

Abstract
Many DNA-binding assays such as electrophoretic mobility shift assays (EMSA), chemiluminescent assays, chromatin immunoprecipitation (ChIP)-based assays, and multiwell-based assays are used to measure transcription factor activity. However, these assays are nonquantitative, lack specificity, may involve the use of radiolabeled oligonucleotides, and may not be adaptable for the screening of inhibitors of DNA binding. On the other hand, using a quantitative DNA-binding enzyme-linked immunosorbent assay (D-ELISA) assay, we demonstrate nuclear protein interactions with DNA using the RUNX2 transcription factor that depend on specific association with consensus DNA-binding sequences present on biotin-labeled oligonucleotides. Preparation of cells, extraction of nuclear protein, and design of double stranded oligonucleotides are described. Avidin-coated 96-well plates are fixed with alkaline buffer and incubated with nuclear proteins in nucleotide blocking buffer. Following extensive washing of the plates, specific primary antibody and secondary antibody incubations are followed by the addition of horseradish peroxidase substrate and development of the colorimetric reaction. Stop reaction mode or continuous kinetic monitoring were used to quantitatively measure protein interaction with DNA. We discuss appropriate specificity controls, including treatment with non-specific IgG or without protein or primary antibody. Applications of the assay are described including its utility in drug screening and representative positive and negative results are discussed.
Keywords: Cellular Biology, Issue 78, Transcription Factors, Vitamin D, Drug Discovery, Enzyme-Linked Immunosorbent Assay (ELISA), DNA-binding, transcription factor, drug screening, antibody
Introduction
DNA-binding assays have utility in measuring the ability of transcription factors to interact with DNA. Assays for DNA binding include electrophoretic mobility shift assays (EMSA) that depend on radiolabeled oligonucleotides 1 or chemiluminescence assays 2. Chromatin immuneprecipitation (ChIP) based assays 3 as well as assays employing 96-well formats 4 have also been described. However, the EMSA is a non-quantitative assay that requires the use of radiolabeled oligonucleotides. When nuclear proteins associate with the specific nucleotide promoter sequences, binding complexes are retarded on polyacrylamide gels and the specific transcription factor can be validated with an antibody "supershift". We have developed a quantitative DNA-binding assay using an enzyme-linked immunosorbent format (D-ELISA), which is able to measure the interaction of RUNX2 with DNA-binding sequences corresponding to defined promoter elements in RUNX2 target genes. Use of an anti-RUNX2 antibody provides specificity to the assay and the lack of radiolabel distinguish this assay from the traditional gel shift assay 5. Detection of binding complexes is possible with the use of a secondary antibody coupled to horseradish peroxidase (HRP), which converts an HRP substrate, tetramethyl benzidine (TMB) to a colored product for spectrophotometric analysis. The assay reported here can incorporate the use of mutated DNA oligonucleotides as controls and can be used for detection of competitive or non-competitive inhibitors of DNA binding. The screening of novel anti-tumor compounds is also possible with this assay.
Protocol
Several steps are performed ahead of time and several reagents are prepared and stored prior to the procedure: (1) cell culture and protein isolation, (2) preparation of oligonucleotide, (3) preparation of 96-well plates, (4) nuclear extract incubation overnight. Procedure requires 2 days because of the overnight incubation of nuclear protein with DNA oligonucleotides.
1. Preparation of Buffers
1.1 Nuclear protein isolation
Hypotonic Buffer: To prepare 100 ml of hypotonic buffer, add 1 ml (1 M HEPES, pH 7.9), 150 μl (1 M MgCl2), 333 μl (3 M KCl) to 98.3 ml H2O. Final Concentrations: 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl. For Hypotonic buffer + NP40 (for use in step 2.11): Add 0.5 ml (10% NP40) to 9.5 ml Hypotonic buffer for final 0.5% NP40.
Low Salt Buffer: To prepare 100 ml of low salt buffer (for use in step 2.15), add 1 ml (1 M HEPES, pH 7.9), 25 ml Glycerol, 150 μl (1 M MgCl2), 666 μl (3 M KCl), 40 μl (0.5 M EDTA) to 73 ml H2O. Final concentrations: 10 mM HEPES, 25% Glycerol, 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA.
High Salt Buffer: To prepare 100 ml of high salt buffer (for use in step 2.15), add 1 ml (1 M HEPES, pH 7.9), 25 ml Glycerol, 150 μl (1 M MgCl2), 26.7 ml (3 M KCl), 40 μl (0.5M EDTA) to 47 ml H2O. Final concentrations: 10 mM HEPES, 25% Glycerol, 1.5 mM MgCl2, 800 mM KCl, 0.2 mM EDTA.
10% NP40 Detergent: 10 ml NP40 to 90 ml H2O
Protease and phosphatase inhibitors [added to hypotonic buffer + NP40 (step 2.11), to low salt buffer (step 2.15), and to high salt buffer (step 2.15) just prior to use]: Add 2.5 μl (1 M DTT) and 50 μl of protease inhibitors (100x) to 5 ml of each buffer for final concentration of 0.5 mM DTT and 1x protease inhibitors. The protease inhibitor 100x stock mixture includes: sodium fluoride (Ser/Thr, acidic phosphatases), sodium orthovanadate (Tyr, alkaline phosphatases), β-glycerophosphate (Ser/Thr phosphatases), sodium pyrophosphate (Ser/Thr phosphatases), Aprotinin (Ser proteases), Bestatin (amino-peptidases), E64 (Cysteine proteases), Leupeptin (Ser/Cys proteases), EDTA (metalloproteases).
1.2 Preparation of assay plates
Plate fixing solution: To prepare 500 ml of solution, add 5.25 g sodium carbonate to 500 ml H2O, mix well and adjust pH to 9.7 for a final concentration of 0.1 M sodium carbonate.
Plate blocking solution: To prepare stock poly dI/dC oligonucleotide, resuspend 1 Unit (~50 mg) of poly dI/dC in 1 ml of 10 mM Tris/HCl, pH 7.9 containing 1 mM EDTA for a stock concentration of 50 μg/μl. Stock is diluted to 1 μg/μl prior to use.
1.3 Wash buffers and antibody dilutions
NOTE: Streptavidin wash buffer is used to wash plates in between additions and to dilute primary and secondary antibodies.
To prepare 1 L of Streptavidin wash buffer, add 2.4 g Tris/HCl, 37.5 ml (4 M NaCl), 10 ml (10% BSA), 5 ml (10% Tween-20), to 1 L Millipore-sterile filtered water, pH 7.18. Store at 4 °C. Final concentrations: 20 mM Tris/HCl, 150 mM NaCl, 0.1% BSA, 0.05% Tween-20.
1.4 DNA-binding buffer
NOTE: This buffer is stored at -20 °C.
To prepare 15 ml of DNA binding buffer, add 180 μl (1 M HEPES, pH 7.9), 300 μl (3 M KCl), 12 μl (0.5 M EDTA, pH 8.0), 7.5 μl (1 M DTT), 3.0 ml (60% glycerol), 300 μl (50 μg/μl poly dI/dC) diluted with deionized/distilled H2O (12 ml). Final concentrations: 12 mM HEPES/pH 7.9, 60 mM KCl, 0.4 mM EDTA, 0.5 mM DTT, 12% glycerol, 1 μg/μl poly dI/dC.
2. Cell Culture and Nuclear Protein Isolation
NOTE: The preparation requires about 3 hr. Stimulation of cells will depend on experiment. The number of cells used for each experiment will vary and all volumes reflect a typical experiment using 3 x 107 cells/point. Adjust volumes to accommodate the cell number actually used in the experiment 6.
To prepare nuclear protein for DNA binding assays, culture human cells that express RUNX2 (endothelial, osteosarcoma, breast cancer cells) in appropriate media 6.
Treat subconfluent cells with nocodazole (0.2 μg/ml for 16 hr) to arrest cells at the G2/M cell cycle boundary 6. Under these conditions, the RUNX2 protein is stabilized and maximal DNA binding is achieved. In this way, enough nuclear protein is available for multiple assays and inhibitors can be compared from the same preparation.
For each step, pre-cool the centrifuge to 4 °C and prepare buffers and chill 20 min at 4 °C before cell harvest.
Chill cells (4 °C) and conduct procedures on ice.
Centrifuge cells at 1,000 rpm for 5 min in a 15 ml polypropylene round-bottom tube.
Wash 1x with ice-cold PBS (Ca2+ and Mg2+ free).
Centrifuge again and discard PBS supernatant.
Add 500 μl of hypotonic buffer without protease inhibitors, being careful to completely resuspend the cell pellet.
Transfer contents to a 1.5 ml Eppendorf tube.
Immediately centrifuge at 5,500 rpm for 5.5 min; discard the supernatant and KEEP the cell pellet.
Resuspend the cell pellet in 500 μl of hypotonic buffer containing 0.5% NP40. Add protease and phosphatase inhibitors and 0.5 mM DTT (as described in step 1.1.5).
Allow the sample to rest on ice for 30 min.
Centrifuge at 13,000 rpm for 10 min.
Discard the supernatant (contains the cytosolic proteins).
Resuspend the nuclear pellet in 60 μl each of low salt buffer (to which protease inhibitors and 0.5 mM DTT have been added from step 1.1.5) and high salt buffer (to which protease inhibitors and 0.5 mM DTT have been added from step 1.1.5) for a total of 120 μl. Add 60 μl of low salt buffer to the pellet first and resuspend, then add 60 μl of high salt buffer. Vortex the pellet to aid in resuspension.
NOTE: This step helps get rid of some of the nuclear membrane proteins and set up for the lysis step: low salt first (resuspend pellet, vortex), then high salt (vortex) optimizes this step. Keep extract on ice for 30 min, with vortexing after the first 10 min.
Centrifuge at 12,000 rpm for 15 min; keep the supernatant containing the nuclear protein.
Measure protein concentration (try to keep samples at 2-5 mg/ml), aliquot in appropriate amounts (10 μl/tube) and store at -80 °C.
NOTE: Estimated yield: 30 x 106 cells will yield 5 μg/μl protein in 120 μl. 5 x 106 cells will yield 2.2 μg/μl protein in 40 μl.
3. Preparation of Double Stranded Oligonucleotide
Design complementary oligonucleotides with compatible half sites on the ends. For RUNX2 these are: 5'-CGTATTAACCACAATACTCGCGTATTAACCACAATACTCGCGTATTAACCACAATAC TCG -3'-Biotin (sense) and 5'CGAGTATTGTGGTTAATACGCGAGTATTGTGGTTAATACGCGAGTATT GTGGTTAATACG -3'-Biotin (antisense).
NOTE: Pilot experiments determined that three RUNX2 binding sites and single-end labeled biotin yielded the most reproducible results.
Prepare 5x annealing buffer: Add 300 μl (1 M Tris-HCl, pH 7.6), 30 μl (1 M MgCl2), 10 μl (1 M DTT) to 260 μl H2O (final 600 μl).
To 200 pmol of each single-stranded oligonucleotide (3 μl), add 12 μl of 5x annealing buffer and 45 μl H2O for a total of 60 μl (200 pmol/60 μl).
Heat at 95 °C for 5-10 min in a heating block.
Cool to 65 °C slowly (by setting the temperature to 65 °C, it takes 15 min), then cool to room temperature slowly by turning off the power of the heating block (or by placing in 50 ml of the 65 °C water in a beaker on ice; this takes 15-20 min).
4. Preparation of 96-well Plates
NOTE: Do not use milk proteins in the wash buffers.
Fix plate with fixing solution (0.1 M sodium carbonate): add 300 μl/well.
Incubate for 2 hr by rocking at room temperature (RT), or no rocking at 4 °C (O/N).
Wash plate 3x with streptavidin wash buffer (WB): add 300 μl/well each time.
Add biotin-labeled double-stranded oligonucleotide (3 consensus binding sites per nucleotide) for 2 hr with rocking (1.25 nmol/well; 100 μl/well).
Wash 3x with cold WB: add 300 μl/well and then immediately invert the plate into a sink and blot the edges on a paper towel after each addition.
NOTE: Do not use pipette tips to remove fluid from the plates as this may scrape avidin:biotin:DNA complexes from the wells. Do this for all subsequent wash steps.
5. Incubation with Nuclear Extract
NOTE: Do not substitute salmon or herring sperm DNA for the poly dI/dC blocking buffer. Avoid blocking plates with milk proteins. Both of these blocking agents result in high background values.
Incubate with specific transcription factor prepared from nuclear extract (90 μl/well). Prepare a master mix containing nuclear extract (DNA binding protein; 3-9 μg/well) + poly dI/dC (1 μg/μl from a 50 μg/ul stock) in 1x DNA binding buffer. Volume is as needed for the total number of wells required (90 μl/well).
Any potential inhibitor, such as vitamin D3 (Figure 2) or CADD compound 5221975 (Figure 3), or the appropriate dilution of the solvent control, such as ethanol, is added at this step.
Place plate on rocking platform, O/N at 4 °C.
Wash 3x with WB: add 300 μl/well
6. Addition of Primary Antibody
NOTE: Primary antibody dilutions should be prepared fresh - storage is not recommended.
Dilute RUNX2-specific monoclonal antibody (stock 1 μg/μl) 1/5,000 in Streptavidin wash buffer. Prepare enough for addition to each well of the 96-well plate (90 μl/well) in triplicate.
Incubate for 1 hr at RT on a rocking platform.
Wash 3x with WB; add 300 μl/well.
7. Addition of Secondary Antibody
NOTE: Secondary antibody dilutions can be stored at 4 °C overnight if needed the next day.
Dilute Fab-specific affinity purified-HRP conjugated antibody (7.1 mg/ml stock) to 1:1,400 in Streptavidin wash buffer: 3.5 μl antibody/5.0 ml wash buffer. Prepare enough for addition to each well of the 96-well plate (90 μl/well) in triplicate.
Incubate for 30 min at RT on a rocking platform.
Wash 6x with WB by inverting plate into sink (300 μl/well).
8. HRP Substrate and Product Development
Add 50 μl of TMB substrate per well directly from stock bottle.
Incubate 10-20 min at RT in the dark (stop reaction method) or measure absorbance directly (continuous kinetic monitoring).
9. Reaction Measurement
Stop reaction method: For visual stop reaction, check for color change from clear to blue and stop reaction with 50 μl sulfuric acid.
Measure absorbance at 450 nm with the Biotrak II Visible plate reader Spectrophotometer (Amersham Biosciences; GE Healthcare Biosciences Corp. Piscataway NJ, USA) or a similar instrument.
Continuous kinetic monitoring method: Add substrate after the last wash and just prior to placing in the spectrophotometer (do not stop the reaction).
Measure absorbance at 635 nm with the Bio-Tek Synergy HT Multi-reaction microplate reader Spectrophotometer (Bio-Tek Instruments, Inc; Winooski, VT, USA) or a similar instrument.
Representative Results
The D-ELISA method is highly specific for the designated DNA-binding protein as long as a sequence-specific, double-stranded oligonucleotide containing three copies of the consensus RUNX2 binding site (ACACCA) is used. The primary antibody recognizing the protein factor also enhances the specificity. The secondary antibody contains covalently-linked horseradish peroxidase (HRP) that converts a clear substrate (tetramethyl benzidine) to a colored product for ease of detection (Figure 1). For these reasons, several important background controls need to be included in each assay plate to ensure specific DNA binding. These include measuring the colorimetric product from the secondary antibody-HRP reaction in wells containing either (1) no nuclear protein, (2) no primary antibody, or (3) a non-specific mouse IgG instead of the specific monoclonal antibody. In our routine measurements, the no-protein controls are subtracted from the specific protein and expressed as net reaction product. Further, when computer-assisted drug design (CADD) is used to discover compounds and the drugs are tested in DNA binding assays, the effect of the solvent used to solubilize the compounds must be determined in separate wells. Typically, drug screening will require the use of water, ethanol, or dimethyl sulfoxide to solubilize compounds. Each of these solvents is tested separately and at the appropriate concentrations. Some drug screens identified compounds that have little effect on DNA binding, as shown for the vitamin D receptor ligand, 1α,25-OH Vitamin D3 (Figure 2). This compound did not inhibit DNA binding even at higher concentrations (100 nM to 100 μM). Other drug screens detected compounds that inhibit protein:DNA binding (Figure 3). Based on analyses of kinetic measurements 7, increasing the amounts of the CADD5221975 inhibitor resulted in a dose-dependent, sigmoidal inhibition curve, with inhibitor concentrations above 10-3 M effectively inhibiting DNA binding, while concentrations below 10-11 M showed very little inhibition. The EC50 (concentration at which inhibitor caused a 50% reduction in the binding of RUNX2 to DNA) was 10 nM for this compound. The statistical analyses for this assay have been published previously 6 and, for simplicity, only one well of a triplicate series of wells is shown in representative Figures 2 and 3.
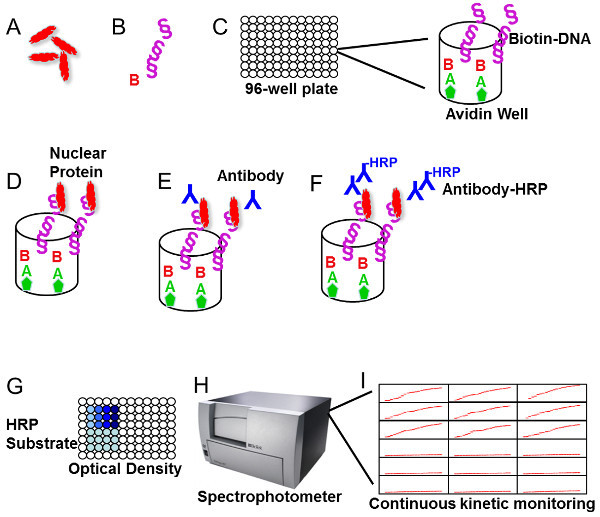 Figure 1. Flow chart of the D-ELISA protocol. (A) Isolate nuclear proteins from mammalian cells. Growing cells are fractionated by detergent lysis and high salt methods to extract proteins tightly associated with DNA. (B) Prepare double stranded oligonucleotide. The RUNX2 binding site ACACCAA in triplicate is prepared with a biotin tag at the 3'-end. (C) Prepare 96-well plates. Plates are obtained with avidin coating, but must be fixed with high pH before adding biotin-labeled oligonucleotides. (D) Incubate nuclear extracts with the DNA. Plates are blocked with non-specific poly-dI/dC prior to addition of nuclear extract proteins. (E) Add primary antibody. Antibody is prepared fresh prior to incubation with plate. (F) Add secondary antibody. Secondary antibody is followed by extensive washing. (G) Add HRP substrate and develop the product. Colorimetric product is visible on the plate. (H) Measure the product absorbance on a spectrophotometer. Absorbance is either 450 nm (stop reaction) or 635 nm (continuous monitoring). (I) Typical continuous readout at 635 nm. Shown are specific reaction and background (no protein) controls in triplicate.
Figure 1. Flow chart of the D-ELISA protocol. (A) Isolate nuclear proteins from mammalian cells. Growing cells are fractionated by detergent lysis and high salt methods to extract proteins tightly associated with DNA. (B) Prepare double stranded oligonucleotide. The RUNX2 binding site ACACCAA in triplicate is prepared with a biotin tag at the 3'-end. (C) Prepare 96-well plates. Plates are obtained with avidin coating, but must be fixed with high pH before adding biotin-labeled oligonucleotides. (D) Incubate nuclear extracts with the DNA. Plates are blocked with non-specific poly-dI/dC prior to addition of nuclear extract proteins. (E) Add primary antibody. Antibody is prepared fresh prior to incubation with plate. (F) Add secondary antibody. Secondary antibody is followed by extensive washing. (G) Add HRP substrate and develop the product. Colorimetric product is visible on the plate. (H) Measure the product absorbance on a spectrophotometer. Absorbance is either 450 nm (stop reaction) or 635 nm (continuous monitoring). (I) Typical continuous readout at 635 nm. Shown are specific reaction and background (no protein) controls in triplicate.
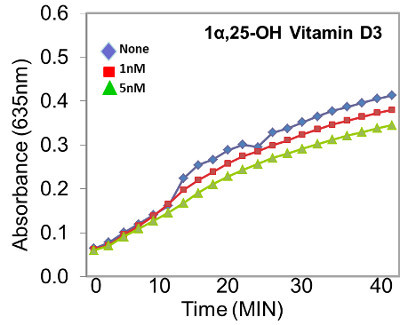 Figure 2. 1α,25-OH Vitamin D3 has little effect on RUNX2 DNA binding. In this representative example, the biologically active vitamin D3, 1α,25-OH Vitamin D3, has little effect on RUNX2 DNA binding. Other vitamin D3 compounds, however, have dramatic effects on DNA binding 6.
Figure 2. 1α,25-OH Vitamin D3 has little effect on RUNX2 DNA binding. In this representative example, the biologically active vitamin D3, 1α,25-OH Vitamin D3, has little effect on RUNX2 DNA binding. Other vitamin D3 compounds, however, have dramatic effects on DNA binding 6.
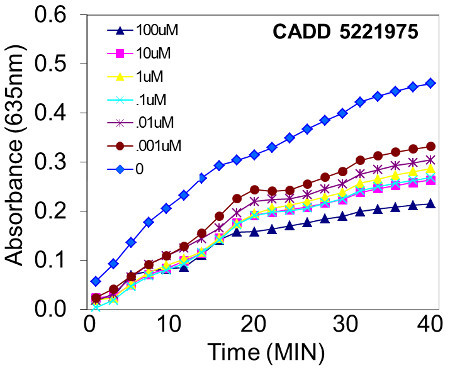 Figure 3. Inhibition of DNA binding by putative RUNX2:DNA active drug. In this representative example, a compound that was identified from a computer-assisted drug design (CADD) screen exhibited a dose-dependent inhibition of RUNX2 DNA binding from 1 nM to 100 μM. Binding inhibition was calculated using established methods 7 and yielded an EC50 (concentration of inhibitor that resulted in a 50% inhibition of DNA binding) of 10 nM.
Figure 3. Inhibition of DNA binding by putative RUNX2:DNA active drug. In this representative example, a compound that was identified from a computer-assisted drug design (CADD) screen exhibited a dose-dependent inhibition of RUNX2 DNA binding from 1 nM to 100 μM. Binding inhibition was calculated using established methods 7 and yielded an EC50 (concentration of inhibitor that resulted in a 50% inhibition of DNA binding) of 10 nM.
Discussion
DNA-binding assays are used to measure the ability of transcription factors to interact with DNA. Assays for DNA binding include electrophoretic mobility shift (EMSA) 1 and chromatin immuneprecipitation (ChIP) based assays 3 as well as assays employing 96-well formats 4 such as chemiluminescent assays 2. The EMSA is non-quantitative and uses radiolabeled (32P) oligonucleotides. These are incubated with nuclear proteins and binding complexes are separated on agarose or polyacrylamide gels. In contrast, the quantitative D-ELISA is able to measure the interaction of RUNX2 with DNA-binding sequences corresponding to defined promoter elements in RUNX2 target genes. The use of an anti-RUNX2 antibody increased the specificity of the assay and distinguishes this assay from the traditional gel shift assay 5. While the D-ELISA is an in vitro assay and cannot survey promoter occupancy in live cells, which is possible with ChIP assays, it can be used to quantitatively screen for compounds that inhibit the DNA-binding complexes. These assays are limited to DNA interaction analyses and cannot predict whether a specific promoter is activated or repressed. Further approaches, such as promoter-luciferase reporter assays, are necessary to define the transcriptional activity of the specific transcription factor.
The general protocol of the D-ELISA method described here was adapted from a previous method used to measure active Nfkappa-B 8. This D-ELISA protocol provides a method for quantitatively measuring protein:DNA binding that is sequence-specific and does not involve the use of radioactivity. If necessary, reaction velocities (Vmax) can also be calculated from continuous kinetic monitoring of the reaction and this may provide additional discrimination of test compounds 7.
RUNX2 and its cofactor Cbf ß were found to be associated with the biotin-labeled oligonucleotides 6, thus validating the specificity of the assay and also emphasizing that it is possible to identify cofactors that might associate with specific DNA-binding transcription factors. With continuous kinetic monitoring, incubation can be extended and less nuclear protein may be needed to detect changes in DNA binding. Therefore, kinetic monitoring is expected to be more sensitive than stop reaction methods. An important application of this kinetic method includes screening for drugs that inhibit or activate transcription factor DNA binding 6.
Other possible problems that might arise in the execution of the assay include the presence of high background values. High background values could be due to: (1) high secondary antibody concentrations, (2) insufficient blocking, (3) the use of salmon sperm DNA as blocker, (4) the absence of a blocking protein step or (5) primary or secondary antibodies with low specificity. If these are encountered, several remedies are possible including: (1) optimizing the concentration of secondary antibody in pilot studies and using lower volume of antibody per well, (2) blocking the plate with a basic sodium carbonate solution (3) using dI/dC as non-specific DNA rather than salmon sperm, which may contain promoters with transcription binding elements, or (4) using different pairs of primary or secondary antibodies from different sources.
On the other hand, low signal strength could be caused by (1) low amount of target protein, (2) the concentrations of primary or secondary antibodies are not optimal, (3) excitation and/or emission wavelengths are not optimal, and (4) antibodies have poor affinity for their substrates. Several remedies to these problems include: (1) As part of the troubleshooting, one can use 30 μl low salt buffer + 30 μl high salt buffer if fewer cells are available and the nuclear transcription factor is present in high amounts. Or one could use 90 μl low salt buffer + 90 μl low salt buffer if more cells are available. More cells are useful when expression of the specific factor to be tested is low. (2) Optimize the concentration of primary antibody to increase signal. (3) Monitor the filters on the instrument to make sure they are set for correct excitation and emission maxima for the TMB substrate; alternative fluorescent or chemiluminescent substrates can also be used. (4) If the primary antibody is of low affinity, increase the time of incubation on the plate.
In terms of drug discovery, the current RUNX2 DNA-binding ELISA has been used to detect enhanced binding of a protein to its DNA target and identified natural compounds (such as vitamin D3) that are selective modulators of DNA binding. Some of these compounds exhibit non-competitive mechanisms of action and alter biological function consistent with an interfacial inhibition paradigm 9. With the recent resolution of genomic DNA sequencing, further applications could include discovery of novel proteins interacting with non-coding ("junk") DNA, which regulates expression of coding regions 10.
Disclosures
We have nothing to disclose.
Acknowledgments
The technical assistance and instrumentation of the University of Maryland Greenebaum Cancer Center Translational Core Facility, especially Drs. Rena Lapidus and Mariola Sadowska, are gratefully acknowledged. The work responsible for the development of this assay was funded in part by NIH RO1CA108846, AHA Grant-in-Aid GRNT2130014, a VA Merit Award to A.P., and by the University of Maryland Cigarette Restitution Funds (CRF) provided to the Marlene & Stewart Greenebaum Cancer Center.
References
- Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya N, Sarno A, Idler IS, et al. High-throughput detection of nuclear factor-kappaB activity using a sensitive oligo-based chemiluminescent enzyme-linked immunosorbent assay. Int. J. Cancer. 2010;127:404–411. doi: 10.1002/ijc.25054. [DOI] [PubMed] [Google Scholar]
- Hartzell DD, Trinklein ND, Mendez J, et al. A functional analysis of the CREB signaling pathway using HaloCHIP-chip and high throughput reporter assays. BMC Genomics. 2009;10:497. doi: 10.1186/1471-2164-10-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori KA, Ahlskog JK, Sistonen L, Nikinmaa M. TransLISA, a novel quantitative, nonradioactive assay for transcription factor DNA-binding analyses. FEBS J. 2009;276:7366–7374. doi: 10.1111/j.1742-4658.2009.07446.x. [DOI] [PubMed] [Google Scholar]
- D'Souza DR, Salib MM, Bennett J, et al. Hyperglycemia Regulates RUNX2 Activation and Cellular Wound Healing through the Aldose Reductase Polyol Pathway. J. Biol. Chem. 2009;284:17947–17955. doi: 10.1074/jbc.M109.002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood KF, D'Souza DR, Mochin MT, et al. Regulation of RUNX2 transcription factor-DNA interactions and cell proliferation by Vitamin D3 (cholecalciferol) prohormone activity. J. Bone Miner Res. 2012;27:913–925. doi: 10.1002/jbmr.1504. [DOI] [PubMed] [Google Scholar]
- Held P. Biotek Instruments Application Note. Biotek; 2007. Kinetic analysis of ß-galactosidase activity using the PowerWave HT and Gen5 data analysis software: basic enzyme kinetic determinations. Available from: http://www.biotek.com/resources/docs/B-Gal_Michaelis-Menten_App_Note.pdf. [Google Scholar]
- Renard P, Ernest I, Houbion A, et al. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E. Genomics. ENCODE project writes eulogy for junk DNA. Science. 2012;337:1159–1161. doi: 10.1126/science.337.6099.1159. [DOI] [PubMed] [Google Scholar]