution of fibrosis in patients with myelofibrosis. Findings that reflect resolution of bone marrow fibrosis with Janus kinase (JAK) inhibitor treatment have not been reported previously. Here we describe the case of a patient with post-PV myelofibrosis who had a marked reduction in bone marrow fibrosis during treatment with the JAK1/JAK2 inhibitor ruxolitinib. To our knowledge, this is the first detailed report of such a case.
Methods
The patient was enrolled in the COMFORT-II trial and was randomized to the ruxolitinib arm.6 COMFORT-II is a randomized, open-label, multicenter study evaluating the safety and efficacy of ruxolitinib 15 or 20 mg twice daily (bid) compared with best available therapy for the treatment of primary myelofibrosis, post-PV myelofibrosis, and post-ET myelofibrosis. The primary endpoint was the proportion of patients who achieved a ≥35% reduction from baseline in spleen volume (i.e., a spleen response) at week 48, as measured by magnetic resonance imaging, while the key secondary endpoint was a spleen response at week 24. Other secondary endpoints included overall survival, histopathological characteristics of bone marrow, and analyses of symptoms and quality of life; dynamics of JAK2 V617F clonal burden was an exploratory endpoint of the trial. The study design and patients’ criteria were fully described previously.6 Of note, the starting dose was determined by the patient’s platelet count at baseline (15 mg bid for patients with a platelet count from 100×109/L to 200×109/L and 20 mg bid for patients with a platelet count >200×109/L), and the dose was titrated for each patient throughout the trial to optimize safety and efficacy. Dose reductions were required for thrombocytopenia and followed a strict protocol-defined dosing regimen. The last data cutoff for the COMFORT-II study was 1 December, 2012; here we report the latest on-study results for this patient as well as additional findings from our institution.
The study protocol was approved by the institutional review board prior to enrollment of patients and the study was conducted in accordance with the principles set forth by the Declaration of Helsinki.
Results
A 74-year old male patient presented to our clinic with constitutional symptoms (night sweats and fever), pruritus, and marked splenomegaly of 26 cm below the left costal margin (spleen volume: 3390 cm3). He had received a diagnosis of PV 10 years previously, in 1999, and had been receiving treatment with hydroxycarbamide since the initial diagnosis. Comorbidities included hypertension and monoclonal gammopathy, both of which were found at the time the PV was diagnosed.
A diagnosis of post-PV myelofibrosis was confirmed, the patient was assigned to the intermediate-2 risk category on the basis of International Prognostic Scoring System (IPSS) criteria3 (age >65 years and presence of constitutional symptoms), and he was enrolled in the COMFORT-II trial6 at a starting dose of ruxolitinib 15 mg bid (platelet count at baseline: 138×109/L). His initial hemoglobin level was 140 g/L, and his white blood cell count was 15.6×109/L. At screening, the patient was found to be JAK2 V617F–positive. Cytogenetic analysis showed an additional abnormality of 46,XY,der(22)t(1;22)(q21;p11.2) [4]/46,XY[3]; the abnormal clone was detected in four out of seven cells analyzed by G-banding, with an unbalanced translocation between chromosomes 1 and 22 that resulted in partial trisomy 1q – a recognized finding in myelofibrosis.7
After the initiation of ruxolitinib treatment, the pateint’s splenomegaly improved dramatically (Figure 1): a 30% reduction in palpable spleen length was observed at week 4 (the first spleen assessment). However, the patient became mildly thrombocytopenic (Figure 2) with a platelet count of 86×109/L, and the dose of ruxolitinib was reduced to 10 mg bid as per study protocol. Platelet counts recovered with this dose reduction, and the patient has remained on treatment at a dose of 10 mg bid. At this dose, splenomegaly progressively resolved. The smallest palpable spleen length measurement, 2 cm (a 90% reduction from baseline), was recorded after 108 weeks of ruxolitinib treatment. The patient was classified as having a spleen response, as defined per protocol, with a 42% reduction in spleen volume at week 24, a 58% reduction at week 48, and a 75% reduction at the latest assessment by magnetic resonance imaging at week 144 (Figure 3). In terms of symptoms, both fatigue and intractable pruritus were present at study entry; within 48 h of the initiation of ruxolitinib treatment, the pruritus had resolved (and has remained negligible) and the fatigue had improved markedly.
Figure 1.
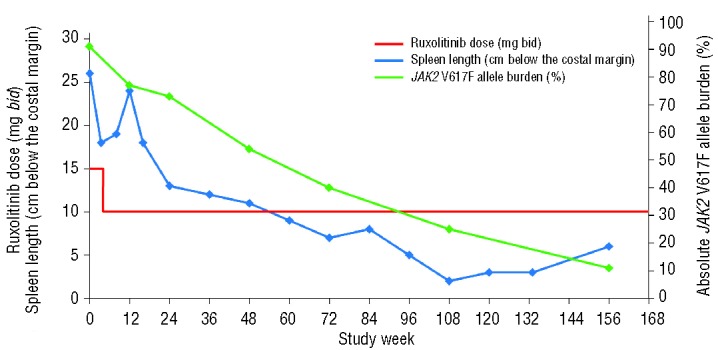
Ruxolitinib dose, palpable spleen length, and JAK2 V617F allele burden over time. bid, twice daily.
Figure 2.
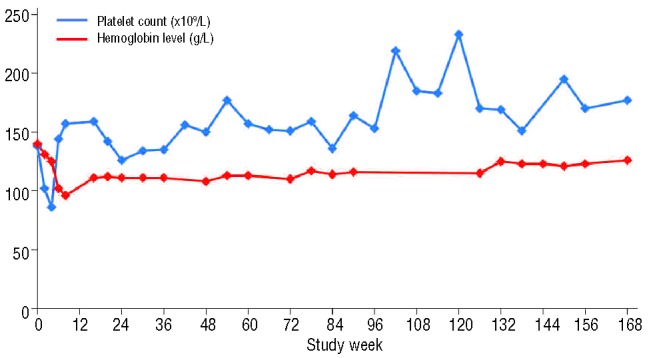
Platelet counts and hemoglobin levels over time.
Figure 3.
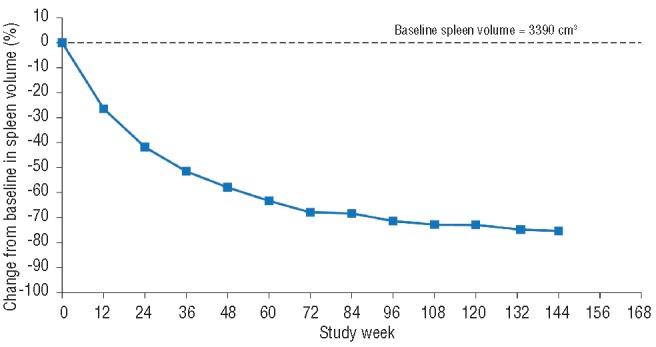
Percentage change from baseline in spleen volume over time.
After 168 weeks of ruxolitinib treatment, morphological analysis of the bone marrow indicated a significant improvement (Figure 4). At the pre-trial baseline assessment in 2009, analysis of hematoxylin and eosin–stained trephine core sections showed maximally increased cellularity with disordered hematopoiesis and pleomorphic megakaryocytes, most of which were large and had abnormally lobated nuclei. Many formed tight clusters, and CD61 immunohistochemical analysis (highlighting megakaryocytes and platelets) emphasized the increase, pleomorphism, and clustering. There was a diffuse and dense increase in reticulin with extensive intersections (Gomori reticulin stain) and focal formation of collagen (Martius Scarlet Blue trichrome stain), but no osteosclerosis [overall World Health Organization (WHO) score: 3].8 After 48 weeks of ruxolitinib treatment, these abnormalities were still apparent, cellularity had decreased, and reticulin fibers were less prominent; however, patches of dense reticulin remained with extensive intersections. After more prolonged treatment (168 weeks), hematoxylin and eosin staining showed normal overall cellularity and more uniformly dispersed megakaryocytes (best seen with CD61 immunostaining). Occasional atypical megakaryocytes were still present, but clustering was minimal and most megakaryocytes had normal morphological characteristics. CD61 immunostaining confirmed that megakaryocytes were more dispersed, were not present in tight clusters, and had limited pleomorphism; platelet shedding into the marrow interstitium was minimal compared with that at baseline. Silver staining showed complete normalization of the fiber pattern (WHO score: 0), and only normal perivascular collagen was evident with Martius Scarlet Blue staining. In parallel with this, the red cell changes characteristic of myelofibrosis (tear drop poilkilocytes and nucleated red cells) also resolved, and the lactate dehydrogenase level normalized, as documented in centrally reviewed laboratory findings.
Figure 4.

Histologic characteristics of bone marrow at baseline and after 48 and 168 weeks of ruxolitinib treatment (a full description is provided in the text). H&E, hematoxylin and eosin.
Of interest, JAK2 V617F allele burden was much reduced with ruxolitinib treatment, from an absolute allele burden of 91% at baseline to approximately 11% at week 156, which is an 88% reduction. This reduction occurred gradually over the course of treatment (Figure 1). The cytogenetic abnormality persisted despite the resolution of fibrosis.
Ruxolitinib treatment was generally well tolerated by this patient. Hematologic adverse events included thrombocytopenia, which resolved after dose reduction, and anemia. These adverse events are expected in the context of JAK1/JAK2 inhibitor therapy, but experience from the COMFORT studies has shown that they are manageable in most patients, and the incidence decreases after 6 months of treatment.9 There was a gradual decline in hemoglobin levels, from 140 g/L at baseline to 96 g/L at day 78. However, levels recovered shortly thereafter to ≥108 g/L for the remainder of treatment (Figure 2); the patient has not required any transfusions. Non-hematologic adverse events included two lower respiratory tract infections (on study days 112 and 826) that resolved with antibiotic treatment. Other adverse events of interest that were considered unrelated or unlikely to be related to treatment included basal cell carcinoma (resolved by Moh surgery) and squamous cell carcinoma (resolved by excision of the lesion on the right side of the chest).
Discussion
Dysregulation of the JAK/STAT pathway is a hallmark of myelofibrosis,10,11 and the resulting overexpression of several pro-inflammatory cytokines has been implicated in the progression of fibrosis.12 Given the reported effects of ruxolitinib treatment on various pro-inflammatory cytokines,6,13,14 one might expect an improvement in bone marrow fibrosis in some patients. At the time of the primary analyses of the COMFORT studies, no major changes in the morphological characteristics of bone marrow were observed (after 6 months and 1 year of treatment in COMFORT-I and -II, respectively).6,13 Reduced-intensity allogeneic hematopoietic stem cell transplantation has been shown to resolve fibrosis within 6 to 12 months in patients with myelofibrosis15; the duration of treatment may need to be longer to observe significant improvements in fibrosis in the context of JAK1/JAK2 inhibitor therapy, where the pathway cannot be completely repressed. For example, in one retrospective analysis of patients at the MD Anderson Cancer Center receiving ruxolitinib in the phase I/II study, 15% of patients (10/67) had an improvement in fibrosis at 24 months and 24% (4/17) had an improvement at 48 months (assessed histologically on the basis of the WHO fibrosis scoring). For comparison, 10% of patients (3/31) in a control group who received hydroxycarbamide showed an improvement after 24 months, and no patients (0/20) showed an improvement at 48 months.16 Of note, in that analysis, 53% of ruxolitinib-treated patients showed stabilization in morphological characteristics of bone marrow at 48 months compared with 35% of hydroxycarbamide-treated patients. Published literature on interferon therapy is also notable for occasional reports of resolution of histopathological features in either PV (as documented in 3 patients by Larsen and colleagues17) or myelofibrosis; Silver and colleagues18 reported morphological remission in two of 17 patients with early myelofibrosis (WHO grade 1 or 2 fibrosis and IPSS low or intermediate-1 risk at diagnosis). A large French group19 collated data regarding responses to interferon in myelofibrosis but did not document histological responses.
Here we report for the first time the resolution of bone marrow fibrosis with normalization of hematopoiesis in a patient receiving ruxolitinib treatment in COMFORT-II. A pre-requisite for entry into the COMFORT studies was the presence of advanced disease (IPSS score intermediate-2 or above) and the studies have shown that ruxolitinib prolongs survival in patients with myelofibrosis compared with placebo and best available therapy, which suggests a potential disease-modifying effect.9,20 Longer-term follow up of the COMFORT study cohorts should help to determine whether ruxolitinib treatment can indeed improve the morphological characteristics of bone marrow in other patients with myelofibrosis and affect the natural course of the disease; if it does, it will be of great interest to define what the particular characteristics of such responding patients might be.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Tefferi A. Essential thrombocythemia, polycythemia vera, and myelofibrosis: current management and the prospect of targeted therapy. Am J Hematol. 2008;83(6): 491–7 [DOI] [PubMed] [Google Scholar]
- 2.Vannucchi AM. Management of myelofibrosis. Hematol Am Soc Hematol Educ Program. 2011;2011:222–30 [DOI] [PubMed] [Google Scholar]
- 3.Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13): 2895–901 [DOI] [PubMed] [Google Scholar]
- 4.Mesa RA, Schwager S, Radia D, Cheville A, Hussein K, Niblack J, et al. The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009;33(9): 1199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLornan DP, Mead AJ, Jackson G, Harrison CN. Allogeneic stem cell transplantation for myelofibrosis in 2012. Br J Haematol. 2012; 157(4): 413–25 [DOI] [PubMed] [Google Scholar]
- 6.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9): 787–98 [DOI] [PubMed] [Google Scholar]
- 7.Andrieux J, Demory JL, Caulier MT, Agape P, Wetterwald M, Bauters F, et al. Karyotypic abnormalities in myelofibrosis following polycythemia vera. Cancer Genet Cytogenet. 2003;140(2): 118–23 [DOI] [PubMed] [Google Scholar]
- 8.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8): 1128–32 [PubMed] [Google Scholar]
- 9.Verstovsek S, Mesa R, Gotlib J, Levy R, Gupta V, DiPersio J, et al. Long-term outcome of ruxolitinib treatment in patients with myelofibrosis: durable reductions in spleen volume, improvements in quality of life, and overall survival advantage in COMFORT-I, [abstract 800]. 54th ASH Annual Meeting and Exposition, Atlanta, GA; 2012 Dec 8–11 [Google Scholar]
- 10.Vannucchi AM. Management of myelofibrosis. Hematol Am Soc Hematol Educ Program. 2011;2011:222–30 [DOI] [PubMed] [Google Scholar]
- 11.Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;188(7): 1723–35 [DOI] [PubMed] [Google Scholar]
- 12.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24(2): 133–45 [DOI] [PubMed] [Google Scholar]
- 13.Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9): 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363 (12): 1117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroger N, Kvasnicka M, Thiele J. Replacement of hematopoietic system by allogeneic stem cell transplantation in myelofibrosis patients induces rapid regression of bone marrow fibrosis. Fibrogenesis Tissue Repair. 2012;5 (Suppl 1): S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kvasnicka HM, Thiele J, Bueso-Ramos C, Hou K, Cortes J, Kantarjian H, Verstovsek S. Exploratory analysis of the effect of ruxolitinib on bone marrow morphology in patients with myelofibrosis [abstract 703]. J Clin Oncol. 2013;31 (Suppl). [Google Scholar]
- 17.Larsen TS, Iversen KF, Hansen E, et al. Long term molecular responses in a cohort of danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res. 2013;37(9): 1041–5 [DOI] [PubMed] [Google Scholar]
- 18.Silver RT, Vandris K, Goldman JJ. Recombinant interferon-alpha may retard progression of early primary myelofibrosis: a preliminary report Blood. 2011;117(24): 6669–72 [DOI] [PubMed] [Google Scholar]
- 19.Ianotto JC, Boyer-Perrard F, Gyan E, Laribi K, Cony-Makhoul P, Demory JL, et al. Efficacy and safety of pegylated-interferon alpha-2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol. 2013;162(6): 783–91 [DOI] [PubMed] [Google Scholar]
- 20.Cervantes F, Kiladjian J, Niederwieser D, Sirulnik A, Stalbovskaya V, McQuitty M, et al. Long-term efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis [abstract 801]. 54th ASH Annual Meeting and Exposition, Atlanta, GA; 2012 Dec 8–11 [Google Scholar]