

Abstract
The formation of 5-hydroxymethylcytosine (5hmC), a key intermediate of DNA demethylation, is driven by the ten eleven translocation (TET) family of proteins that oxidize 5-methylcytosine (5mC) to 5hmC. To determine whether methylation/demethylation status is altered during the progression of Alzheimer’s disease (AD), levels of TET1, 5mC and subsequent intermediates, including 5hmC, 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) were quantified in nuclear DNA from the hippocampus/parahippocampal gyrus (HPG) and the cerebellum of 5 age-matched normal controls, 5 subjects with preclinical AD (PCAD) and 7 late-stage AD (LAD) subjects by immunochemistry. The results showed significantly (p < 0.05) increased levels of TET1, 5mC, and 5hmC in the HPG of PCAD and LAD subjects. In contrast, levels of 5fC and 5caC were significantly (p < 0.05) decreased in the HPG of PCAD and LAD subjects. Overall, the data suggest altered methylation/demethylation patterns in vulnerable brain regions prior to the onset of clinical symptoms in AD suggesting a role in the pathogenesis of the disease.
Keywords: Alzheimer’s disease, preclinical Alzheimer’s disease, 5-hydroxymethylcytosine, 5-methylcytosine
1. Introduction
Methylation of cytosine residues at the 5′ position (5mC) by members of the DNA methyltransferase (DNMT) family of proteins is the predominant and most widely characterized epigenetic modification. Cytosine methylation plays an important role in multiple cellular processes, including retrotransposon silencing, genomic imprinting, X-chromosome inactivation, regulation of gene expression, and maintenance of epigenetic memory (Bird, 2002). Regarded as a relatively stable epigenetic modification, DNA methylation status is considered static following wide-spread and dynamic alterations in methylation patterns during embryonic development (Hajkova et al., 2010; Iqbal et al., 2011; Pastor et al., 2011; Wu et al., 2011), differentiation (Ficz et al., 2011; Pfaffeneder et al., 2011; Tahiliani et al., 2009), and reprogramming of somatic cells (Bhutani et al., 2010; Koh et al., 2011).
Active demethylation of the Arabidopsis genome is achieved through a methylcytosine specific DNA glycosylase, although no mammalian 5mC glycosylase has been identified to date and a mechanistic role for active demethylation of the human genome remains unclear. However, dynamic regulation of methylation patterns throughout the lifespan in the human cortex (Bakulski et al., 2012; Siegmund et al., 2007) suggests demethylation occurs in the absence of a human methylcytosine specific DNA glycosylase. Initially proposed by Iyer et al. (Iyer et al., 2009), the enzymatic capacity of both human and mouse ten eleven translocation 1 (TET1) proteins to catalyze the oxidation of 5mC to the sixth base, 5-hydroxymethylcytosine (5hmC), in an iron (II) and α-ketoglutarate dependent manner, has been demonstrated (Ito et al., 2010; Tahiliani et al., 2009). Not only does the oxidation of 5mC by the TET family of DNA hydroxylases represent a potential active demethylation pathway, it also represents a method of epigenetic modification (Wu and Zhang, 2011). Further studies indicate that subsequent oxidation of the sixth base yields multiple derivatives including, 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (He et al., 2011; Ito et al., 2011). 5mC derivatives including 5hmC, and 5fC represent possible substrates for targeted removal through initiation of the base excision repair (BER) pathway (He et al., 2011; Maiti and Drohat, 2011) (Figure 1 adapted from (Maiti and Drohat, 2011). Conventional understanding of genomic methylation has largely been associated with gene repression via recognition of the methyl-CpG binding domain of methyl-CpG binding proteins and recruitment of co-repressors, whereas demethylation is associated with active gene transcription (Suzuki and Bird, 2008).
Fig. 1.
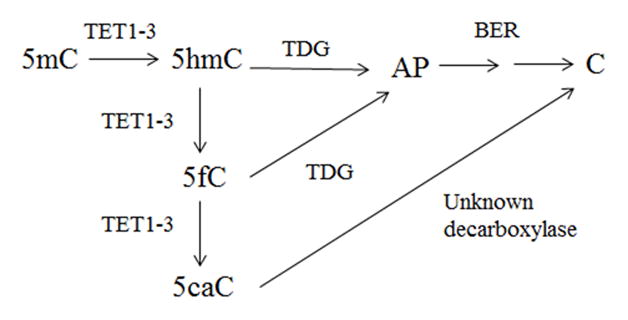
Potential mechanism for active DNA methylation involving TET-mediated oxidation of 5mC, followed by TDG initiated BER pathway via recognition of 5hmC and 5fC (AP is an abasic site) (adapted from (Maiti and Drohat, 2011)).
Globisch et al. reported that 5hmC, the oxidation product of 5mC, is wide spread throughout mouse tissue but it is significantly higher in the central nervous system (Globisch et al., 2010) with the highest levels observed in the hippocampus and cortex (Munzel et al., 2010), areas associated with memory and higher cognitive function. Tissue distribution of 5hmC reported by Li et al. and Nestor et al. indicate levels of 5hmC are more pronounced in the brain in human tissue as well (Li and Liu, 2011; Nestor et al., 2012).
Alzheimer’s disease (AD), an insidious and progressive neurodegenerative disease, is characterized by cognitive decline, neuronal loss, and two hallmark pathological features, neurofibrillary tangles and senile plaques. Previous studies show an age-dependent hypomethylation of the promoter region (-224-101) of the amyloid precursor protein (APP) (Tohgi et al., 1999) and hypomethylation of APP in the brain of a single late-stage AD (LAD) subject but not in an age-matched normal control (NC) or diseased control subject (West et al., 1995). Furthermore, in vitro hypomethylation of presenilin 1 (PS1) a component of the γ-secretase complex led to increased cleavage of APP and increased production of Aβ in a neuroblastoma cell line (Fuso et al., 2005). Collectively, these data suggest alterations in the methylation status of the genome may contribute to the pathogenesis of AD.
The current study was designed to evaluate both the potential and relative oxidation of 5mC in relation to AD disease progression. Levels of 5hmC were initially quantified in nuclear DNA (nDNA), RNA, and mitochondrial DNA (mtDNA) isolated from the superior and middle temporal gyrus (SMTG) and cerebellum (CER) of LAD subjects and NC subjects. Overall levels of 5hmC were the highest in nDNA, therefore subsequent studies of 5mC and derivatives focused on nDNA. Levels of TET1 and thymine DNA glycosylase (TDG) were quantified in the nuclear fraction by Western blot analysis. To allow comparison of vulnerable and non-vulnerable brain regions during the progression of AD, nuclear protein fractions and nDNA were prepared from the hippocampus/parahippocampal gyrus (HPG) and the cerebellum (CER) of subjects with preclinical AD (PCAD), LAD, and NC subjects. The sample size, 7 LAD subjects, 5 PCAD subjects, and 5 NC subjects are reflective of limited tissue availability of the vulnerable HPG brain region.
PCAD subjects were chosen for analysis because they represent a discrete stage of disease progression allowing an investigation of potential for altered methylation status early in the disease progression. While identification of PCAD subjects is difficult, those included in the current study were characterized by normal ante-mortem neuropsychological test scores with significant AD associated pathology at autopsy (Galvin et al., 2005; Schmitt, 2000). Previous studies of AD progression indicate that multiple α, β-unsaturated aldehydes derived during lipid peroxidation (Aluise et al., 2010; Bradley et al., 2010a; Bradley et al., 2010b) and nucleic acid oxidation, and mRNA transcript levels of oxoguanine glycosylase 1 (Lovell et al., 2011) are significantly elevated in subjects with PCAD suggesting that perturbations of cellular processes are present before significant cognitive deficits are detected. It has been hypothesized that if subjects characterized as PCAD lived longer they would have eventually developed clinical AD (Smith et al., 2008). In addition to PCAD subjects, LAD subjects were included in the current study to determine if dynamic alterations of methylation status were progressive in later stage of AD.
2. Materials and methods
2.1 Brain sampling
Tissue specimens were obtained through the neuropathology core of the University of Kentucky Alzheimer’s Disease Center (UK-ADC) under IRB approved protocols in accordance with ethical standards in the 1964 Declaration of Helsinki. Tissue specimens of short postmortem interval (PMI) autopsies from 7 LAD subjects (3 men [M]: 4 women [W]), 5 PCAD subjects (0 M: 5 W), and 5 age-matched NC subjects (2 M: 3 W) were flash frozen in liquid nitrogen and maintained at −80°C until processed for analysis.
All subjects were followed longitudinally in the UK-ADC research clinic and underwent neuropsychological testing and physical and neurological examinations annually. All NC subjects had neuropsychological test scores in the normal range and showed no evidence of memory decline. Although there are not well defined criteria for the identification of PCAD, the UK-ADC tentatively describes PCAD subjects as those with sufficient AD pathologic alterations at autopsy to meet intermediate or high NIA-RI criteria, moderate or frequent neuritic plaque scores according to the Consortium to Establish a Registry for AD (CERAD) with Braak scores of III–IV, and ante-mortem psychometric test scores in the normal range when corrected for age and education (Schmitt, 2000). LAD subjects met both the clinical criteria for probable AD and standard histopathological criteria for the diagnosis of AD (Hyman et al., 2012).
Neuropathological examination of multiple sections of neocortex, hippocampus, and CER using the modified Bielschowsky stain, hematoxylin and eosin stain, and Aβ and α-synuclein immunostains were performed for all subjects. Braak staging scores were determined using the Gallyas stain on sections of entorhinal cortex, hippocampus, and amygdala and the Bielschowsky stain on neocortex. Histopathological examination of NC brains showed only age-associated changes and Braak staging scores of I–II. PCAD subjects had moderate or frequent neuritic plaque scores with Braak staging scores in the range from III–IV (Schmitt, 2000). AD subjects met accepted criteria for the histopathological diagnosis of AD with Braak staging scores of VI. Demographic data for all subjects in the study are shown in Table 1.
Table 1.
Subject Demographics
| Group | Mean ± SEM Age (y) |
Sex | Mean ± SEM PMI (h) |
Median Braak Score |
|---|---|---|---|---|
| NC | 84.6 ± 4.1 | N=5; 2 M; 3W | 2.7 ± 0.4 | I |
| PCAD | 89.4 ± 3.1 | N=5; 0M; 5W | 2.9 ± 0.2 | IV* |
| LAD | 77.4 ± 2.5 | N=7; 3 M; 4W | 3.25 ± 0.2 | VI* |
P < 0.05 compared to NC subjects
NC = age-matched normal control; PCAD = preclinical Alzheimer’s disease; PMI = postmortem interval; SEM = standard error of the mean
2.2 Tissue processing: nuclear protein fraction
Nuclear protein extracts were prepared as using EpiQuik™ Nuclear Extraction Kit I (Epigentek, Farmingdale, NY, USA) as described by the manufacturer. Briefly, tissue samples (~0.04 g) were homogenized in pre-extraction buffer containing DTT, incubated on ice for 15 min and centrifuged at 12,000 rpm for 10 min at 4°C. Nuclear pellets were resuspended in extraction buffer containing protease inhibitor cocktail and DTT, incubated on ice for 15 min, and centrifuged at 14,000 rpm for 10 min at 4°C. Nuclear fractions were collected and stored at −80°C until used for analysis.
2.3 Tissue processing: nuclear deoxyribonucleic acid (nDNA)
Tissue samples were prepared as described by Timmons et al. (Timmons et al., 2011). Briefly, brain specimens were homogenized on ice using a Teflon-coated Dounce homogenizer (Wheaton Industries, Millville, NJ, USA) in isolation buffer (IB) containing 10 mM Tris, 0.32 M sucrose, and 0.25 mM EDTA (pH 7.4) for a final tissue to buffer ratio of ~0.1 g/ml. Homogenates were transferred to Nalgene brand polypropylene copolymer centrifuge tubes with polypropylene screw caps (Thermo Fisher Scientific, Rochester, NY, USA) and centrifuged at 2,000 xg for 3 min at 4°C in a fixed angle rotor. The resultant supernatant was saved for mitochondrial isolation and the nuclear pellet was resuspended in half the original IB volume, rehomogenized, and centrifuged at 2,000 xg for 3 min at 4°C. The two supernatant fractions and two nuclear pellets were pooled. The method of Mecocci et al. (Mecocci et al., 1993) with modification (Wang et al., 2006) was used to isolate nDNA. The combined nuclear pellet was resuspended in 5 mL digestion buffer (DB) containing 0.5%SDS, 0.05 M Tris, 0.1 M EDTA, CaCl2 (pH 7.5) with 0.5 mg/mL proteinase K and incubated in water bath at 57°C overnight. NaCl (160 μL/10 mL, 5 M) was added and the solution was extracted 3X with a Tris-buffered phenol containing 5.5 mM 8-hydroxyquinoline and 3X with isoamyl alcohol/chloroform (4% [v/v]). 8-hyroxyquinoline was used to minimize artifactual oxidation as previously described by Wang et al. (Wang et al., 2006). The aqueous layer was collected, NaCl (800 μL/10 mL, 5 M) and an equal volume of cold absolute ethanol were added, and the DNA precipitated overnight at −20°C. Following centrifugation, the nDNA pellet was washed 3X with 60% ethanol and air-dried. The isolated nDNA was dissolved in autoclaved distilled/deionized water and the concentration and purity determined using a NanoDrop 1000 Spectrophotometer (NanoDrop, Wilmington, DE, USA).
2.4 Tissue processing: mitochondrial deoxyribonucleic acid (mtDNA)
The method of Sims and Anderson (Sims and Anderson, 2008) with modification by Timmons et al. (Timmons et al., 2011) was used to prepare an enriched mitochondrial fraction. Pooled supernatant collected during isolation of the nuclear fraction was centrifuged at 20,000 xg for 10 min at 4°C. The resulting crude mitochondrial pellet was resuspended in IB (10 mL) and centrifuged at 2,000 xg for 3 min at 4°C. The resulting supernatant was centrifuged at 20,000xg for 20 min at 4°C. Crude free and synaptosomal mitochondria were resuspended in 3 mL of Precoll (GE Healthcare Life Sciences, Piscataway, NJ, USA): IB (19% [v/v]) and transferred to a polyallomer centrifuge tubes (Beckman Coulter Inc., Brea, CA, USA) and centrifuged at 19,600 xg for 30 min at 4°C in Beckman Coulter L-80 Ultracentrifuge equipped with a SW 55 Ti swinging bucket rotor. The supernatant was discarded and the pellet containing purified free mitochondria and synaptosomes was transferred to a 2.0 mL polypropylene microcentrifuge tube (Thermo Fisher Scientific) and washed in 1 mL phosphate buffered saline (PBS) and centrifuged at 19,600 xg for 10 min at 4°C in a Thermo IEC Micromax 851 Centrifuge (Thermo Fisher Scientific) with a fixed angle rotor. The supernatant was discarded and the pellet resuspended in PBS containing digitonin (1% [w/v]) and placed on ice for 10 min. Percoll: IB (3 mL, 28% [v/v]) was added to the digitonin treated sample and centrifuged at 19,600 xg for 10 min at 4°C. The supernatant was discarded and the pellet containing free and synaptic mitochondria was washed 3X in PBS. Purified mitochondria were resuspended in digestion buffer (1 mL) containing 2% SDS, 0.1M EDTA, CaCl2 (pH 7.5) and 0.5 mg/mL proteinase K for 4 hours in 37°C water bath. NaCl (160 μL/10mL, 5 M) was added and the solution extracted 3X with tris-buffered phenol containing 8-hydroxyquinoline (5.5 mM) and 3X with isoamyl alcohol/chloroform (4% [v/v]). mtDNA was precipitated with sodium acetate (800 μL/10 mL) and two volumes of cold absolute ethanol overnight at −20°C. Following centrifugation at 14,000 xg for 20 min at 4°C the mtDNA pellet was washed 3X with 60% ethanol, and air dried. The isolated mtDNA was dissolved in autoclaved distilled/deionized water and the concentration and purity determined using a NanoDrop 1000 Spectrophotometer (Nanodrop).
2.5 Western blotting of nuclear, cytosolic, and mitochondrial fractions
To verify the purity of mitochondria isolated using our extraction protocol, Western blot analysis was carried out on representative total homogenate, and nuclear, cytosolic, and mitochondrial fractions on NuPAGE Novex 4–12% Bis Tris Midi Gel (Invitrogen, Green Island, NY, USA) and transferred to nitrocellulose membranes. Membranes were blocked in 5% dry milk in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TTBS) with 3.3% goat serum (Invitrogen) for 1 h at room temperature and blots were probed with either rabbit anti-lamin B at a 1:3750 dilution (nuclear marker; Santa Cruz Biotechnology; Santa Cruz, CA, USA), mouse anti-MAP2 at a 1:3750 dilution (cytosolic marker; Abcam, Cambridge, MA, USA), rabbit anti-porin at a 1:3750 dilution (mitochondrial marker; EMD Chemical, Gibbstown, NJ, USA) overnight at 4°C. Blots were washed 3× 10 min each with TTBS at room temperature and incubated with horseradish peroxidase conjugated goat anti-rabbit secondary antibody for porin and lamin B and goat ant-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) at a 1:7500 dilution for 2 h at room temperature. Blots were washed 3× 10 min each in TTBS. Bands were visualized using enhanced chemiluminescence per manufacturer’s instructions (Thermo Scientific) and intensities were quantified using Scion Image Analysis Software (Scion, Fredrick, MD, USA).
2.6 Polymerase chain reaction of mitochondrial DNA and nuclear DNA
To verify mtDNA purity, PCR was performed with 500 ng mtDNA and nDNA using primers for APOE, a nuclear coded protein and MT-ND2, mitochondrial encoded NADH dehydrogenase 2, using GoTaw PCR Core System (Promega, Madison WI, USA). Primer sequences were 5′GGCGCTCGGATGGCGCTGAG-3′ (sense primer) and 5′-GCACGGCTGTCCAAGGAGCTGCAGGC-3′ (reverse primer) for APOE (Tsukamoto et al., 1993); and 5′-CGTCACACAAGCAACAGCCTCAATT -3′ (sense primer) and 5′-TGTGCAGTGGGATCCCTTGAGTTA-3′ for MT-ND2 (Dzitoyeva et al., 2012) (Integrated Device Technology, Santa Clara, CA, USA). PCR products were separated on 2% low-melt agarose gel containing ethidium bromide as previously described (Addya et al., 1997).
2.7 Tissue processing: ribonucleic acid (RNA)
Tissue samples were prepared as described by Chomczynski et al. (Chromczynski and Sacchi, 1987). Briefly, tissue specimens (~100 mg) were homogenized in 1 ml of TRIReagentR in a polypropylene microcentrifuge tube and incubated at room temperature for 5 min. Chloroform (200 μL) was added, mixed by hand, incubated for 5 min at room temperature, and centrifuged at 9,000 xg for 15 min at 4°C. Isopropyl alcohol (500 μL) was added to the resulting clear aqueous phase containing RNA, incubated at room temperature for 10 min and centrifuged at 9000xg for 10 min at 4°C. RNA pellets were washed in 75% ethanol, air dried, and stored at −80°C until analysis. Samples were resuspended in autoclaved distilled/deionized water and the concentration and purity determined using a NanoDrop 1000 Spectrophotometer (Nanodrop). Representative RNA isolates were submitted to the UK MircoArray Core Facility for electrophoretic analysis.
2.8 Quantification of cytosine modifications by dot blot analysis
To verify the presence of 5hmC in representative samples of nDNA and RNA prepared from the SMTG, samples (50 ng and 100 ng) were pretreated either with either DNase-1 free RNase-1 or RNase-I free DNase-I (0.5 μg/μl PBS, Roche, Mannheim, Germany) for 2 h at 37°C. Levels of 5hmC were determined by dot blot immunochemistry using a Scheicher & Schuell Dot-Blot apparatus as described by Saiki et al. (Saiki, 1986). Briefly, pretreated nDNA and RNA were mixed with aqueous NaOH (0.05M) and boiled for 5 min in a total volume of 60 μL. The samples were loaded in triplicate onto Hybond-N+ positively charged nylon membrane (GE Healthcare Life Sciences) under vacuum. Following air-drying, blots were blocked for 1 h at room temperature in 5% dry milk in TTBS and 3.3% goat serum (Invitrogen). Blots were probed with anti-5hmC (1:10,000) (Active Motif, Carlsbad, CA, USA) overnight at 4°C. Blots were washed 3× 10 min each with TTBS at room temperature and incubated with horseradish peroxidase conjugated goat anti-rabbit secondary antibody for 5hmC (1:20,000) (Jackson ImmunoResearch) for 2 h at room temperature. Blots were washed 3× 10 min each in TTBS. Dots were visualized using enhanced chemiluminescence per manufacturer’s instructions(Thermo Scientific) and intensities quantified using Scion Image Analysis Software (Scion).
To determine relative levels of 5hmC in nDNA, RNA, or mtDNA, equal amounts of each nucleic acid isolated from the SMTG and CER were determined as previously described. Dots were visualized using enhanced chemiluminescence per manufacturer’s instructions (ThermoScientific) and intensities quantified using Scion Image Analysis Software (Scion, Fredrick, MD, USA). The immunostaining intensities of replicates were averaged and average dot intensities for nDNA, RNA, and mtDNA samples were normalized to mean nDNA levels for each blot.
Total levels of 5mC, 5hmC, 5fC, and 5caC in nDNA from the HPG and CER were determined by dot blot immunochemistry as previously described. Blots were probed with either: anti-5mC (1:1000), anti-5hmC (1:10,000), anti-5fC (1:1,000), or anti-5caC (1:1000) antibodies (Active Motif) overnight at 4°C. Blots were washed 3× 10 min each with TTBS at room temperature and incubated with horseradish peroxidase conjugated goat anti-mouse secondary antibody for 5mC (1:2000), or goat anti-rabbit secondary antibody for 5hmC (1:20,000), 5fC (1:2000), and 5caC from (1:2000) (Jackson ImmunoResearch) for 2 h at room temperature. Blots were washed 3 × 10 min each in TTBS. Dots were visualized using enhanced chemiluminescence per manufacturer’s instructions (Thermo Scientific) and intensities were quantified using Scion Image Analysis Software (Scion). The immunostaining intensities of replicates were averaged and average dot intensity for each PCAD, LAD and NC subject was normalized to mean NC levels for each blot.
To verify the specificity of 5mC, 5hmC, 5fC, and 5caC antibodies used in this study, PCR products of the APC gene promoter were amplified with either cytosine, 5mC, or 5hmC, and a 38 bp DNA oligonucleotide containing twelve 5-carboxylcytosine residues (Active Motif) were loaded in duplicate and subjected to dot blot analyses as described above. To determine a linear response of 5mC, 5hmC, 5fC, and 5caC immunoreactivity, increasing amounts of PCR products of the APC gene promoter amplified with either 5mC, or 5hmC, and a 38 bp DNA oligonucleotide containing 12 5caC residues (Active Motif) were loaded in duplicate and subjected to dot blot analyses as described above. To verify a linear response of 5mC, 5hmC, 5fC, and 5caC immunoreactivity increasing amounts of a representative nDNA sample were loaded in duplicate and subjected to dot blot analyses as described above. To verify a linear response of 5hmC immunoreactivity increasing amounts of a representative RNA sample were loaded in duplicate and subjected to dot blot analyses as described above.
2.9 Western Blotting of TET1 and TDG
Protein samples (15 μg) were separated on NuPAGE Novex 4–12% Bis Tris Midi Gel (Invitrogen) and transferred to nitrocellulose membranes. The membranes were blocked for 1 h at room temperature in 5% dry milk in TTBS and 3.3% goat serum or rabbit serum (Invitrogen) and incubated overnight with anti-TET1 (1:5000) (Active Motif) or anti-TDG (1:500) (Epigentek) antibodies. Blots were washed 3× 10 min each with TTBS at room temperature and incubated with horseradish peroxidase conjugated rabbit anti-goat secondary antibody for TET1 (1:1000) or goat anti-mouse secondary antibody for TDG (1:1000) (Jackson ImmunoResearch) for 2 h at room temperature. Blots were washed 3× 10 min each in TTBS and visualized using enhanced chemiluminescence per manufacturer’s instructions (Thermo Scientific). After development, membranes were stripped and re-probed with primary rabbit anti-lamin B (1:1000) (Santa Cruz Biotechnology) and intensities were quantified using Scion Image Analysis Software. Optical densities of TET1 and TDG normalized to optical densities levels of lamin B.
2.10 Statistics
All data were tested for normality using the Wilkes-Shapiro test. Braak staging scores demonstrated non-normal distributions and were analyzed using the Mann-Whitney U-test and are reported as median values with range. All other data demonstrated a normal distribution. Normalized dot blot values of 5-methylcytosine, 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine, normalized values of TET1 and TDG, age, and PMI were compared using ANOVA and are reported as mean ± standard error of mean (SEM). All statistical comparisons were carried out using Sigma Plot. Statistical significance was set at p ≤ 0.05.
3. Results
Subject demographic data are shown in Table 1. There were no significant differences in age or PMI between NC and PCAD or LAD subjects. Median Braak staging scores were significantly higher in PCAD subjects (IV) and LAD subjects (VI) compared to age-matched NC subjects (I).
3.1 Antibody specificity
Antibodies used in the current study, particularly the 5-hmC antibody developed by Active Motiff, have been used successfully for a variety of applications including cellular mapping of mC derivatives (Globisch et al., 2010; Haffner et al., 2011), immunoprecipitation for MeDIP and MeDIP-seq (Nestor et al., 2012; Stroud et al., 2011), and identification (Ito et al., 2010) and relative quantification (Nestor et al., 2012) of oxidized 5mC. To assess antibody specificity, APC gene promoter PCR products amplified with either cytosine, 5mC or 5hmC or a 38 bp oligonucleotide with twelve 5caC residues were subjected to dot blot analysis and probed with the 5mC, 5hmC, 5fC, and 5caC antibodies (Fig. 2). The 5mC antibody recognized only the APC gene promoter PCR product amplified with 5mC. Similarly the 5hmC antibody recognized only the APC gene promoter PCR product amplified with 5hmC, and the 5caC antibody recognized only the 38 bp oligonucleotide containing 12 5caC. In addition, the 5fC antibody showed no cross reactivity with DNA standards containing cytosine, 5mC, 5-hmC, or 5caC residues. Significant positive linear responses between immunostaining intensity and increasing amounts of DNA standards were observed for the 5mC antibody (Fig. 3A) (r = 0.96, p < 0.05), the 5hmC antibody (Fig. 3B), and the 5caC antibody (Fig. 3C) (r = 0.95, p < 0.05). Significant positive linear responses between immunostaining intensity and increasing amounts of representative DNA isolated from the SMTG were observed for the 5mC antibody (Fig. 4A) (r= 0.99, p < 0.05), the 5hmC antibody (Fig. 4B) (r = 0.99, p < 0.005), the 5fC antibody (Fig. 4C) (r = 0.99, p < 0.05), and the 5caC antibody (Fig. 4D) (r = 0.99, p < 0.01).
Fig. 2.

Specificity of monoclonal antibodies against 5-methylcytosine, 5-hydroxymethylcytosine, 5-forymylcytosine, and 5-carboxylcytosine. The APC gene promoter PCR product amplified with 5-methylcytosine loaded in duplicate in columns 1–2, the APC gene promoter PCR product amplified with 5-hydroxymethylcytosine loaded in duplicate in columns 3–4, a 38 bp oligonucleotide containing 12 5-carboxylcytosine residues loaded in duplicates in columns 5–6, and the APC gene promoter PCR product amplified with cytosine loaded in duplicates in columns 7–8 and incubated with either monoclonal mouse anti-5-methylcytosine (A), monoclonal rabbit anti-5-hydroxymethylcytosine (B), monoclonal rabbit anti-5-formylcytosine (C), and monoclonal rabbit anti-5-carboxylcytosine (D).
Fig. 3.
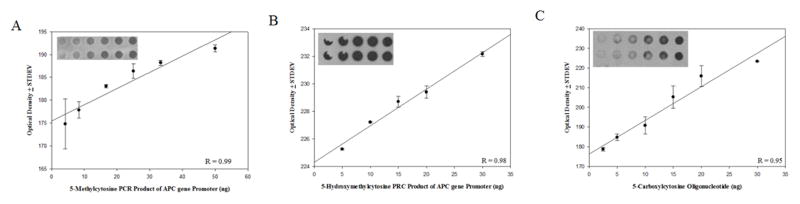
Evaluation of concentration dependent monoclonal antibody responses. 5-methylcytosine (r = 0.96, p < 0.05) (A), 5-hydroxymethylcytosine (r = 0.98, p < 0.05) (B), and 5-carboxylcytosine (r = 0.95, p < 0.05) (C) showed a significant positive linear response between immunostaining and increasing amounts of DNA standards.
Fig. 4.

Evaluation of concentration dependent monoclonal antibody responses. A representative SMGT nDNA 5-methylcytosine (r = 0.99, p < 0.05) (A), 5-hydroxymethylcytosine (r = 0.99, p < 0.005) (B), 5-forymylcytosine (r = 0.99, p < 0.05) (C), and 5-carboxylcytosine (r = 0.99, p < 0.01) (D) showed a significant positive linear response between immunostaining and increasing amounts of DNA.
3.2 Levels of 5hmC are significantly higher in nuclear DNA compared to RNA and mitochondrial DNA in both LAD and NC subjects
Western blot analysis of representative samples of total homogenate, and nuclear, cytosolic, and mitochondrial-fractions for lamin B, a nuclear protein, MAP2, a cytosolic protein, and porin, a mitochondrial transport protein, showed no cross contamination of proteins between the nuclear, cytosolic, and mitochondrial fractions (Fig. 5A, 5B, and 5C). PCR amplification of APOE from a representative subject in each brain region studied was only observed for nDNA fractions (Fig. 5D), whereas MT-ND2 amplification was detected only in mtDNA fractions (Fig. 5E). Sharp peaks representing the eukaryotic 18S and 28S ribosomal subunits, the absence of smaller well-defined peaks between the two ribosome subunits, and RNI of 9.8 suggest high quality isolates (Fig. 5F).
Fig. 5.
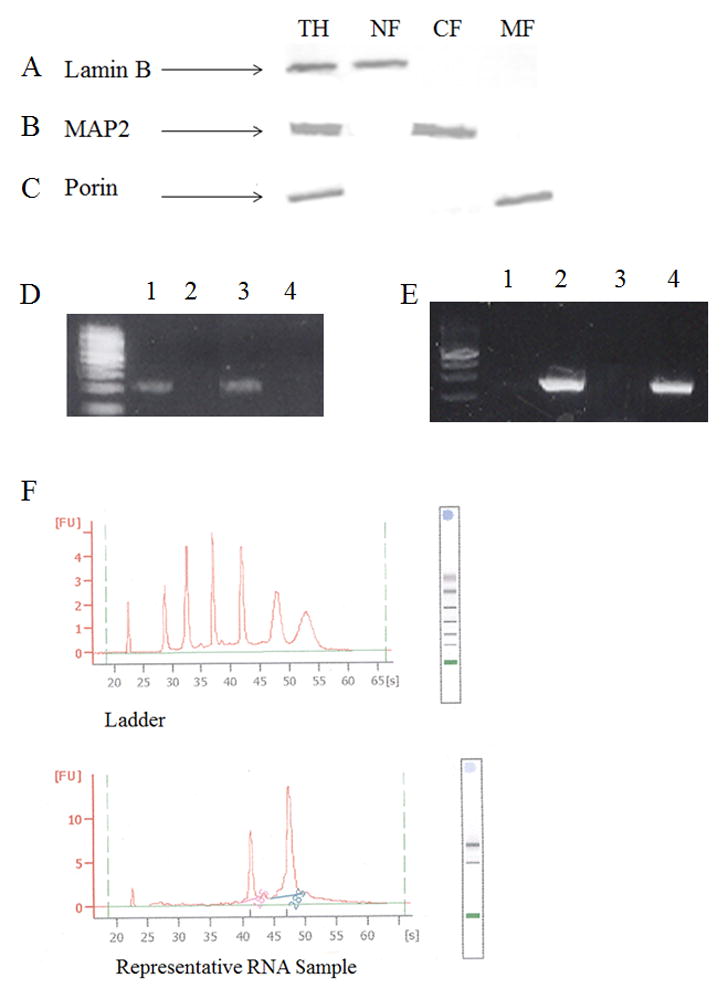
Purity of nDNA and mtDNA. Representative Western blot analysis of total homogenate (TH; Row 1), nuclear- (NF; Row 2), cytosolic- (CF; Row 3), and mitochondrial-fractions (NF; Row 4) probed for lamin B a nuclear envelope protein (A), MAP2 a microtubular associated protein (B), and Porin a voltage dependent anion channel found on the outer membrane of the mitochondria (C). Representative PCR amplification product of APOE (D) and MT-ND2 (E) of mtDNA (Lane 1) and nDNA (Lane 2) from SMTG and mtDNA (Lane 5) and nDNA (Lane 6) from CER. Representative RNA electropherogram (F).
Immunostaining intensity of 5hmC in a representative RNA sample pretreated with DNase-1 free RNase-1 was minimal compared to RNA pretreated with RNase-1 free DNase (Fig. 6). Similarly, immunostaining of 5hmC in a representative nDNA sample pretreated with RNase-1 free DNase-1 was minimal compared to nDNA pretreated with DNase-1 free RNase-1 (Fig. 6). Mean levels of 5hmC were significantly lower (p < 0.001) in RNA (16.8 ± 6.8 % of nDNA) and mtDNA (16.1 ± 1.9 % of nDNA) compared to nDNA (100.0 ± 2.0 % of nDNA) in the SMTG of NC subjects (Fig. 7A). Additionally, mean levels of 5hmC were significantly lower (p < 0.001) in RNA (26.7 ± 7.2 % of nDNA) and mtDNA (18.7 ± 6.2 % of nDNA) compared to nDNA (100.0 ± 0.8 % of nDNA) in the SMTG of LAD subjects (Fig. 7A).
Fig. 6.
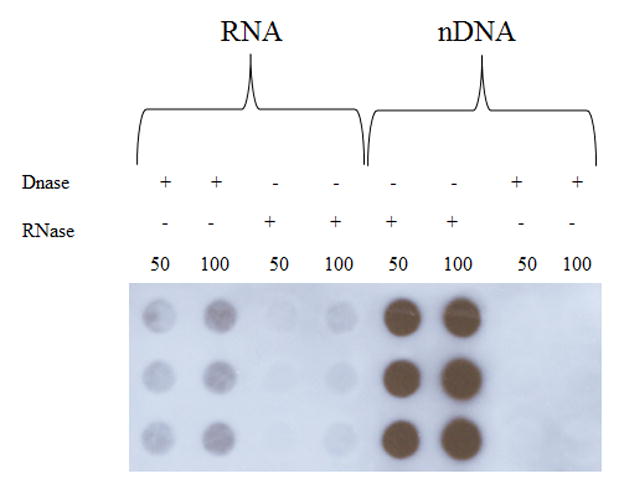
Immunostaining intensity of RNA and DNA pretreated with either DNase-1 free RNase-1 or RNase-I free DNase-I.
Fig. 7.
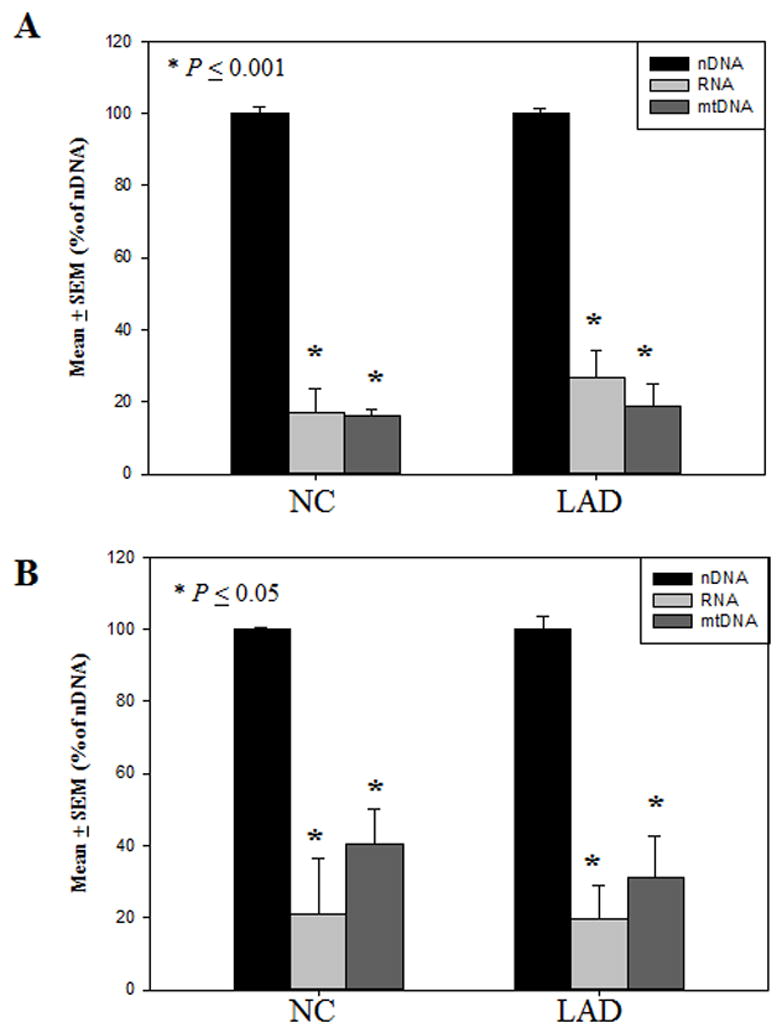
Levels of 5-hydroxymethylcytosine expressed as mean ± standard error of the mean (SEM) (% nDNA) in normal control (NC) subjects and late-stage Alzheimer’s disease (LAD). Levels of 5-hydroxymethylcytosine were significantly (P < 0.001) lower in the RNA and mitochondrial DNA (mtDNA) compared to the nuclear DNA (nDNA) in the superior and middle temporal gyrus (SMTG) of NC and LAD subjects (A). Levels of 5-hydroxymethylcytosine were significantly (p ≤ 0.05) lower the RNA and mitochondrial DNA (mtDNA) compared to the nuclear DNA (nDNA) in the cerebellum (CER) of NC and LAD subjects (B).
Similarly, mean levels of 5hmC were significantly lower (p ≤ 0.05) in RNA (20.9 ± 15.7 % of nDNA) and mtDNA (40.5 ± 9.6 % of nDNA) compared to nDNA (100.0 ± 0.6 % of nDNA) in the CER of NC subjects (Fig. 7B). Additionally, mean levels of 5hmC were significantly lower (p < 0.001) in RNA (31.0 ± 11.6 % of nDNA) and mtDNA (18.7 ± 6.2 % of nDNA) compared to nDNA (100.0 ± 3.6 % of nDNA) in the CER of LAD subjects (Fig. 7B).
Although, levels of 5hmC were significantly higher in nDNA, levels of 5hmC were significantly higher (p < 0.05) in RNA and trended toward a significant increase (p < 0.1) in mtDNA of LAD subjects compared to NC subjects in the SMTG. There were no significant difference in the levels of 5hmC in mtDNA or RNA of the CER of LAD subjects compared to NC.
3.3 Levels of 5mC and 5hmC are elevated whereas levels 5fC and 5caC are decreased in the HPG of PCAD and LAD subjects
Mean levels of 5mC were significantly elevated in the HPG (p < 0.0005) of both PCAD (106.1 ± 0.7 % of NC) and LAD subjects (105.2 ± 0.2 % of NC) compared to NC subjects (100.0 ± .3 % of NC) (Fig. 8A). In contrast, no significant differences in levels of 5mC were observed in the non-vulnerable CER of PCAD (99.2 ± 0.7 % of NC) or LAD subjects (101.0 ± 1.3 % of NC) compared to NC subjects (100.0 ± 0.7 % of NC) (Fig. 8A). Similar to levels of 5mC, levels of 5hmC were significantly elevated in the HPG but not the CER of PCAD and LAD subjects. A more dramatic (p < 0.00005) elevation of 5hmC levels was detected in the HPG of PCAD (119.8 ± 0.8 % of NC) and LAD subjects (116.3 ± 1.5 % of NC) compared to NC subjects (100.0 ± 1.5 % of NC) (Fig. 8B). Levels of 5hmC in the CER were not significantly altered in PCAD (97.1 ± 1.4 % of NC) or LAD subjects (104.0 ± 5.1 % of NC) compared to NC subjects (100.0 ± 1.2 % of NC) (Fig. 8B).
Fig. 8.

Levels of 5-methylcytosine and subsequent derivatives expressed as mean ± standard error of the mean (SEM) (% of normal control [NC]) in the hippocampus/parahippocampal gyrus (HPG) of NC, preclinical Alzheimer’s disease (PCAD), and late-stage Alzheimer’s disease (LAD). Levels of 5-methylcytosine were significantly (p < 0.0005) increased in the HPG of PCAD and LAD subjects compared with NC subjects (A), 5-hydroxymethylcytosine levels were significantly (p < 0.00005) increased in the HPG of PCAD and LAD subjects compared with NC subjects (B), 5–formylcytosine levels were significantly decreased in the HPG of PCAD and LAD subjects compared with NC subjects (C), and 5-carboxylcytosine levels were significantly decreased in (P < 0.001) in the HPG of PCAD and LAD subjects compared with NC subjects (D).
In contrast to levels of 5mC and 5hmC, levels of 5fC and 5caC showed a modest but significant reduction in the HPG of both PCAD and LAD subjects. Mean global levels of 5fC were significantly (p < 0.05) decreased in the HPG of PCAD (99.3 ± 0.1 % of NC) and LAD subjects (99.4 ± 0.09 % of NC) compared to age-matched NC subjects (100.0 ± 0.2 % of NC) (Fig. 8C). Levels of 5fC in the CER of PCAD subjects (98.9 ± 0.09 % of NC) were significantly (p < 0.05) reduced and trended toward a significant decrease (p < 0.10) in LAD subjects (99.3 ± 0.1 % of NC) compared to NC subjects (100.0 ± 0.4 % of NC) (Fig, 8C). In the HPG, mean levels of 5caC were significantly (p< 0.001) reduced in PCAD (91.4 ± 1.5 % of NC) and LAD subjects (93.5 ± 1.0 % of NC) compared to NC subjects (100.0 ± 0.4 % of NC) (Fig. 8D). In contrast, levels of 5caC were not significantly altered in the CER of PCAD subjects (98.8 ± 0.4 % of NC) or LAD subjects (100.2 ± 0.9 % of NC) compared to NC subjects (100.0 ± 0.2 % of NC) (Fig. 8D).
3.4 Increased expression of TET1 in both vulnerable and non-vulnerable brain regions
In contrast to levels of cytosine base modifications, mean levels of TET1 were significantly increased in both the HPG and CER in AD. Mean levels of TET1 were significantly elevated in the HPG of PCAD (p < 0.01) (157.8 ± 9.6 % of NC) and LAD subjects (p < 0.005) (202.7 ± 12.5 % of NC) compared to NC subjects (100.0 ± 13.5 % of NC) (Fig. 9A). Similarly, levels of TET1 were significantly higher in the CER of LAD subjects (p < 0.005) (113.3 ± 0.8 % of NC) compared to NC subjects (100.0 ± 2.7 % of NC) (Fig. 9A). Although levels of TET1 in the CER of PCAD subjects (107.0 ± 1.4 % of NC) were not significantly increased, they trended toward significance (p < 0.1) compared to NC subjects (Fig. 9A).
Fig. 9.

Levels of protein expression expressed as mean ± standard error of the mean (SEM) (% of normal control [NC]) in the hippocampus/parahippocampal gyri (HPG) of NC, preclinical Alzheimer’s disease (PCAD), and late-stage Alzheimer’s disease (LAD). Levels of TET1 normalized to lamin-B were significantly (p < 0.01) increased in the HPG of PCAD and LAD subjects compared with NC subjects (A), thymine DNA glycosylase (TDG) normalized to lamin B were trending toward significance (p< 0.1) increased in the HPG of LAD subjects compared with NC subjects (B).
3.5 Expression of TDG
In contrast to expression levels of TET1, levels of TDG were comparable throughout disease progression in both vulnerable and non-vulnerable brain regions. Mean levels of TDG were not significantly altered in the HPG of PCAD subject (101.4 ± 3.1 % of NC) but trended toward significance in the HPG of LAD subjects (p < 0.1) (111.3 ± 4.3 % of NC) compared to NC subjects (100.00 ± 4.2 % of NC) (Fig. 9B). Mean levels of TDG detected in the CER of PCAD subjects (97.3 ± 3.7 % of NC) or LAD subjects (102.5 ± 4.0 % of NC) were not significantly different compared to NC subjects (100.00 ± 1.3 % of NC) (Fig. 9B).
4. Discussion
While earlier studies of DNA modifications have focused on changes in brain in LAD patients, more recent studies suggest cellular alterations may occur early in the pathogenesis of AD and may represent the best hope for therapeutic interventions. To this end, PCAD subjects represent an intermediate stage between normal aging and LAD. PCAD subjects in the current study were asymptomatic from a clinical perspective but did have significant AD associated pathology. Multiple studies have demonstrated that pronounced oxidative damage is associated with the earliest pathologically detectable stage of AD progression including the generation of reactive α, β-unsaturated aldehydes during lipid peroxidation and nucleic acid oxidation (Bradley et al., 2010a; Bradley et al., 2010b; Lovell et al., 2011). The current study includes tissue samples from longitudinally followed, well characterized NC, PCAD, and LAD subjects with short postmortem intervals. While this preliminary study was designed to investigate potential alterations in relative levels of 5mC and subsequent derivatives during AD pathogenesis, future studies are needed to quantify absolute levels of oxidized 5mC derivatives by LC/MS in a larger sample size.
Primordial germ cell reprogramming is associated with wide-spread active demethylation during chromatin remodeling (Hajkova et al., 2008; Hajkova et al., 2002). Multiple initiation pathways for removal of the stable methylation modification have been proposed including direct deamination of 5mC by activation-induced cytidine deaminase (AID) yielding a G:T mismatch followed by TDG-activated BER (Bhutani et al., 2010); direct deamination of 5hmC by AID to 5-hydroxymethyluracil (5hmU) yielding at G:U mismatch followed by TDG and the single-stranded mono-functional uracil DNA N-glycosylase –activated BER (Boorstein et al., 2001); or further derivatization of 5hmC yielding 5fC and 5caC (Ito et al., 2011; Pfaffeneder et al., 2011) that are both recognized by TDG initiating the BER pathway (He et al., 2011; Maiti and Drohat, 2011). Both 5fC and 5caC have been detected in mouse embryonic stem (mES) cells during wide-spread remodeling (He et al., 2011; Ito et al., 2011). Depletion of TDG in mES cells led to a marked increase in genomic accumulation of 5caC (He et al., 2011). Collectively these studies suggest that TET mediated oxidation of 5mC and subsequent removal of derivatives by the TDG-initiated BER pathway is a valid and active demethylation pathway. In the current study, global levels of 5mC, 5hmC, 5fC, and 5caC, and levels of TET and TDG proteins were quantified relative to age-matched NC subjects throughout the progression of AD.
Although 5hmC was detected in the nDNA, RNA and mtDNA, mean levels were higher in nDNA compared to mtDNA and RNA in the SMTG and CER. Thus, due to limited tissue specimens from the HPG, further analyses were carried out using nDNA, although the possibility that levels of 5mC oxidation derivatives are altered in the mtDNA or RNA during the progression of AD cannot be eliminated. Indeed we observed a significant increase in levels of 5hmC in the RNA and a trend toward a significant increase in the mtDNA. However, Nunomura et al. reported no significant difference in RNA immunostaining intensities of 8-hydroxyguanine (8OHG), a prominent marker of nucleic acid oxidation (Nunomura et al., 2012). In contrast, Dzitoyeva et al. (Dzitoyeva et al., 2012) demonstrated that levels of 5hmC in the frontal cortex of aged mice were significantly decreased in mtDNA and coincided with increased levels of mRNA transcripts for NADH dehydrogenase subunits.
Significantly higher levels of 5hmC, the product of TET mediated 5mC oxidation, were observed in the HPG of PCAD and LAD subjects suggesting initiation of active demethylation in the HPG is an early in disease progression. Levels of TET1 were dramatically increased in the HPG of both PCAD and LAD subjects suggesting increased expression may account for the marked increase of 5mC oxidation. The catalytic activity of nuclear isolated TET proteins remains to be determined in the HPG and CER of the current study subjects. However, the data illustrate a pronounced disconnect between levels of TET1 protein and levels of 5hmC, suggesting the activity of the nuclear located TET proteins may be compromised.
In contrast to levels of 5hmC, the formyl and carboxyl derivatives were modestly decreased in the HPG of PCAD and LAD subjects. The modest decrease of 5fC and 5caC may be a function of extremely low levels (20 and 3 residues per 106C, respectively) (Ito et al., 2011). In the absence of a removal mechanism, it was expected that levels of formyl and carboxyl derivatives would accumulate in the genomic DNA, although to a lesser extent than 5hmC, as a result of decreased TET activity toward the 5hmC and 5fC derivatives (Ito et al., 2011). However, the decreased substrates of the TDG-activated BER pathway may be the result of quick turnover (Globisch et al., 2010; He et al., 2011) as suggested by increasing values of kmax by factors of ~50,000x for G:5fC mismatch and ~12,000 x for G:5caC relative to G:T mismatch (Maiti and Drohat, 2011). Yet, increased levels of TDG only trended toward significance in the HPG of LAD subjects, but total levels may not be the contributing factor given the increased efficiency toward derivative guanine mismatch pairs. Although the current study was carried out in bulk tissue, Iwamoto et al. (Iwamoto et al., 2011) reported that hypomethylation was primarily associated with NeuN+ population of nuclei from human frontal cortex and was distinctively distinguishable from bulk tissue methylation profiles. Additionally, immunostaining of 5hmC was primarily localized in the nuclei of fully differentiated neurons of the dentate gyrus (Globisch et al., 2010) and 5mC immunostaining was primarily localized in the nuclei of cortical (Mastroeni et al., 2009) and NFT bearing neurons (Mastroeni et al., 2010).
Oxidation of nucleic acids is a well-documented phenomenon associated with both normal aging and neurodegeneration, including AD pathogenesis (Gabbita et al., 1998; Loft et al., 2008; Lovell et al., 1999; Lovell et al., 2011; Moreira et al., 2008; Nunomura et al., 2012; Wang et al., 2006). Therefore, non-enzymatic oxidation of 5mC to 5hmC by the hydroxyl radical may interfere with quantification of active demethylation products (Castro et al., 1996). Indeed, Müzel et al. reported increased levels of 5hmC in 90-day old mice compared to 1-day old mice (Munzel et al., 2010). In addition, a series of knockout experiments in mES cells showed levels of 5hmC were dependent on existing levels of 5mC (Ficz et al., 2011) and increased levels of both 5mC and 5hmC in aged mouse brains in the absence of increased guanine oxidation (Munzel et al., 2010). Furthermore, multiple studies of oxidative damage to nucleic acids have failed to demonstrate a significant increase in the oxidized cytosine product, 5-hydroxycytosine, during AD disease progression (Gabbita et al., 1998; Wang et al., 2006). Rather, the age-specific epigenetic drift observed by Müzel et al. is a common phenomenon in human brain but is more pronounced in the brains of LAD subjects (Wang et al., 2008). Furthermore, significant derivations in the methylation patterns of monozygotic twins suggest that age-specific epigenetic drift is influenced by both internal and external factors (Fraga et al., 2005).
Decreased gene expression due to the presence of 5mC in the promoter region is mediated through the recognition of 5mC by the methyl-CpG binding domain of methyl-CpG binding proteins, which recruits histone modifying enzymes. Oxidation products of both guanine and 5mC inhibit binding of the methyl-CpG binding domain of methyl-CpG binding protein 2 (Valinluck et al., 2004). Similarly, the presence of 8OHG in oligonucleotides results in decreased methylation suggesting that its presence also interferes with the activity of the DNMT family of proteins (Weitzman et al., 1994). It has been proposed that occurrence of genome wide oxidation of methyl cytosine may potentially alter expression. While studies are limited, a few have suggested possible roles for altered methylation/demethylation in the amyloidgenic processing of APP. For example, hypomethylation of the APP gene from the frontal cortex was reported in a subject with LAD but not an age-matched NC subject or diseased control subject with Pick’s disease (West et al., 1995). Additionally, alteration of s-adenosylmethionine/homocysteine cycle has been suggested as a possible risk factor for AD (Seshadri et al., 2002) through modulation of PS1 expression (Fuso et al., 2005; Scarpa et al., 2003). In vitro hypomethylation of the γ-secretase, presenilin 1 (PS1) led to increased cleavage of APP and increased production of Aβ in neuroblastoma cell lines under reduced S-adenosylmethionione availability in folate and vitamin B12 deficient medium (Fuso et al., 2005). In contrast, administration of s-adenosylmethionine and subsequent methylation of PS1 resulted in down-regulation of PS1 gene expression followed by reduced Aβ production (Scarpa et al., 2003). However, measurements of s–adenosylmethionine and s-adenosylhomocyosteine in the cerebrospinal fluid of LAD subjects were not significantly different from those quantified in age-matched NC subjects (Mulder et al., 2005).
While the current study demonstrates increased oxidation of 5mC during the progression of AD, it fails to identify where within the genome the oxidation is occurring. Genomic mapping of immunoprecipitated 5hmC DNA from the frontal lobe demonstrated an enrichment in both the promoter and intragenic regions (Jin et al., 2011). The presence of 5mC in promoter regions is associated with decreased gene expression (Jin et al., 2011) and transcripts (Robertson et al., 2011), whereas enrichment of 5hmC in the intragenic regions is associated with significantly higher expression levels (Jin et al., 2011). These data suggest that the outcome of 5mC oxidation with respect to transcription and expression is dependent on genomic location but clustered in the promoter and intragenic regions (Nestor et al., 2012). Gene ontology pathway analysis found significant enrichment of 5hmC in pathways associated with neurodegenerative orders and hypoxic response (Song et al., 2011).
Multiple studies have documented altered genetic profiles (Dunckley et al., 2006; Liang et al., 2008; Wang et al., 2008) and protein expression in LAD subjects (Liang et al., 2008). Although an earlier study demonstrated no significant differences in the percentage of methylation in CCGG sites in LAD subjects (Schwob et al., 1990), several more recent studies report significant alterations of cytosine methylation. Studies by Mastroeni et al. (Mastroeni et al., 2010; Mastroeni et al., 2009)quantified lower levels of methylation in the entorhinal cortex and the neocortex in LAD (Mastroeni et al., 2010; Mastroeni et al., 2009). Additionally, decreased methylation in a monozygotic twin with LAD suggests alterations of the methylome may prove to play a key role in AD (Mastroeni et al., 2009). While studies from Mastroeni et al. demonstrated global decreases in methylation, identification of differentially methylated CpG sites across multiple genes in LAD suggest that the phenomenon is bidirectional (Bakulski et al., 2012).
Other studies suggest altered methylation may play an active role in the memory formation. Miller et al. (Miller et al., 2010)demonstrated that during contextual fear conditioning, methylation was increased in the anterior cingulate cortex of rats up to 30 days post conditioning (Miller et al., 2010). Whereas rapid demethylation and de novo methylation of in low CpG density regions of genes related to neuronal plasticity in the dentate granule cells of adult mice following electroconvulsive stimulation (Guo et al., 2011), pharmacological disruption of methylation resulted in the loss of remote memory implicating a role for methylation in the preservation of long-term memory (Levenson et al., 2006; Miller et al., 2010). However, both active and rapid methylation of the memory suppressor gene protein phosphatase 1 (PP1) by the DNMT family of proteins (Feng et al., 2010) and demethylation of the synaptic plasticity gene reelin and brain derived neurotrophic factor (BDNF) (Lubin et al., 2008) occurred during contextual fear conditioning suggesting that both processes are essential for memory formation (Miller and Sweatt, 2007).
5. Conclusions
Overall, the current study suggests altered methylation status of genomic DNA is detectable not only in LAD, but also early during the pathogenesis of AD (PCAD). Furthermore, accumulation of both methylated and oxidized methyl substrates occur in an area associated with memory and higher cognitive functioning, the HPG. Although this study is limited to relative quantification in bulk tissue, it warrants further investigation. The detection of altered methylation/demethylation status in a brain region vulnerable to AD pathology before the onset of clinical symptoms in PCAD subjects and a sustained phenomenon in LAD suggest a role in the pathogenesis of neurodegeneration in AD.
Highlights.
5hmC is more pronounced in nDNA than mtDNA or RNA
Levels of 5mC and 5hmC are increased in PCAD and LAD HPG compared to NC HPG
Levels of 5fC and 5caC are decreased in PCAD and LAD HPG compared to NC HPG
Levels of TET1 are increased in PCAD and LAD HPG compared to NC HPG
Acknowledgments
This research was supported by NIH grants 5P01-AG05119 and P30-AG028383. The authors thank the UK-ADC Clinical, Neuropathology and Biostatistics Cores for tissue procurement and neuropathologic data. The authors also thank Ms. Sonya Anderson for subject demographic data and Ms. Paula Thomason for editorial assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addya K, Wang YL, Leonard DG. Optimization of Apolipoprotein E Genotyping. Mol Diagn. 1997;2:271–276. doi: 10.1054/MODI00200271. [DOI] [PubMed] [Google Scholar]
- Aluise CD, Robinson RA, Beckett TL, Murphy MP, Cai J, Pierce WM, Markesbery WR, Butterfield DA. Preclinical Alzheimer disease: brain oxidative stress, Abeta peptide and proteomics. Neurobiol Dis. 2010;39:221–8. doi: 10.1016/j.nbd.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, Albin RL, Hu H, Rozek LS. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29:571–88. doi: 10.3233/JAD-2012-111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–7. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Boorstein RJ, Cummings A, Jr, Marenstein DR, Chan MK, Ma Y, Neubert TA, Brown SM, Teebor GW. Definitive identification of mammalian 5-hydroxymethyluracil DNA N-glycosylase activity as SMUG1. J Biol Chem. 2001;276:41991–7. doi: 10.1074/jbc.M106953200. [DOI] [PubMed] [Google Scholar]
- Bradley MA, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free radical biology & medicine. 2010a;48:1570–6. doi: 10.1016/j.freeradbiomed.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley MA, Xiong-Fister S, Markesbery WR, Lovell MA. Elevated 4-hydroxyhexenal in Alzheimer’s disease (AD) progression. Neurobiology of aging. 2010b doi: 10.1016/j.neurobiolaging.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro GD, Diaz Gomez MI, Castro JA. 5-Methylcytosine attack by hydroxyl free radicals and during carbon tetrachloride promoted liver microsomal lipid peroxidation: structure of reaction products. Chem Biol Interact. 1996;99:289–99. doi: 10.1016/0009-2797(95)03680-6. [DOI] [PubMed] [Google Scholar]
- Chromczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dunckley T, Beach TG, Ramsey KE, Grover A, Mastroeni D, Walker DG, LaFleur BJ, Coon KD, Brown KM, Caselli R, Kukull W, Higdon R, McKeel D, Morris JC, Hulette C, Schmechel D, Reiman EM, Rogers J, Stephan DA. Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging. 2006;27:1359–71. doi: 10.1016/j.neurobiolaging.2005.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. 2012;33:2881–91. doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, Fan G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–30. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. Journal of neurochemistry. 1998;71:2034–40. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Powlishta KK, Wilkins K, McKeel DW, Jr, Xiong C, Grant E, Storandt M, Morris JC. Predictors of preclinical Alzheimer disease and dementia: a clinicopathologic study. Archives of neurology. 2005;62:758–65. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–51. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, Toubaji A, Argani P, Iacobuzio-Donahue C, Nelson WG, Netto GJ, De Marzo AM, Yegnasubramanian S. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–37. doi: 10.18632/oncotarget.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–81. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–7. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A. 2011;108:3642–7. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–33. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, Saito T, Geschwind DH, Kato T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21:688–96. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39:5015–24. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–13. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–73. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Li W, Liu M. Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids. 2011;2011:870726. doi: 10.4061/2011/870726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang WS, Dunckley T, Beach TG, Grover A, Mastroeni D, Ramsey K, Caselli RJ, Kukull WA, McKeel D, Morris JC, Hulette CM, Schmechel D, Reiman EM, Rogers J, Stephan DA. Altered neuronal gene expression in brain regions differentially affected by Alzheimer’s disease: a reference data set. Physiol Genomics. 2008;33:240–56. doi: 10.1152/physiolgenomics.00242.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loft S, Hogh Danielsen P, Mikkelsen L, Risom L, Forchhammer L, Moller P. Biomarkers of oxidative damage to DNA and repair. Biochemical Society transactions. 2008;36:1071–6. doi: 10.1042/BST0361071. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. Journal of neurochemistry. 1999;72:771–6. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Soman S, Bradley MA. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev. 2011;132:443–8. doi: 10.1016/j.mad.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–86. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–8. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–37. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4:e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative Damage to Mitochondrial DNA Shows marked Age-dependent Increases in Human Brian. American Neurological Association. 1993;34:609–15. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- Miller CA, Gavin CF, White JA, Parrish RR, Honasoge A, Yancey CR, Rivera IM, Rubio MD, Rumbaugh G, Sweatt JD. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–6. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–69. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA, Perry G. Nucleic acid oxidation in Alzheimer disease. Free radical biology & medicine. 2008;44:1493–505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Mulder C, Schoonenboom NS, Jansen EE, Verhoeven NM, van Kamp GJ, Jakobs C, Scheltens P. The transmethylation cycle in the brain of Alzheimer patients. Neurosci Lett. 2005;386:69–71. doi: 10.1016/j.neulet.2005.03.073. [DOI] [PubMed] [Google Scholar]
- Munzel M, Globisch D, Bruckl T, Wagner M, Welzmiller V, Michalakis S, Muller M, Biel M, Carell T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed Engl. 2010;49:5375–7. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–77. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Tamaoki T, Motohashi N, Nakamura M, McKeel DW, Jr, Tabaton M, Lee HG, Smith MA, Perry G, Zhu X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol. 2012;71:233–41. doi: 10.1097/NEN.0b013e318248e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–7. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA, Hagemeier C, Carell T. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed Engl. 2011;50:7008–12. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- Robertson J, Robertson AB, Klungland A. The presence of 5-hydroxymethylcytosine at the gene promoter and not in the gene body negatively regulates gene expression. Biochem Biophys Res Commun. 2011;411:40–3. doi: 10.1016/j.bbrc.2011.06.077. [DOI] [PubMed] [Google Scholar]
- Saiki RK, Bugawan TL, Horn GT, Mukkis KB, Erlich HA. Analysis of enzymatically amplified beta-globin and hla-dq alpha DNA with allele-specific olgionucleotide probes. Nature. 1986;324:163–166. doi: 10.1038/324163a0. [DOI] [PubMed] [Google Scholar]
- Scarpa S, Fuso A, D’Anselmi F, Cavallaro RA. Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Lett. 2003;541:145–8. doi: 10.1016/s0014-5793(03)00277-1. [DOI] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclincal” AD revisited: Neuropathology of cognitively older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- Schwob NG, Nalbantoglu J, Hastings KE, Mikkelsen T, Cashman NR. DNA cytosine methylation in brain of patients with Alzheimer’s disease. Ann Neurol. 1990;28:91–4. doi: 10.1002/ana.410280117. [DOI] [PubMed] [Google Scholar]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med. 2002;346:476–83. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Anderson MF. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat Protoc. 2008;3:1228–1238. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
- Smith CD, Chebrolu H, Markesbery WR, Liu J. Improved predictive model for pre-symptomatic mild cognitive impairment and Alzheimer’s disease. Neurol Res. 2008;30:1091–6. doi: 10.1179/174313208X327973. [DOI] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, Wang J, Zhang L, Looney TJ, Zhang B, Godley LA, Hicks LM, Lahn BT, Jin P, He C. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons MD, Bradley MA, Lovell MA, Lynn BC. Procedure for the isolation of mitochondria, cytosolic and nuclear material from a single piece of neurological tissue for high-throughput mass spectral analysis. J Neurosci Methods. 2011;197:279–82. doi: 10.1016/j.jneumeth.2011.02.027. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70:288–92. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- Tsukamoto K, Watanabe T, Matsushima T, Kinoshita M, Kato H, Hashimoto Y, Kurokawa K, Teramoto T. Determination by PCR-RFLP of apo E genotype in a Japanese population. J Lab Clin Med. 1993;121:598–602. [PubMed] [Google Scholar]
- Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–8. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. Journal of neurochemistry. 2006;96:825–32. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci U S A. 1994;91:1261–4. doi: 10.1073/pnas.91.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci. 1995;6:141–6. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–84. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–52. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]