

Abstract
The discovery that the Ten-Eleven Translocation (TET) hydroxylases cause DNA demethylation has fundamentally changed the notion of how DNA methylation is regulated. Clonal analysis of the hematopoetic stem cell compartment suggests that TET2 mutations can be early events in hematologic cancers and recent investigations have shown TET2 mutations in diffuse large B-cell lymphoma. However, the detection rates and the types of TET2 mutations vary, and the relation to global methylation patterns has not been investigated. Here, we show TET2 mutations in 12 of 100 diffuse large B-cell lymphomas with 7% carrying loss-of-function and 5% carrying missense mutations. Genome-wide methylation profiling using 450K Illumina arrays identified 315 differentially methylated genes between TET2 mutated and TET2 wild-type cases. TET2 mutations are primarily associated with hypermethylation within CpG islands (70%; P<0.0001), and at CpG-rich promoters (60%; P<0.0001) of genes involved in hematopoietic differentiation and cellular development. Hypermethylated loci in TET2 mutated samples overlap with the bivalent (H3K27me3/H3K4me3) silencing mark in human embryonic stem cells (P=1.5×10−30). Surprisingly, gene expression profiling showed that only 11% of the hypermethylated genes were down-regulated, among which there were several genes previously suggested to be tumor suppressors. A meta-analysis suggested that the 35 hypermethylated and down-regulated genes are associated with the activated B-cell-like type of diffuse large B-cell lymphoma in other studies. In conclusion, our data suggest that TET2 mutations may cause aberrant methylation mainly of genes involved in hematopoietic development, which are silenced but poised for activation in human embryonic stem cells.
Introduction
Promoter cytosine methylation (5-methylcytosine) is the most common mechanism of tumor suppressor inactivation in cancer.1 The 5-methylcytosine silencing mark is a reversible epigenetic modification that is inherited through consecutive cell divisions when DNA methyltransferases (DNMT) copy the DNA methylation pattern to newly synthesized DNA strands during replication. A vast amount of data has shown that patterns of DNA methylation are perturbed in hematologic cancers. The demonstration that the Ten-Eleven Translocation (TET) proteins can convert 5-methylcytosine to 5-hydroxymethylcytosine and lead to DNA demethylation2–4 underlies the proposal of a model for how DNA methylation fidelity is maintained, and how inactivation of these enzymes can lead to promoter hypermethylation.5
Mutations and translocations of a spectrum of epigenetic regulators have been implicated in hematologic cancers. Interestingly, many of these changes are present across disease entities and are involved in the pathogenesis of tumors of both myeloid and lymphoid origin, indicating they may be essential for the initiation of malignant hematopoiesis. Histone methyltransferases are disrupted by translocations of the mixed lineage leukemia (MLL) gene in acute lymphoid and acute myeloid leukemias,6 but are frequently mutated in diffuse large B-cell lymphoma (DLBCL),7 and while chronic myeloproliferative neoplasms and myelodysplastic syndromes carry inactivating mutations of EZH2,8 activating mutations are observed in B-cell lymphoma.9
Similarly, the TET proteins have been implicated in both myeloid and more recently lymphoid cancers. TET1 was first identified as an MLL translocation partner in acute myeloid leukemia,10 however the functionally related TET2 is frequently mutated in a variety of myeloid cancers.11 In addition, myeloid cancers have IDH1 and IDH2 mutations that are reported to be mutually exclusive to TET2 mutations and cause production of the onco-metabolite 2-hydroxyglutarate, a competitive inhibitor of α-ketoglutarate that is essential for TET2 catalytic activity.11–13 While an association between promoter hypermethylation and IDH mutations was confirmed,12,14 the data on the role of TET2 mutations in myeloid cancers are divergent.12,15–18
Early studies showed that patients with concomitant myeloproliferative and lymphoid malignancies have deletion of chromosome 4q24, where TET2 is located,19 and recent studies found that lymphoma-associated TET2 mutations are also observed in early hematopoietic progenitors with myeloid colony-forming capacities.20 In addition, acute myeloid leukemia/myelodysplastic syndromes arising secondary to lymphoma were demonstrated to carry the same TET2 mutation as the preceding lymphoma, indicating a common cell of origin. While TET2 and IDH2 mutations were found in a large fraction of angioimmunoblastic T-cell lymphoma,21,22 the results on TET2 mutations in DLBCL obtained from exome/genome sequencing studies are inconsistent. One previous study identified loss-of-function mutations only,20 another only missense mutations,23 while a third study did not detect any mutations at all.24
Using a mutation screening assay focused on TET2, we found TET2 mutations in 12% of DLBCL, with 5% carrying missense and 7% carrying nonsense, splice-site or frame-shift mutations. While several studies show that global DNA methylation changes delineate subtypes of lymphoid tumors,25–29 the mechanisms that direct DNA methylation to specific gene promoters in lymphoid tumors are largely unknown.29 To gain insight into the relation between TET2 mutations and DNA methylation in DLBCL we performed genome-wide comprehensive methylation profiling using 450K Illumina bead arrays. We found an association between distinct TET2 mutations and an aberrant hypermethylation signature that occurs predominantly at promotor CpG-islands. These results suggest that TET2 mutations may contribute to hypermethylation of a subset of genes in DLBCL.
Methods
The selection of DLBCL cases and controls, and tissue handling are described in the Online Supplementary Material. The study was approved by the ethical committee.
Detection of TET2 mutations
The entire coding sequences and all splice sites of the TET2 gene (exons 3–11) were scanned for mutations by polymerase chain reaction (PCR) combined with denaturing gradient gel electrophoresis (DGGE)30 using the primer sets and conditions listed in Online Supplementary Table S1A.
Global methylation profiling (Infinium Human Methylation 450 Assay)
The 12 DLBCL with TET2 mutations (TET2mut) were matched to 18 TET2 wild-type (TET2wt) cases with respect to age, sex, lactate dehydrogenase level, clinical stage, performance score and International Prognostic Index (IPI). Twenty-six of 30 cases had more than 80% tumor cells; however, it has been reported that as few as 50% tumor cells can be used for optimal DNA methylation processing.31 Global methylation profiling of these cases and peripheral blood B-lymphocytes was performed on 450K Infinium arrays (Illumina Inc). This platform comprehensively interrogates methylation status of 450,000 CpG in the human genome corresponding to all NCBI RefSeq genes, which include CpG in the promoters, enhancers, and gene bodies as well as CpG located outside the coding regions. In addition, probes have been mapped to CpG islands as well as shores and shelves of CpG islands (www.Illumina.com).
The Infinium DNA methylation assay was performed at the Genomic Core at the USC Epigenome Center, Los Angeles (USA). Briefly, 1 μg of DNA was bisulfite-converted using the Zymo EZ DNA methylation kit (Zymo Research). The effectiveness of bisulfite conversion was checked by the MethyLight assay, as described elsewhere.32 Each bisulfite-converted DNA sample was whole genome amplified followed by enzymatic end-point fragmentation, precipitation and re-suspension. The re-suspended samples were hybridized to Human Methylation 450 BeadChip as described previously.33 BeadArrays were scanned and the raw data were extracted as idat files. We used methylumi (http://www.bioconductor.org/packages/devel/bioc/html/methylumi.html) to extract β values from idat files. Measurements in which the fluorescent intensity was not statistically significantly above the background signal (detection P-value >0.05) were removed from the data set. DNA methylation β values were reported as a DNA methylation score ranging from 0 (non-methylated) to 1 (completely methylated).
For details on methodology for data filtering and normalization of DNA methylation data, bioinformatic analysis, gene expression profiling, Ingenuity pathway and DAVID (Database for annotation, visualization and integrated discovery) analysis, and comparison to ChIP-seq data corresponding to trimethylation of histone H3 lysine 4 (H3K4me3) and histone H3 lysine 27 (H3K27me3) in human embryonic stem cells (hESC) from ENCODE, see the Online Supplementary Information on materials and methods.
Results
To investigate the involvement of TET2 in the lymphoid compartment in more detail, we analyzed 100 primary DLBCL for TET2 mutations. Given the large size of TET2, we applied a mutation-screening assay based on PCR-DGGE with 38 PCR-amplified segments covering all coding exons and splice sites of TET2. This assay allows detection of infrequent mutant alleles (down to 5%) in a mixture with wild-type DNA from infiltrating normal cells, as can be seen in DLBCL. We excluded from further analysis all sequence variants that were polymorphisms in public databases, or that were recurrent in 500 alleles from the Danish population.
Mutation analysis of TET2 in diffuse large B-cell lymphoma cell lines
We initially screened a panel of five DLBCL cell lines (Farage, DB1, RL, HT and Toledo) for TET2 mutations, and found two previously unreported heterozygous missense mutations, which both cause a non-conservative substitution of a basic to a non-polar residue (Table 1 and Online Supplementary Figure S1). The significance of H679P found in the HT cell line is unknown; however, in Toledo, the R1972W is predicted to change a highly conserved residue in the catalytic double strand β-helix –2OG-Fe(II)-dependent dioxygenase (DSBH) domain of TET2.
Table 1.
TET2 mutations in primary cases of DLBCL and DLBCL cell lines.
Mutation analysis of TET2 in primary diffuse large B-cell lymphomas
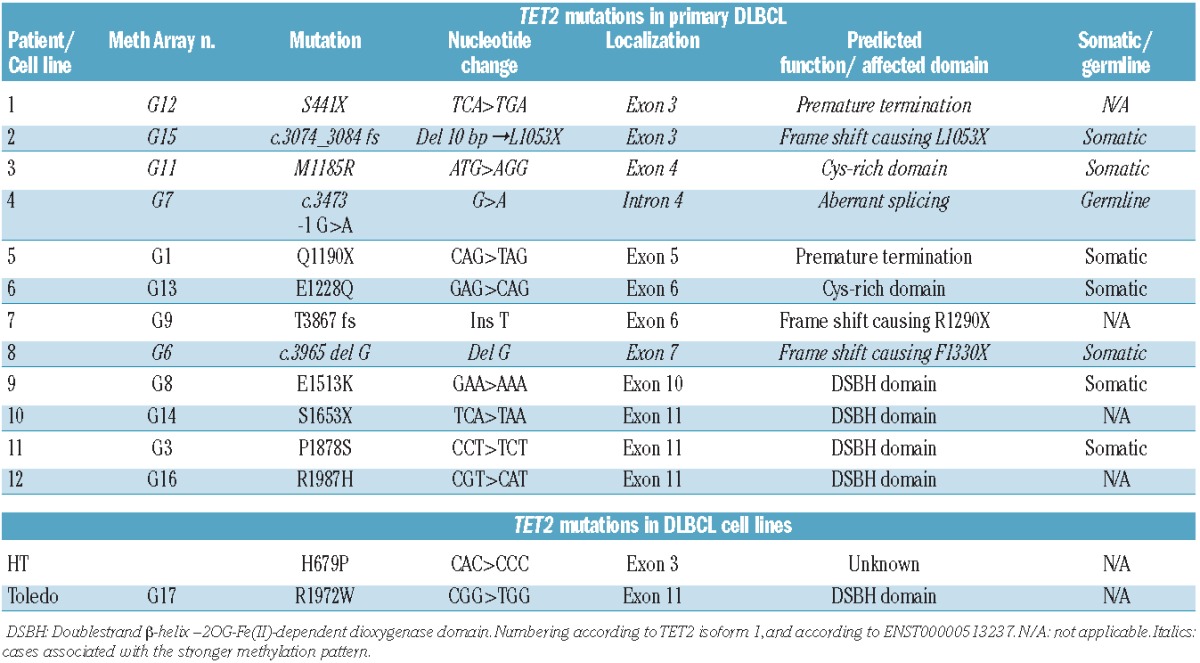
Analyses of 100 primary DLBCL identified 17 sequence variants among which 12 were likely to be of functional significance (Table 1). The latter included five missense, three nonsense, three frame-shift mutations, and one splice-site mutation. The mutations were distributed throughout the entire TET2 genomic sequence with clustering of missense mutations in the DSBH and cysteine-rich domains as previously observed in myeloid tumors (Figure 1). Seven somatic mutations represent bona fide loss-of-function mutations. These mutations are all predicted to cause pre-termination signals that deplete the dioxygenase domain. Three of the loss-of-function TET2 mutations were not present in normal control tissue, in three cases normal control tissue was not available, and in one patient the splice-site mutation c.3473 -1G>A (affecting the invariant acceptor splice site of intron 3) was also present in the bone marrow that did not show any morphological signs of lymphoma, but was hypercellular and hyperproliferative. Non-hematopoietic tissue was not available from this patient.
Figure 1.
TET2 point mutations in primary DLBCL. Distribution and types of TET2 point mutations in primary DLBCL. Twelve mutations were observed in 12 of a total of 100 cases.
Three of the missense mutations locate to conserved residues in the DSBH domain and are predicted to affect the oxygenase activity of TET2, and two are predicted to change the conserved cysteine-rich domain (Figure 1 and Online Supplementary Figure S2). Among the five missense mutations, four were not present in normal control tissue, and in one case normal control tissue was not available. Sanger sequencing of all 12 mutations and the available controls for eight of the 12 cases are shown in Online Supplementary Figure S2.
Mutations in different epigenetic regulators
In the 100 cases of DLBCL no mutations were observed at commonly mutated loci in myeloid cancers IDH1 R132, IDH2 R140 or R172, or in DNMT3A exons 20–22, for which reason no further analyses of these genes were performed.
TET2 mutant cases are associated with a distinct epigenetic profile
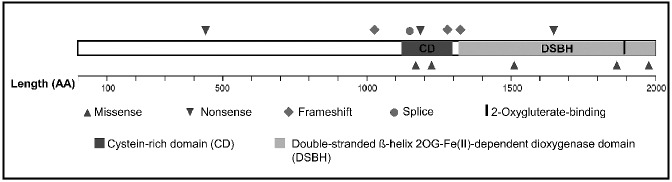
We next performed genome-wide DNA methylation profiling using the Illumina 450K Infinium arrays and compared 12 TET2mut DLBCL to 18 matched TET2wt DLBCL and normal CD19+ peripheral blood B-lymphocytes. A subset of differentially methylated regions identified on the arrays was re-analyzed using methylation-specific melting analysis, which confirmed the results (Online Supplementary Figure S3). To identify the CpG dyads showing most significant methylation in TET2mut relative to TET2wt samples, a mean β value of TET2mut and TET2wt samples was determined for each group. Illumina has reported that a 0.2 difference separation cut-off of β values can lead to the detection of differential methylation with 99% confidence in 450K or 27K Chips.33 Using both the stringent criteria of Δβ value ≥0.2 between TET2mut and TET2wt as well as a false discovery rate (FDR)-adjusted P<0.05, a set of 578 probes was selected (Online Supplementary Table S2). This threshold led to the identification of 315 genes hypermethylated in the group of TET2mut samples.
A supervised hierarchial clustering of these differentially methylated probes using Euclidean distance and complete linkage separated the samples into three groups. Cluster A with the highest methylation intensity (mean Δβ = 0.5) consisted of four DLBCL with loss of function mutations (G15, G12, G7, and G6) and one DLBCL case with a missense mutation in the cysteine-rich domain M1185R (G11). Cluster B, with intermediate methylation intensity (mean Δβ value = 0.3), contained samples with missense mutations in the cysteine-rich and DSBH domains (G13,G16) and nonsense mutations (G14, G9), together with two wild-type cases (G20,G21). Cluster C, with low β values, contained the vast majority of wild-type samples, and normal CD19+ peripheral blood lymphocytes (G18). Three mutant samples cluster along with wild-type samples (one loss-of-function mutation (G1) and two missense mutations in the DSBH domain (G3 and G8) (Figure 2).
Figure 2.
Hierarchical clustering of β methylation values from 578 CpG sites showing differential methylation in TET2mut and TET2wt samples. These 578 most variable probes were selected as having a FDR-adjusted P<0.05, and a mean β value difference of ≥0.2 between TET2mut and TET2wt samples (explained in the Online Supplementary Design and Methods section). Columns represent samples; rows represent CpG sites. Euclidean distance and complete linkage were used to study the cluster pattern of methylation probes. β values are represented using a pseudocolor scale from 0 to 1 as per color bar. TET2mut samples are separated into two distinct groups, one with higher mean β values and a second group with lower mean β values. Three TET2mut samples, G1, G3, and G8, cannot be distinguished from TET2wt. G18W= Normal CD19+ B cells.
Since the focus of the present investigation was to identify signature genes associated with TET2mut, we did not further analyze the differential methylation pattern between the three subclusters.
Biological relevance of differentially methylated genes in TET2mut cases
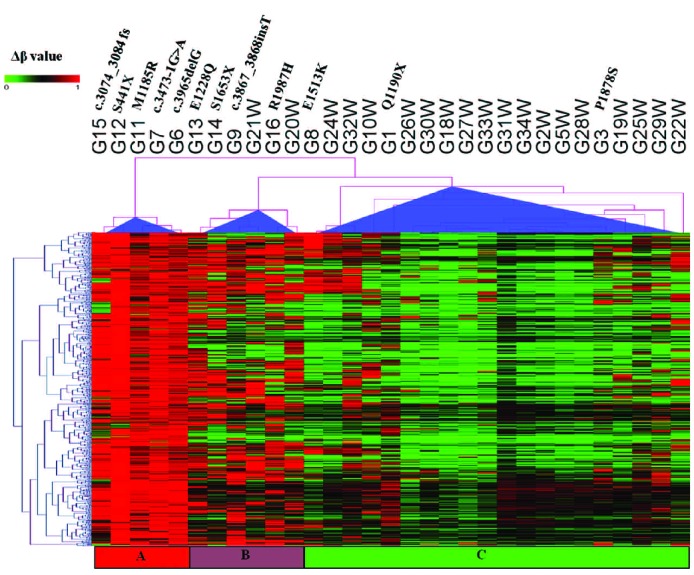
We used 315 genes corresponding to the 578 probes identified as differentially methylated (Figure 2 and Online Supplementary Table S2) to perform gene set enrichment analysis using Ingenuity pathway analysis. This analysis of differentially methylated genes revealed several important functional classes of genes associated with cancer in general and with hematologic malignancies in particular. Cancer, hematologic system development and dysfunction, and hematopoiesis are among the most significantly ranked functional categories based on −log (P value) (Figure 3).
Figure 3.
Functional gene ontology for differentially methylated TET2 signature genes. Seven top biological functions were identified and ranked by their P value (Y-axis).
To understand the interaction of enriched functional molecules in our dataset better, we also studied the networks affected among the differentially methylated genes. High scoring gene networks, identified as described in the Online Supplementary Materials and Methods are listed in Online Supplementary Table S3. The high scoring gene networks include cellular and hematologic system development and function, hematopoeisis, and cancer.
Significantly hypermethylated CpG sites in TET2mut cases locate to areas involved in transcriptional regulation
Among the top 578 most varying probes, which can differentiate between TET2mut and TET2wt samples, we observed a significant enrichment of probes located in promoters within −200 bp of transcription start sites (TSS), as well as in gene bodies (P<0.0001: Fisher’s exact test, Figure 4A), while only 16% of probes are intergenic (Figure 4A). In addition, a preference for CG-rich areas was seen with 70% of probes being located within CpG islands (P<0.0001), 18% of probes at the shores (within 2 Kb from a CpG island), 1% of probes at the shelves ( >2Kb from a CpG island) (Figure 4B), and only 11% outside these regions (Figure 4B).
Figure 4.
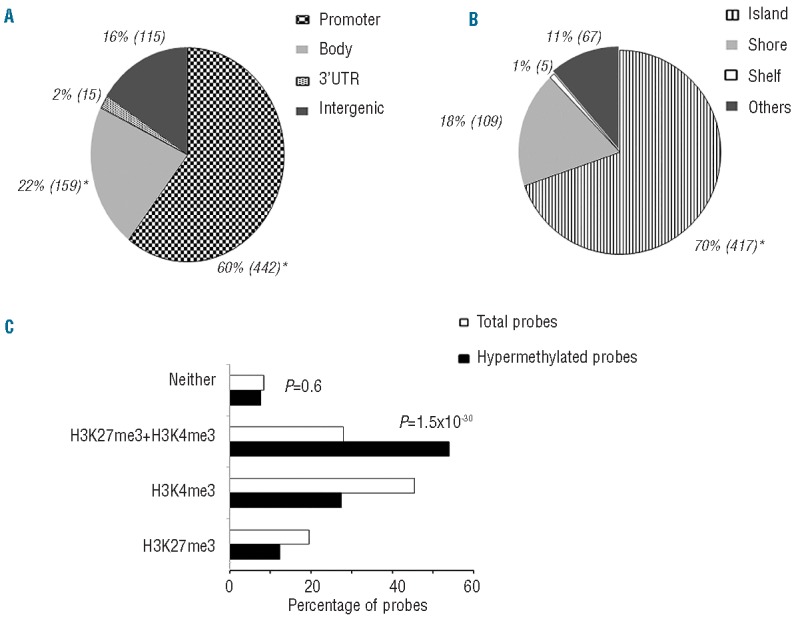
Relative distribution of the 578 “TET2 signature probes”. Distribution of TET2 signature probes (n=578) in the context of (A) functional genomic distribution classified in different groups: Promoter, Gene body, 3’UTR, and Intergenic and of (B) CpG neighborhood classified into Island, Shore, Shelf, and Other/Open sea. There was a significant enrichment of probes corresponding to promoters and gene bodies, and of probes mapping to CpG islands, determined by Fisher’s exact test (P<0.0001, marked as *). Numbers in parentheses indicate the number of probes in the corresponding genomic region. (C) TET2 differentially methylated loci correspond to the bivalent mark, H3K4me3/H3K27me3 enriched regions. Chip-seq data for hES cells were downloaded from Encode and peak (P<0.01) coordinates were identified if they fell within hypermethylated regions or not. Of the 578 probes a majority of the differentially methylated loci match to the bivalent mark, H3K4me3 and H3K27me3 (53.4%). This enrichment is highly statistically significant from the total number of probes on the 450K platform mapping to these histone marks (H3K27me3 + H3K4me3, P = 1.5×10−30; Fisher’s exact test).
A significant proportion of TET2 signature genes in diffuse large B-cell lymphoma are targeted by polycomb in human embryonic stemcells
Polycomb group (PcG) proteins are chromatin regulators with a crucial role in establishing and maintaining epigenetic memory during development and cellular differentiation. It has been widely accepted that Polycomb target genes in hES cells that are associated with histone H3 trimethylated on lysine 27 (H3K27me3) alone, or H3K27me3 in combination with histone H3 trimethylated on lysine 4 (H3K4me3), are susceptible to undergo hypermethylation in DLBCL as well as in other cancers.29,34,35 We therefore overlaid the H3K27me3, H3K4me3, and H3K27/H3K4me3 enriched genomic coordinates from the published Encode dataset with the 578 hypermethylated probes in the TET2mut DLBCL. We found that 5-methylctyosine-hypermethylated loci in the TET2mut samples overlap with 53.4% of the probes enriched for the bivalent mark H3K27me3/H3K4me3 in hES. In fact, 27.1% of these loci overlap with the H3K4me3 histone mark alone, and 12.1% with the H3K27me3 mark only, while only 7.4% of hypermethylated loci in our dataset do not overlap with either histone mark. As shown in Figure 4C, there is highly significant enrichment of the bivalent histone mark (H3K27me3/H3K4me3) (P=1.5×10−30) relative to all the probes, suggesting that TET2 target loci are significantly enriched for the bivalent mark (signifying genes poised for activation) in hES cells.
Integrative analysis of gene expression and DNA methylation
Since promoter DNA methylation is known to lead to transcriptional silencing, we next examined the extent to which DNA hypermethylation affects gene expression in TET2mut samples. Using the criteria described in the Online Supplementary Materials and Methods, we compared DNA methylation and gene expression data from a subset of four DLBCL with TET2 mutations and high DNA methylation intensity (cluster A, Figure 2) with those of five TET2wt samples with low DNA methylation intensity from cluster C.
First, a simple hierarchial clustering based on the gene expression alone (fold change ≥1.2, P<0.01) of these cases showed a clear separation of TET2mut versus TET2wt samples into two distinct groups (Online Supplementary Figure S4) suggesting that TET2mut genes may represent a biological entity and that genes identified as differentially expressed can serve as signature genes to discriminate between TET2mut and TET2wt cases.
Secondly, to integrate differentially methylated genes with gene expression data, methyation probe level data were summarized to gene level based on Entrez gene identifiers, and merged with gene expression data based on common Entrez gene identifiers. Of a total of 315 differentially methylated genes, 305 genes were present on the Affymetrix expression array (Figure 2 and Online Supplementary Table S2). A majority (n=222) of the 305 genes (73%) did not show any differential gene expression, however a set of 35 (11%) genes, which were hypermethylated (|Δβ| ≥0.2) in TET2mut cases, were down-regulated (fold change ≥1.2; P<0.05) (Figure 5A; Online Supplementary Table S4). The majority of these 35 genes (27/35) were hypermethylated at the promotor CpG sites. Of the remaining 35 genes, seven have CpG probes mapped to the gene body while one of the genes showed methylation in the 3’UTR. Interestingly, 26 of these 35 down-regulated genes showed a highly differential methylated pattern (|Δβ| ≥0.4). However, only five of the differentially methylated genes showed differential expression of ≥2 (P<0.01). By comparison to the previously published gene sets GSE11318 and GSE23501, these 35 genes, which are hypermethylated and down-regulated, are associated with the activated B-cell-like type of DLBCL (Online Supplementary Figure S5).
Figure 5.
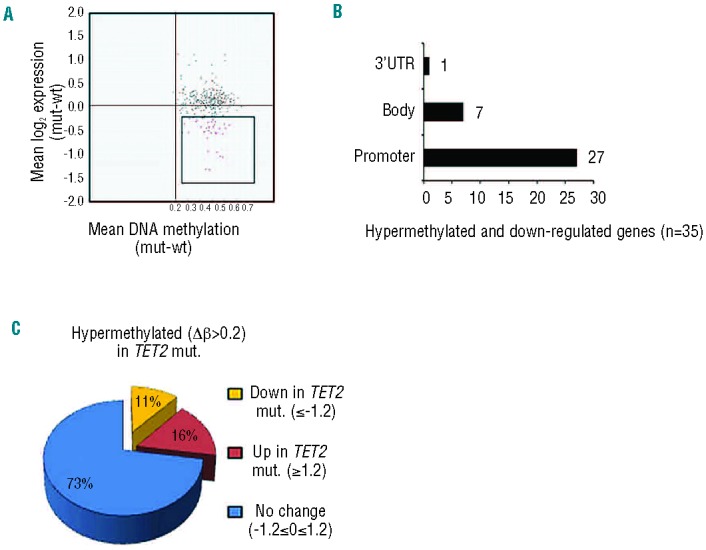
Integrated analysis of gene expression and DNA methylation between TET2mut and TET2wt cases. (A) Mean DNA methylation β-value differences between four TET2mut and five TET2wt samples were plotted on the X-axis, and mean log2- transformed gene expression value differences were plotted on the Y-axis to generate a starburst plot for each of the 305 TET2 signature genes. Of these genes, 35 genes were found to be hypermethylated (red dots) and downregulated. (B) The majority of these genes (n=27) were hypermethylated in the promoter regions. (C) A large proportion of 305 hypermethylated genes showed no change (73%) in gene expression.
Matching these 35 genes to the tumor suppressor data base,36 three genes qualified as tumor suppressors (ETS1, CDK2AP1 and HTAIP2). However, among the promotor hypermethylated and down-regulated genes, EPHB1, ZIK1, GRASP, FEZ1 and DYRK237–41 have been shown to be methylated in other cancers. EPHB1 belongs to the family of Epherin receptors, which is involved in normal hematopoietic development and tumorigenesis, and has been suggested to be a tumor suppressor gene silenced by promoter methylation in acute lymphocytic leukemia.37 Silencing of DYRK2 increases cell proliferation in human cancer cells due to the escape of c-Jun and c-Myc from ubiquitination-mediated degradation.41 Other genes include FAN1, which codes FANCD2/FANCI-associated nuclease 1. This nuclease is recruited to sites of DNA damage by monoubiquitinated FANCD2, is required for the maintenance of chromosomal stability, and plays a key role in DNA repair.42 The HTATIP2 gene, also called TIP30, encodes an oxidoreductase required for tumor suppression by promoting apoptosis and inhibiting angiogenesis. It has been shown that TIP30 stabilizes p53 mRNA and binds directly to the DNA-binding and C-terminal domain of p53.43 Accordingly, the silencing of these genes is likely to influence lymphomagenesis directly.
Clinical features of TET2 mutant cases
Patients with TET2 mutations did not differ significantly from patients without TET2 mutations with respect to sex, age, lactate dehydrogenase concentration, performance score, clinical stage or IPI (Online Supplementary Table S5). No differences in overall survival were observed between TET2mut and TET2wt cases (Online Supplementary Figure S6), and the addition of rituximab did not influence this finding.
Discussion
The mechanisms leading to alterered DNA methylation in hematopoietic cancers are not well understood, and while many studies have been conducted in myeloid cancers, only few have focused on lymphoid tumors. The analyses of an association of TET2 mutations with DNA hypermethylation in myeloid cancers have led to varying conclusions, which most likely is due to the use of different methylation detection platforms and different tumor cell content in the samples.12,15–18 The previous studies of the relation between global DNA methylation patterns and TET2 mutations were performed with methylation detection platforms that mainly cover promoter CpG-islands. Here, we have investigated in more detail the genomic regions that attained increased levels of DNA methylation in the TET2mut cases in diagnostic samples of DLBCL using the 450K Illumina platform, which, in addition to promoter CpG islands, gives information on the methylation patterns at CpG sites in shores, shelves, gene bodies, enhancers and intergenic regions.
Our first result was that 12% of DLBCL carry TET2 mutations. Using a TET2-focused approach, we found a higher frequency of TET2 mutations in DLBCL than previously described by deep sequencing.20,23,24 One study reported that 2% of miscellaneous types of B-cell tumors and 5.7% of DLBCL carry TET2 loss-of-function mutations.20 In a second study, RNA-sequencing of 127 non-Hodgkin’s lymphoma, including 97 DLBCL, revealed only TET2 missense mutations,23 and in a third study whole-exome screening of 55 primary DLBCL did not detect any TET2 mutations.24
In the current study we identified both loss-of-function and missense mutations in one cohort of patients, at frequencies comparable to those observed in two of the above mentioned, individual studies.20,23 This discrepancy in the frequencies of mutation types may be explained by the different methodology used including sequence coverage and the percent tumor involvement of the samples. In addition, several chromosomal aberrations, including pluriploidy, can be present in DLCBL, which can lead to “dilution” of mutated alleles, an effect that can complicate mutation detection.
Secondly, we found that these mutations correlate with hypermethylation of a subset of genes preferentially involved in networks that regulate hematopoietic differentiation, development, and cell cycle regulation. The “TET2 signature probes” showed a clear preference for TSS and gene bodies, and the significantly hypermethylated probes were located at or near the TSS. In addition, we observed a significant enrichment of differentially methylated probes in CG-rich areas, particularly at CpG islands. We and others have previously shown that the formation of hydroxymethylcytosine is decreased upon abrogation of TET expression,2,5,16 and have suggested that TET proteins oppose aberrant DNA methylation to prevent gene silencing by promoter methylation. Taken together this suggests that TET2 plays a major role in keeping transcriptional initiation sites free of methylation, as previously proposed.5 We did not observe a complete separation of TET2mut and TET2wt samples based upon methylation profile, which is consistant with the heterogenous nature of DLBCL. Although we did not detect IDH1, IDH2 or DNMT3a mutations in these samples, it is highly likely that disruption of additional epigenetic regulators may cause aberrant DNA methylation in DLBCL, which is a subject for further study.
Surprisingly, only 35 of 305 hypermethylated TET2 signature genes were downregulated. A fraction of these are reported as tumor suppressors involved in cell cycling and DNA repair, and are silenced by methylation in other cancers. However, the majority of methylated genes show no difference in expression between methylated and unmethylated tumors. This could be due to the expression of these genes in the bystander cells in the lymphoma even though we ensured that the tumor cell content was >80%. Alternatively, they may already be silenced by different mechanisms. It has previously been shown that many genes that are already silenced by H3K27me3 in normal germinal center B cells are frequently aberrantly DNA hypermethylated in DLBCL.34 Interestingly, the genes with a “TET2 hypermethylation signature” coincide with genes having the bivalent mark in hES-cells. These genes have previously been linked to cancer-specific DNA methylation in several studies. Notably, we found that a total of 53.4% of the “TET2 methylation signature genes” carried the bivalent H3K4me3/H3K27me3 silencing mark in hES cells. Thus, it can be speculated that TET2 inactivation leads to promoter methylation of already silenced genes, thereby causing permanent silencing, which would ensure that the genes are not activated at a later stage in differentiation. Interestingly, a significant fraction of the “TET2 signature genes” are involved in cellular development and hematopoiesis, suggesting they may normally play an active role at some stage of development. Thus, we hypothesize that TET2 mutations do not define the lymphoma sub- or phenotype, but may prohibit the expression of genes crucial for normal lymphopoiesis.
The current classification of lymphoid malignancies is based on the idea that malignant tumors have morphological and gene expression patterns that resemble those of lymphocytes at distinct stages of normal B- and T-cell differentiation, which has fostered the so-called “cell of origin” concept.44–47 At present, the exact developmental step at which TET2 mutations occur in lymphoid disorders is not known. However, the “cell of origin concept” is being challenged by the observations that lymphoma-associated TET2 mutations are found in common hematopoietic progenitors of the same patients,20 and that Tet2-deficient mice, in addition to the expected myeloproliferation,48 show expansion of lymphoid cells of both B- and T-cell lineage.20,49
TET2 mutations do not associate with a particularly aggressive phenotype in DLBCL, and no influence is seen on overall survival. By contrast, our observations support the idea that TET2 alterations could potentially prime or maintain the malignant clone. The facts that TET2mut cases coincide with a very distinct gene expression profile and that mice transplanted with TET2-deficient hematopoietic stem cells accumulate abnormal B cells20 argue for a biological role of TET2 mutations in DLBCL. However, we obviously cannot exclude that some mutations may be simple passenger events.
In conclusion, the current study and previously published data suggest that TET2 mutations may lead to the accumulation of methylation errors in genomic regions involved in transcriptional regulation of a subset of genes targeted by TET2. This seems to preferentially occur in a set of genes involved in hematopoietic differentiation and cell cycling, which are, for the vast majority of genes, not differentially expressed between methylated and unmethylated cases in tumor samples. Accordingly, these genes may play a role at a different stage of tumor development. Since there are now indications that epigenetic therapy may be particularly efficient in TET2mut cases,50 these observations may potentially prompt the design of clinical trials using hypomethylation therapy in TET2mut subsets of lymphoid malignancies, in which these drugs have not yet been implemented.
Taken together, these observations suggest that epigenetic defects in hematopoietic progenitor cells may, at least in some instances, precede several types of hematologic malignancy. This is not only interesting from a biological viewpoint, but may potentially change our notion of how hematopoietic cancers develop, and could possibly have importance for future clinical and therapeutic decision-making.
Acknowledgments
Funding: This study was supported by grants from Copenhagen University (FA), Rigshospitalets Research Foundation (FA, KG), The Danish Cancer Society (KG, KH, ER, UR), The Novo Nordisk Foundation (KG, KH), The Lundbeck Foundation (KG), The Danish Strategic Research Foundation (KG, KH), The Danielsen Foundation, The Danish National Research Foundation grant number DNRF82 (KH) and the European Research Council (KH). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors thank E Andres Housmann of Brown State University for important discussion on RPMM clustering.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10): 726–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethyl-cytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929): 930–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310): 1129–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333 (6047): 1300–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473(7347): 343–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11): 823–33 [DOI] [PubMed] [Google Scholar]
- 7.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9): 830–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42(8): 665–7 [DOI] [PubMed] [Google Scholar]
- 9.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42(2): 181–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia. 2003;17(3): 637–41 [DOI] [PubMed] [Google Scholar]
- 11.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9): 599–612 [DOI] [PubMed] [Google Scholar]
- 12.Figueroa ME, bdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6): 553–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1): 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deneberg S, Guardiola P, Lennartsson A, Qu Y, Gaidzik V, Blanchet O, et al. Prognostic DNA methylation patterns in cytogenetically normal acute myeloid leukemia are predefined by stem cell chromatin marks. Blood. 2011;17(118): 5573–82 [DOI] [PubMed] [Google Scholar]
- 15.Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics. 2012;7(2): 201–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;9(468): 839–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nischal S, Bhattacharyya S, Christopeit M, Yu Y, Zhou L, Bhagat TD, et al. Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res. 2013;73(3): 1076–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez C, Martinez-Calle N, Martin-Subero JI, Segura V, Delabesse E, Fernandez-Mercado M, et al. TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS One. 2012;7(2): e31605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viguie F, Aboura A, Bouscary D, Ramond S, Delmer A, Tachdjian G, et al. Common 4q24 deletion in four cases of hematopoietic malignancy: early stem cell involvement? Leukemia. 2005;19(8): 1411–5 [DOI] [PubMed] [Google Scholar]
- 20.Quivoron C, Couronne L, Della VV, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20(1): 25–38 [DOI] [PubMed] [Google Scholar]
- 21.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de LL, Jais JP, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8): 1901–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7): 1466–9 [DOI] [PubMed] [Google Scholar]
- 23.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360): 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109(10): 3879–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JH, Li Y, Guo J, Pei L, Rauch TA, Kramer RS, et al. Genome-wide DNA methylation maps in follicular lymphoma cells determined by methylation-enriched bisulfite sequencing. PLoS One. 2010;5(9). pii: e13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood. 2010;116(20): e81–e89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leshchenko VV, Kuo PY, Shaknovich R, Yang DT, Gellen T, Petrich A, et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116 (7): 1025–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115(2): 296–305 [DOI] [PubMed] [Google Scholar]
- 29.Martin-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, et al. New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood. 2009;113(11): 2488–97 [DOI] [PubMed] [Google Scholar]
- 30.Guldberg P, Grønbæk K, Worm J, Thor SP, Zeuthen J. Mutational analysis of oncogenes and tumor suppressor genes in human cancer using denaturing gradient gel electrophoresis. Methods Mol Med. 2002;68:125–39 [DOI] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22): 2059–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods Mol Biol. 2009;507:325–37 [DOI] [PubMed] [Google Scholar]
- 33.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4): 288–95 [DOI] [PubMed] [Google Scholar]
- 34.Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116(24): 5247–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter J, Ammerpohl O, Martin-Subero JI, Montesinos-Rongen M, Bibikova M, Wickham-Garcia E, et al. Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer. 2009;9:455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao M, Sun J, Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res. 2013;41(Database issue): D970–D976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, et al. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood. 2010;115(12): 2412–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borinstein SC, Conerly M, Dzieciatkowski S, Biswas S, Washington MK, Trobridge P, et al. Aberrant DNA methylation occurs in colon neoplasms arising in the azoxymethane colon cancer model. Mol Carcinog. 2010;49(1): 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beggs AD, Jones A, El-Bahwary M, Abulafi M, Hodgson SV, Tomlinson IP. Whole-genome methylation analysis of benign and malignant colorectal tumours. J Pathol. 2013;229(5): 697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Zhu Z, Sun X, Dong XY, Wei J, Gu F, et al. Down-regulation of tumor suppressor gene FEZ1/LZTS1 in breast carcinoma involves promoter methylation and associates with metastasis. Breast Cancer Res Treat. 2009;116(3): 471–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taira N, Mimoto R, Kurata M, Yamaguchi T, Kitagawa M, Miki Y, et al. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J Clin Invest. 2012;122(3): 859–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai F, Hu K, Wu Y, Tang J, Sang Y, Cao J, et al. Human KIAA1018/FAN1 nuclease is a new mitotic substrate of APC/C(Cdh1). Chin J Cancer. 2012;31(9): 440–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SH, Ju SK, Lee TY, Huh SH, Han KH. TIP30 directly binds p53 tumor suppressor protein in vitro. Mol Cells. 2012;34(5): 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC press; 2008 [Google Scholar]
- 45.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769): 503–11 [DOI] [PubMed] [Google Scholar]
- 46.Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002; 346(25): 1937–47 [DOI] [PubMed] [Google Scholar]
- 47.Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105 (36): 13520–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1): 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011;108(35): 14566–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itzykson R, Kosmider O, Cluzeau T, Mansat-De MV, Dreyfus F, Beyne-Rauzy O, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7): 1147–52 [DOI] [PubMed] [Google Scholar]