

Abstract
In vitro and in vivo resistance to prednisolone are predictive for an adverse prognosis in pediatric precursor B-acute lymphoblastic leukemia. Causes of resistance are still poorly understood. In this study, we observed that prednisolone exposure of prednisolone-sensitive patients’ leukemic cells decreased anti-apoptotic MCL1 protein levels by 2.9-fold, while MCL1 protein expression in prednisolone-resistant leukemic patients’ cells was unaffected (P<0.01). Locked nucleic acid oligonucleotides directed against MCL1 reduced MCL1 protein levels by 82±16% (P<0.05) in leukemic cells, decreased proliferation by 9-fold and sensitized to prednisolone up to 80.8-fold, compared to a non-silencing-control locked nucleic acid (P<0.05). Remarkably, we discovered that MCL1-silencing up-regulated the glucose consumption of leukemic cells by 2.5-fold (P<0.05), suggesting a potential rescue mechanism mediated by glycolysis. Targeting glycolysis by 2-deoxyglucose synergistically inhibited leukemic survival by 23.2-fold in MCL1-silenced cells (P<0.05). Moreover, 2-deoxyglucose and MCL1 locked nucleic acid concomitantly sensitized leukemic cells to prednisolone compared to MCL1 locked nucleic acid or 2-deoxyglucose alone (P<0.05). In conclusion, these results indicate the need to target both MCL1 and glycolysis simultaneously to inhibit leukemic survival and sensitize acute leukemia patients towards prednisolone.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common cancer in children. Although cure rates have greatly improved over recent years, treatment is still ineffective in 20% of patients. Unsuccessful treatment can be ascribed to resistance of primary leukemic cells to anti-leukemic drugs.1 Poor prognosis is particularly associated with resistance to prednisolone in childhood ALL, both in vivo and in vitro.2 To increase current survival rates, it is, therefore, necessary to overcome prednisolone resistance.
To identify therapeutic targets to overcome prednisolone resistance, we previously performed microarray analysis on primary BCP-ALL cells of pediatric patients.3 This indicated a role in prednisolone resistance for MCL1, an anti-apoptotic member of the BCL2 family that is frequently over-expressed in a variety of cancers, and that contributes to cancer cell survival and apoptosis resistance.4,5 Functional studies revealed that silencing of MCL1 sensitized leukemic cells to prednisolone.6,7
In addition, we and others discovered that glycolysis is increased in prednisolone-resistant leukemic cells.8,9 Microarray analysis on primary precursor B-acute lymphoblastic leukemia (BCP-ALL) cells of pediatric patients indicated an increased expression of several glycolytic enzymes and glucose transporters in prednisolone resistant patients.3 Furthermore, 2-deoxyglucose (2-DG), an inhibitor of glycolysis, sensitized both leukemic cell lines and patients’ ALL cells to prednisolone.8,9
For three reasons, we now hypothesize that anti-apoptosis sustained by MCL1, and glycolysis are linked and concomitantly induce drug resistance in leukemia. 1) Cellular respiration and apoptosis are closely related survival pathways both associated with prednisolone resistance,6–9 and other targeted molecular leukemia therapies, such as imatinib.10 2) Increased glucose metabolism has been directly linked to MCL1 stabilization and attenuation of apoptosis.11,12 3) BCL2 family members can, besides their apoptotic function, adjust oxidative phosphorylation.13,14
In the present study, we show that silencing of MCL1 by specifically designed locked nucleotide acid antisense oligonucleotides against MCL1 mRNA (MCL1 LNA) inhibited cell survival and sensitized to prednisolone in both BCP-ALL and T-ALL leukemic cells. Moreover, we discovered higher glucose consumption in ALL cells after MCL1 silencing by both shMCL1 and MCL1 LNAs. Most importantly, we demonstrate that 2-DG treatment of MCL1-silenced cells decreased glucose consumption and synergistically reduced leukemic survival. Moreover, MCL1 LNA and 2-DG concomitantly reversed prednisolone resistance in leukemic cells. These data provide evidence that MCL1 and glycolysis should be targeted simultaneously to effectively inhibit leukemic survival and to reverse prednisolone resistance in ALL.
Methods
Cell culture and primary cells
Leukemic cells from children with newly diagnosed ALL were isolated from bone marrow aspirates and prednisolone resistance was assessed, as previously described.1,3 Informed consent was given by patients as approved by the local institutional review board. Only samples with 90% or over leukemic cells upon processing were used in the present study. Reh, 697, Sem, Jurkat, Loucy and HEK293T cells were obtained from DMSZ. The leukemic cell lines were cultured in RPMI+Glutamax (Gibco) and HEK293T cells in DMEM+Glutamax (Gibco) at 37°C in humidified air containing 5% CO2. Cell viability and cell count were determined by a trypan blue exclusion staining assay and analyzed by MACSQuant.
LNA transfection and 2-deoxyglucose treatment
Cell lines were cultured in the presence of either 10 μM locked nucleotide acid oligonucleotides directed against MCL1 (MCL1 LNA), i.e. SPC4120 (MCL1 LNA-a), SPC4342 (MCL1 LNA-b), SPC4343 (MCL1 LNA-c), or a non-silencing control oligonucleotide LNA, i.e. SPC3088. Twenty-four hours after LNA transfection, cells were cultured with and without 0.5 mM 2-deoxyglucose (Sigma). Online Supplementary Figure S1A and B illustrates that 0.5mM 2-deoxyglucose only modestly hampers cell count, while it has a quantifiable effect on glucose consumption. After 96 h, culture medium was replaced by fresh medium containing fresh LNA +/− 2-DG.
Apoptosis measurement
AnnexinV/PI double positive and AnnexinV single positive cells were measured on the BD FACS Calibur flow cytometer.
Quantitative RT-PCR
MCL1 mRNA levels were quantified by incorporation of SYBR Green (Thermo Scientific) by quantitative real-time PCR (Applied Biosystems 7900HT). Primers for MCL1 were; 5′-GGAGGAGGACGAGTTGTAC-3′ (forward) and 5′-AAG GCA CCA AAA GAA ATG-3′ (reverse).
Reverse phase protein array
Primary leukemic cells were cultured for 48 h with 0 μg/mL, 1 μg/mL or 250 μg/mL prednisolone. Protein was isolated and lysates were spotted twice in triplicate on glass-backed nitrocellulose-coated array slides by the facility of Dr. EF Petricoin, George Mason University, Manassas, USA. Antibodies used were: MCL1 antibody (Sigma HPA008455), Bcl-XL (Cell Signaling 2762), BCL-2 (Cell Signaling 2872) and p53 (Cell Signaling 9282).
Western Blot and immunoblotting
Twenty micrograms of protein were used as input for Western blot analysis using anti-MCL1 (Sigma HPA008455), anti BCL-2 (Cell Signaling 2872), anti-β-actin (Abcam ab6276), anti-Clathrin (Santa Cruz Sc12734), IRDye 800CW-labeled anti-rabbit and IRDye 680CW-labeled anti-mouse (Li-Cor IRDye). Protein levels were quantified using the Odyssey system
In vitro MTT drug-resistance assay
Cytotoxicity of cells towards prednisolone (Bufa Pharmaceutical Products) was determined by the in vitro 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) drug-resistance assay, as described previously.1
Glucose-consumption assay
Glucose levels were measured with the glucose assay kit (Sigma, GAGO-20). To calculate glucose consumption, values were compared with plain RPMI glucose levels and corrected for cell growth.
Synergistic effect
Synergistic effects were calculated from equi-effective drug concentrations by the equitation postulated by Berenbaum.15
Statistical analyses
Prednisolone exposure effects within either prednisolone resistant or sensitive patients was analyzed with a Kruskall-Wallis test. A Mann-Whitney U test was used to compare resistant to sensitive patients. The Mann-Whitney U test was also applied to analyze the effects of shMCL1. MCL1 LNA experiments were compared with a t-test. P<0.05 was considered statistically significant.
Additional information regarding methods can be find in the Online Supplementary Appendix.
Results
Downregulation of MCL1 by prednisolone is impaired in prednisolone-resistant leukemic cells of patients
Glucocorticoids are known to induce apoptosis by downregulation of anti-apoptotic BCL-2 family members independent of p53.16,17 We analyzed protein expression of BCL-2 family members and p53 in 3 prednisolone sensitive and 3 resistant primary patient samples after exposure to prednisolone for 48 h (Figure 1 and Online Supplementary Figure S2A–D). After in vitro prednisolone exposure, the expression of MCL1 in leukemic cells of in vitro prednisolone sensitive pediatric ALL patients significantly decreased by 2.9-fold (P<0.01). Whereas, in prednisolone resistant ALL patient cells, these levels did not change upon prednisolone exposure. Prednisolone did not affect the expression levels of BCL-XL, BCL2 nor p53 (Figure 1 and Online Supplementary Figure S2A–D). A similar decrease in MCL1 but not in BCL2 was seen using an extended dilution series of prednisolone (Online Supplementary Figure S2E).
Figure 1.
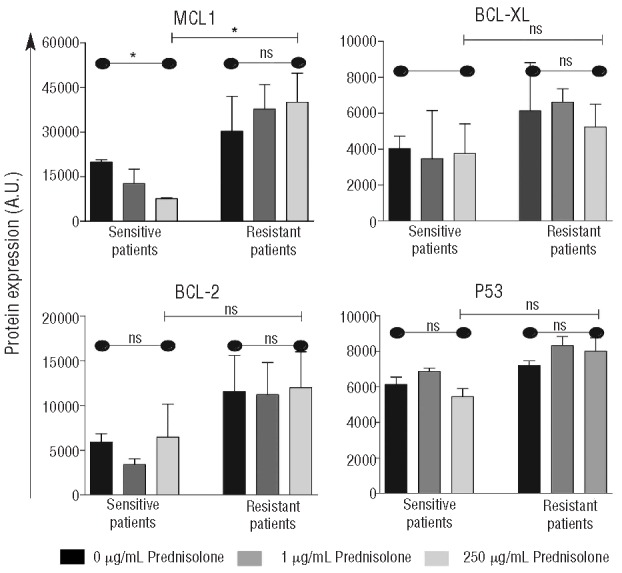
Downregulation of MCL1 by prednisolone is impaired in prednisolone-resistant leukemic cells of patients. Leukemic cells of 3 in vitro prednisolone sensitive and 3 in vitro prednisolone-resistant patients, were treated in vitro for 48 h with 0 μg/mL, 1 μg/mL or 250 μg/mLprednisolone. Protein expression levels of MCL1, BCL-XL, BCL-2 and p53 were analyzed by reverse phase protein array. Bar indicates the mean plus SEM of 3 independent patient samples. (A Kruskal-Wallis test was used to compare 0, 1, 250 μg/mL data points indicated by - - - and a Mann-Whitney U test was used to compare data between sensitive and resistant patients indicated by
 *P<0.05; **P<0.01. A.U.: arbitrary units.
*P<0.05; **P<0.01. A.U.: arbitrary units.
MCL1 is a potent target to inhibit leukemic survival and to sensitize to prednisolone in pediatric ALL
Three different newly-developed LNA oligonucleotides directed against MCL1 were up to 90% effective in silencing MCL1 in five distinct leukemic BCP-ALL and T-ALL cell lines, i.e. MLL-AF4+ BCP-ALL (SEM), ETV6-RUNX1+ BCP-ALL (REH), E2A-PBX1+ BCP-ALL (697), ETP-ALL (Loucy) and tetraploid T-ALL (Jurkat) cells (P<0.01) (Online Supplementary Figures S3 and 4). MCL1 LNA-b provided the most potent and reproducible knockdown of these three MCL1 LNAs. Knockdown achieved by the three MCL1 LNAs was comparable to the knockdown obtained after stable lentiviral transduction of two short hairpin RNA directed against MCL1, i.e. shMCL1a and -b (P<0.05) (Online Supplementary Figure S5). The MCL1 LNAs inhibited leukemic survival up to 90%, increased apoptosis up to 60% and sensitized to prednisolone up to 80.8-fold in five distinct leukemic cell lines, all compared to a non-silencing LNA control (P<0.05) (Online Supplementary Figures S6–8). These MCL1 LNA results were comparable to shMCL1 results (Online Supplementary Figures S9) and indicate that targeting MCL1 may be clinically important.
MCL1-silenced cells up-regulate glycolysis. Targeting glycolysis in MCL1 silenced cells synergistically inhibits leukemic survival and concomitantly reverses prednisolone resistance
To test our hypothesis that MCL1 and glycolysis cooperate in prednisolone resistance, we examined the consumption of glucose in five distinct MCL1 silenced cell lines. A significant increase in glucose consumption, on average 149%, was observed upon MCL1 silencing by MCL1 LNA (P<0.01 MCL1 LNA-b) (Figure 2) and MCL1 LNA-a and LNA-c (Online Supplementary Figures S10) and by shMCL1 (P<0.01) (Online Supplementary Figures S11), compared to non-silencing LNA Controls.
Figure 2.
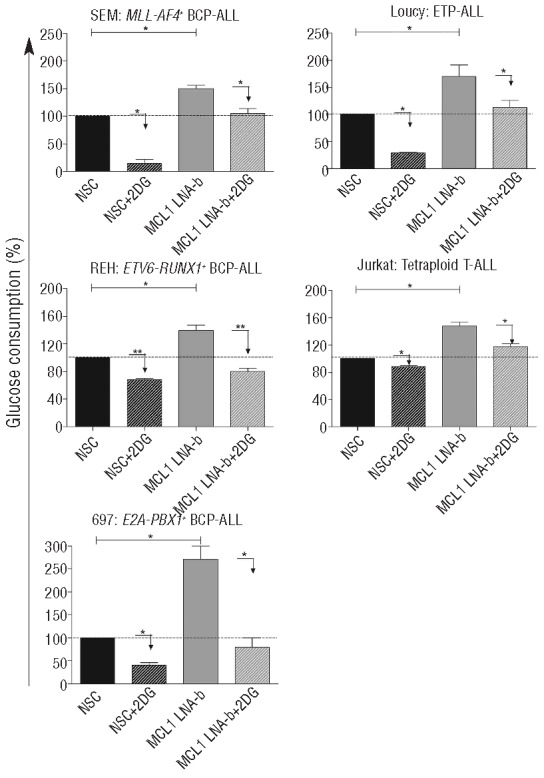
MCL1-silenced cells up-regulate glycolysis, which can be reduced by 2-DG. Glucose consumption of MLL-AF4+ BCP-ALL, ETV6-RUNX1+ BCP-ALL, E2A-PBX1+ BCP-ALL, ETP-ALL and tetraploid T-ALL cell line after treatment with MCL1 LNA-b and/or a non-silencing control LNA (NSC) with or without 0.5mM 2-DG was examined with a glucose consumption assay. To calculate glucose consumption, values were compared with glucose levels in plain RPMI medium and corrected for cell growth. Data are presented as means plus SEM of 3 (ETV6-RUNX1+ BCP-ALL and Tetraploid T-ALL) or 2 independent experiments (*P<0.05, **P<0.01).
This finding suggested that MCL1 silenced cells might increase their glycolysis to rescue from apoptosis. This prompted us to investigate the effect of targeting both MCL1 and glycolysis on leukemic cell survival and prednisolone cytotoxicity. Co-exposure of MCL1 LNA and 2-DG significantly reduced glucose consumption (P<0.05; MCL1 LNA-b, Figure 2; MCL1 LNA-a and MCL1 LNA-c, Online Supplementary Figures S10). In all cell lines, co-treatment of cells with MCL1 LNA and 2-DG synergistically inhibited leukemic cell survival by 30–75%, compared to MCL1 LNA or 2-DG alone, except in the tetraploid T-ALL (P<0.05; Fsyn <1, Figure 3A and Online Supplementary Figures S12). Furthermore, the addition of prednisolone decreased leukemic survival even more (P<0.05; Figure 3B and Online Supplementary Figures S13). Incubation of leukemic cells with both MCL1 LNA and 2-DG concomitantly, albeit moderately, sensitized to prednisolone up to 1.48-fold compared to MCL1 LNA or 2-DG alone (P<0.05; Figure 3C and Online Supplementary Figures S14). The synergism of MCL1 LNAb, 2-DG and prednisolone was best visible in the intermediate responsive cells, i.e. MLL-AF4+, ETV6-RUNX1+ and ETP-ALL cell line, since the sensitive E2A-PBX1+ cell line was already prone to die by monotherapy alone and the highly resistant tetraploid cell line will most likely need higher amounts of the drugs to demonstrate an effect. Overall, these data indicate that MCL1-silenced cells up-regulate glycolysis and that targeting glycolysis in MCL1 silenced cells synergistically inhibits leukemic survival and concomitantly reverses prednisolone resistance.
Figure 3.
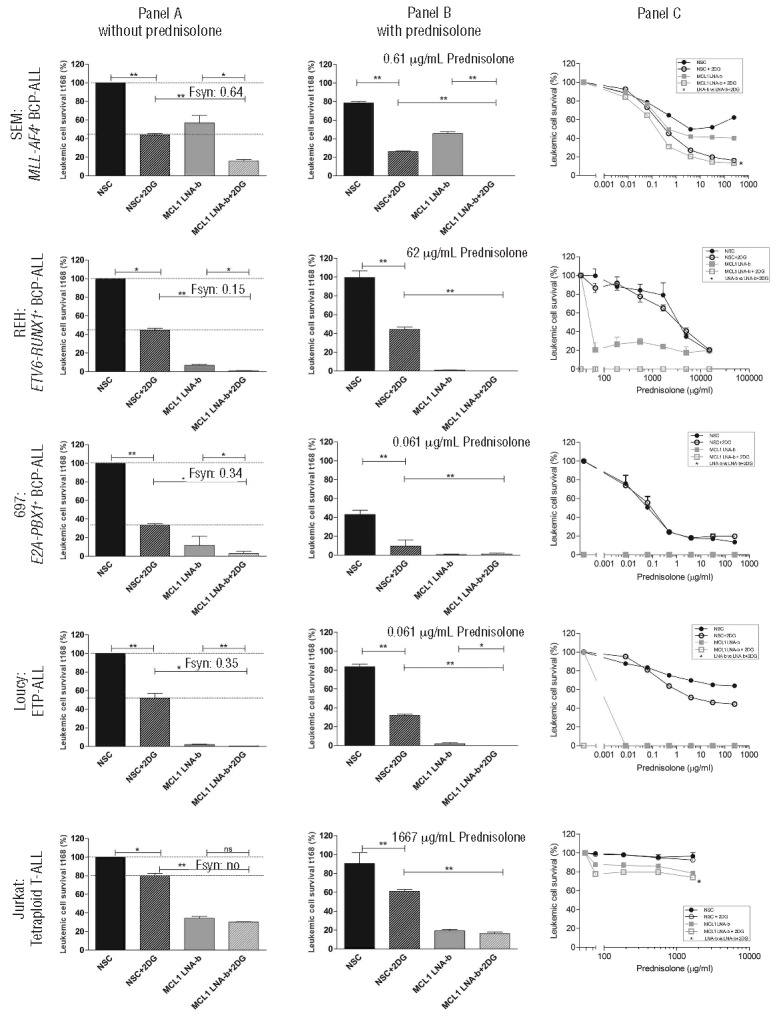
MCL1 silencing together with glycolysis inhibition synergistically inhibits leukemic cell survival and concomitantly sensitizes to prednisolone. (A) Leukemic cell survival of a MLL-AF4+ BCP-ALL, ETV6-RUNX1+ BCP-ALL, E2A-PBX1+ BCP-ALL, ETP-ALL and tetraploid T-ALL cell line after treatment with MCL1 LNA-b and/or 0.5mM 2-DG for 168 h, compared to non-silencing control LNA (NSC, set at 100%). Data are presented as means plus SEM of 3 (ETV6-RUNX1+ BCP-ALL and Tetraploid T-ALL) or 2 independent experiments (*P<0.05, **P<0.01, ***P<0.001). Fsyn represents the synergy factor, where <1 indicates synergy. (B) Leukemic cell survival after prednisolone exposure for three days, i.e. 96 till 168 h after start of MCL1 LNA-b and/or 0.5mM 2-DG in equivalent cell lines as in panel (A). Data were compared to a non-silencing control LNA (NSC) without prednisolone (see also panel (A), set to 100%), to visualize the total effect on cell survival after prednisolone. Data are presented as means plus SEM of 3 (ETV6-RUNX1+ BCP-ALL and Tetraploid T-ALL) or 2 independent experiments (*P<0.05, **P<0.01, ***P<0.001). (C) Sensitivity of leukemic cells to prednisolone was measured in a 3-day MTT assay from t96 to t168 after treatment with MCL1 LNA-b and/or a non-silencing control LNA (NSC) with or without 0.5mM 2-DG. Cell survival depicted on the y-axis was corrected for cell death induced by MCL1 knockdown and 0.5mM 2-DG itself in the absence of prednisolone, to visualize the absolute prednisolone effects. Data are presented as means plus SEM of 3 (ETV6-RUNX1+ BCP-ALL and Tetraploid T-ALL) or 2 independent experiments (*P<0.05, **P<0.01, ***P<0.001).
Discussion
Prednisolone is the spearhead drug used in multi-drug treatment of ALL. Not only is the in vivo and in vitro response to prednisolone a strong prognostic factor for long-term clinical outcome,2 relapsed ALL patients also acquire prednisolone resistance disproportionately to other anti-leukemic agents.18 To date, various mechanisms that sensitize leukemic cells in vitro to prednisolone have been described, including inhibition of the prednisolone interconverter 11β-HSD, knockdown of the glucocorticoid-dependent transcription regulator SMARCA4, inhibition of the voltage-dependent channel hERG1, which signals to ERK/PI3K/Akt survival pathways, knockdown of the calcium scavengers S100A8/S100A9, downregulation of anti-apoptotic MCL1, upregulation of pro-apoptotic BIM, and inhibition of glycolysis.6,8,9,19–22 Sensitizing cells to prednisolone may, therefore, require a multifactorial approach. In this study, we provide evidence that reduction of MCL1 levels and inhibition of glycolysis synergistically inhibits leukemic survival and concomitantly sensitizes to prednisolone in ALL cells.
Downregulation of anti-apoptotic BCL-2 family members is hampered in prednisolone-resistant cells.16 In the present study, we observed that prednisolone exposure decreased the expression of the anti-apoptotic MCL1 in leukemic cells of sensitive patients, whereas that of resistant cells remained unchanged. Prednisolone did not affect the expression of other BCL-2 family members (BCL-XL and BCL-2) nor p53. In line with this are microarray data on primary BCP-ALL, which identified higher anti-apoptotic MCL1 expression in prednisolone-resistant cells, but not higher Bcl-XL, BCL-2 or p53 expression.3 We observed that silencing of MCL1 expression by two means, shMCL1 and MCL1 LNA, induced apoptosis and sensitized to prednisolone in BCP-ALL and T-ALL cell lines. This is consistent with previous findings in other leukemic cell lines that demonstrated prednisolone sensitization after direct knockdown of MCL1 by shMCL1 or indirect downregulation of MCL1 via inhibition of mTOR.6,7 Another study reported that disruption of the complex between beclin-1 and MCL1 by obatoclax and exposure to dexamethasone-activated autophagy-dependent cell-death in otherwise dexamethasone-resistant cells.23 However, inhibition of autophagy only slightly induced resistance to dexamethasone, implying direct anti-apoptotic effects as well.23 Overall, these data indicate MCL1 as a potent therapeutic target to convert glucocorticoid resistance. However, high expression of MCL1 is not predictive for an adverse clinical outcome, suggesting that additional mechanisms co-occur that induce prednisolone resistance in pediatric ALL.6 Here, we observed that targeting MCL1 forces the glycolysis route, thereby rescuing cells from prednisolone-induced apoptosis. It has been shown that BCL2 family members maintain the mitochondrial membrane potential by regulating the permeability transition pore and the ATP/ADP pump, both involved in oxidative phosphorylation.13,14 Knockdown of MCL1 may, therefore, impair oxidative phosphorylation, forcing cells to produce ATP by glycolysis to ensure survival. This hypothesis is supported by our observation that treatment of leukemic cells with azide, a known inhibitor of oxidative phosphorylation, also increases glucose consumption (Online Supplementary Figure S15). Furthermore, an MCL1 amino-terminally truncated isoform was recently discovered that facilitates ATP production, respiration and maintenance of oligomeric ATP synthase in the mitochondria.24 Our three MCL1 LNAs all target the 3′ UTR of MCL1 and, therefore, also diminish this truncated MCL1 isoform. Furthermore, the glycolysis and apoptotic pathways may be connected via activity of the BCL2 family member BAD. The phosphorylation of aminoacid S112 in the BH3 domain of BAD acts like a switch between the metabolic and pro-apoptotic functions ascribed to BAD. BAD resides in a mitochondrial complex together with glucokinase and contributes to the activity of this glucose-metabolizing enzyme.25 Glucokinase mediates the first step in glycolysis by converting glucose into glucose-6-phosphate. Although it has been shown that MCL1 does not bind BAD directly,26 silencing of MCL1 may indirectly induce BAD activity and/or may trigger glycolysis in a similar way as the BAD/glucokinase complex. As a net result, the glycolytic rate (and hence glucose consumption) will increase, and this may provide a rescue mechanism against prednisolone-induced cell death. This speculative functional explanation still awaits further studies. Our study showed that inhibition of both glycolysis and MCL1 synergistically inhibits leukemic cell survival and concomitantly sensitizes leukemic cells to prednisolone. These results indicate the need to target both pathways and suggest that targeting MCL1 as a single target may not yield the desired clinical effect.
We previously demonstrated that ALL cells increase glucose consumption to prevent prednisolone-induced apoptosis.8 Glucocorticoids inhibit intracellular glucose uptake by regulating the expression of glucose transmembrane transporter (GLUT).27–29 Dexamethasone decreases GLUT-1 expression in ALL cells thereby decreasing glycolysis and inducing apoptosis.30 We observed higher expression of GLUT-1 in prednisolone-resistant ALL patients.3 TXNIP, a negative regulator of glucose uptake,30 is correlated to GLUT-1 expression31 and was also found up-regulated after prednisolone treatment in sensitive patients.32 Further studies are needed to demonstrate whether TXNIP and GLUT-1 facilitate the higher glucose metabolism observed after MCL1 knockdown.
Our results suggest that treatment with MCL1 LNA antisense and 2-DG represent a promising approach to decrease leukemic cell survival and to sensitize ALL patients to prednisolone. LNA antisense may offer a more direct and specific way of silencing MCL1 than the current BCL-2 family inhibitors R-(-)-gossypol (AT101) and obatoclax (GX–15–070)33,34 which recently entered clinical phase I/II trials. Both inhibitors can block several members of the BCL-2 family, increasing the chance of side effects in clinical practice. In contrast, LNA antisense specifically target the mRNA expression of one gene. We have shown in this study that MCL1 LNA effectively silences MCL1 mRNA and protein expression in ALL cells, comparable to knockdown with the more stable shMCL1. Notably, no delivery vehicles were necessary to ensure uptake of MCL1 LNA antisense molecules by the ALL cells. Moreover, LNAs are conformationally structured to prevent most of the current hurdles in siRNA treatment, such as delivery, stability of the RNA molecules in circulation, strand bias and off-target effects.35 LNA antisense molecules are currently investigated in phase I early clinical trials for three different target genes: EZN-3042, an inhibitor of Survivin is being investigated in children with relapsed ALL (Clinicaltrials.gov NCT01186328); EZN-4176, which targets the androgen receptor, is being studied in adults with castration-resistant prostate cancer (NCT01337518); and EZN-2968, an HIF-1A-inhibitor, is being examined in advanced solid tumors and lymphoma (NCT00466583). We here provide functional in vitro proof that MCL1 LNA antisense molecules are effective in inhibiting leukemic cell survival and reversing prednisolone resistance and may suggest the need for further investigation of these MCL1-LNAs in clinical trials. However, as our data show that silencing of MCL1 increases glycolysis of cells, neither MCL1 LNA nor BH3 mimetics should be used as single agents. We have shown that this shift in energy metabolism can be exploited to concomitantly sensitize leukemic cells to prednisolone. 2-DG may be a candidate agent since it has therapeutic potential and has been proven to cause chemosensitization in acute leukemia,8 breast cancer,36 and prostate cancer37 cells, and is now used in phase I clinical trials (ClinicalTrials.gov NCT00096707).
In conclusion, MCL1 is a potent target to therapeutically inhibit leukemic survival and to reverse drug resistance in pediatric ALL. However, MCL1-silenced cells up-regulate glycolysis, which may rescue cells from prednisolone-induced apoptosis. These data, therefore, provide evidence for concomitant causes of survival and resistance, and indicate that MCL1 and glycolysis should be targeted simultaneously to reduce leukemic cell survival and prednisolone resistance in ALL.
Acknowledgments
We thank Anja Mølhart Høg (Santaris, Hørsholm, Denmark) for developing the LNA-oligonucleotides and we acknowledge MWJ Luijendijk (Erasmus MC) and EF Petricoin (George Mason University-Manassas USA) for the RPPA analysis.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This work was supported by the Dutch Cancer Society (MdB, RP, EMCR 2005-3313 and AMC 2008-4265) and NIH grant (WEE,MLdB,RP 5R37CA036401). The funding source had no role in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Den Boer ML, Harms DO, Pieters R, Kazemier KM, Gobel U, Körholz D, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21(17): 3262–8 [DOI] [PubMed] [Google Scholar]
- 2.Kaspers GJ, Pieters R, Van Zantwijk CH, Van Wering ER, Van Der Does-Van Den Berg A, Veerman AJ. Prednisolone resistance in childhood acute lymphoblastic leukemia: vitro-vivo correlations and cross-resistance to other drugs. Blood. 1998;92(1): 259–66 [PubMed] [Google Scholar]
- 3.Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJP, Kazemier KM, et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. New Engl J Med. 2004;351(6): 533–42 [DOI] [PubMed] [Google Scholar]
- 4.Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1(5): e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8(16): 1587–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stam RW, Den Boer ML, Schneider P, de Boer J, Hagelstein J, Valsecchi MG, et al. Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2010;115(5): 1018–25 [DOI] [PubMed] [Google Scholar]
- 7.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10(4):331.42. [DOI] [PubMed] [Google Scholar]
- 8.Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJC, et al. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009;113(9):2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eberhart K, Renner K, Ritter I, Kastenberger M, Singer K, Hellerbrand C, et al. Low doses of 2-deoxy-glucose sensitize acute lymphoblastic leukemia cells to glucocorticoid-induced apoptosis. Leukemia. 2009;23 (11):2167.70. [DOI] [PubMed] [Google Scholar]
- 10.Mason EF, Zhao Y, Goraksha-Hicks P, Coloff JL, Gannon H, Jones SN, et al. Aerobic glycolysis suppresses p53 activity to provide selective protection from apoptosis upon loss of growth signals or inhibition of BCR-Abl. Cancer Res. 2010;70(20):8066.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27(12):4328.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradelli LA, Bénéteau M, Chauvin C, Jacquin MA, Marchetti S, Muñoz-Pinedo C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene. 2010;29(11):1641.52. [DOI] [PubMed] [Google Scholar]
- 13.Chen ZX, Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007;14(9):1617.27. [DOI] [PubMed] [Google Scholar]
- 14.Vander Heiden MG, Chandel NS, Schumacker PT, Thompson CB. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Molecular Cell. 1999;3(2):159.67. [DOI] [PubMed] [Google Scholar]
- 15.Berenbaum MC. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin Exp Immunol. 1977;28(1):1.18. [PMC free article] [PubMed] [Google Scholar]
- 16.Laane E, Panaretakis T, Pokrovskaja K, Buentke E, Corcoran M, Söderhäll S, et al. Dexamethasone-induced apoptosis in acute lymphoblastic leukemia involves differential regulation of Bcl-2 family members. Haematologica. 2007;92(11): 1460–9 [DOI] [PubMed] [Google Scholar]
- 17.Brady HJ, Salomons GS, Bobeldijk RC, Berns AJ. T cells from baxalpha transgenic mice show accelerated apoptosis in response to stimuli but do not show restored DNA damage-induced cell death in the absence of p53. EMBO J. 1996;15(6): 1221–30 [PMC free article] [PubMed] [Google Scholar]
- 18.Klumper E, Pieters R, Veerman AJ, Huismans DR, Loonen AH, Hählen K, et al. In vitro cellular drug resistance in children with relapsed/refractory acute lymphoblastic leukemia. Blood. 1995;86(10): 3861–8 [PubMed] [Google Scholar]
- 19.Sai S, Nakagawa Y, Yamaguchi R, Suzuki M, Sakaguchi K, Okada S, et al. Expression of 11beta-hydroxysteroid dehydrogenase 2 contributes to glucocorticoid resistance in lymphoblastic leukemia cells. Leuk Res. 2011;35(12): 1644–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillozzi S, Masselli M, De Lorenzo E, Accordi B, Cilia E, Crociani O, et al. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood. 2011;117(3): 902–14 [DOI] [PubMed] [Google Scholar]
- 21.Spijkers-Hagelstein JAP, Schneider P, Hulleman E, de Boer J, Williams O, Pieters R, et al. Elevated S100A8/S100A9 expression causes glucocorticoid resistance in MLL-rearranged infant acute lymphoblastic leukemia. Leukemia. 2012;26(6): 1255–65 [DOI] [PubMed] [Google Scholar]
- 22.Abrams MT, Robertson NM, Yoon K, Wickstrom E. Inhibition of glucocorticoid-induced apoptosis by targeting the major splice variants of BIM mRNA with small interfering RNA and short hairpin RNA. J Biol Chem. 2004;279(53):55809.17. [DOI] [PubMed] [Google Scholar]
- 23.Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14(6): 575–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Danial NN, Gramm CF, Scorrano L, Zhang C-Y, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424(6951): 952–6 [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17(3): 393–403 [DOI] [PubMed] [Google Scholar]
- 27.Hajduch E, Hainault I, Meunier C, Jardel C, Hainque B, Guerre-Millo M, et al. Regulation of glucose transporters in cultured rat adipocytes: synergistic effect of insulin and dexamethasone on GLUT4 gene expression through promoter activation. Endocrinology. 1995;136(11): 4782–9 [DOI] [PubMed] [Google Scholar]
- 28.Buentke E, Nordström A, Lin H, Björklund A-C, Laane E, Harada M, et al. Glucocorticoid-induced cell death is mediated through reduced glucose metabolism in lymphoid leukemia cells. Blood Cancer J. 2011;1(7): e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, et al. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia. 2006;20(10): 1731–7 [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Rong YP, Malone MH, Davis MC, Zhong F, Distelhorst CW. Thioredoxin-interacting protein (txnip) is a glucocorticoid-regulated primary response gene involved in mediating glucocorticoid-induced apoptosis. Oncogene. 2006;25(13): 1903–13 [DOI] [PubMed] [Google Scholar]
- 31.Yu F-X, Goh S-R, Dai R-P, Luo Y. Adenosine-containing molecules amplify glucose signaling and enhance txnip expression. Mol Endocrinol. 2009;23(6): 932–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tissing WJE, den Boer ML, Meijerink JPP, Menezes RX, Swagemakers S, van der Spek PJ, et al. Genomewide identification of prednisolone-responsive genes in acute lymphoblastic leukemia cells. Blood. 2007;109(9): 3929–35 [DOI] [PubMed] [Google Scholar]
- 33.Etxebarria A, Landeta O, Antonsson B, Basañez G. Regulation of antiapoptotic MCL-1 function by gossypol: mechanistic insights from in vitro reconstituted systems. Biochem Pharmacol. 2008;76(11): 1563–76 [DOI] [PubMed] [Google Scholar]
- 34.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15–070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. 2007;104(49): 19512–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koch T, Rosenbohm C, Hansen HF, Hansen B, Straarup EM, Kauppinen S. Locked Nucleic Acid: Properties and Therapeutic Aspects. In: Therapeutic Oligonucleotides. Cambridge: Royal Society of Chemistry; 2008 [Google Scholar]
- 36.Zhang F, Aft RL. Chemosensitizing and cytotoxic effects of 2-deoxy-D-glucose on breast cancer cells. J Cancer Res Ther. 2009;5(Suppl 1): S41–3 [DOI] [PubMed] [Google Scholar]
- 37.Sahra IB, Tanti J-F, Bost F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK dependent apoptosis in prostate cancer cells. Autophagy. 2010;6(5) [DOI] [PubMed] [Google Scholar]