

Abstract
Fenretinide is significantly more effective in inducing apoptosis in cancer cells than all-trans retinoic acid (ATRA). The current study uses a genome-wide approach to understand the differential role fenretinide and ATRA have in inducing apoptosis in Huh7 cells. Fenretinide and ATRA-induced gene expressions and DNA bindings were profiled using microarray and chromatin immunoprecipitation with anti-RXRα antibody. The data showed that fenretinide was not a strong transcription regulator. Fenretinide only changed the expressions of 1 093 genes, approximately three times less than the number of genes regulated by ATRA (2 811). Biological function annotation demonstrated that both fenretinide and ATRA participated in pathways that determine cell fate and metabolic processes. However, fenretinide specifically induced Fas/TNFα-mediated apoptosis by increasing the expression of pro-apoptotic genes i.e., DEDD2, CASP8, CASP4, and HSPA1A/B; whereas, ATRA induced the expression of BIRC3 and TNFAIP3, which inhibit apoptosis by interacting with TRAF2. In addition, fenretinide inhibited the expression of the genes involved in RAS/RAF/ERK-mediated survival pathway. In contrast, ATRA increased the expression of SOSC2, BRAF, MEK, and ERK genes. Most genes regulated by fenretinide and ATRA were bound by RXRα, suggesting a direct effect. This study revealed that by regulating fewer genes, the effects of fenretinide become more specific and thus has fewer side effects than ATRA. The data also suggested that fenretinide induces apoptosis via death receptor effector and by inhibiting the RAS/RAF/ERK pathway. It provides insight on how retinoid efficacy can be improved and how side effects in cancer therapy can be reduced.
Keywords: retinoic acid receptor, retinoid x receptor, nuclear receptor, hepatocellular carcinoma, ChIP-Seq
1. Introduction
Retinoic acid (RA) and synthetic retinoids have potential for the treatment and prevention of cancers (1, 2). However, retinoids do have side effects, which limit their clinical usage. For example, all-trans retinoic acid (ATRA) can cause a life-threatening RA syndrome in acute promyelocytic leukemia (APL) patients (3). The hepatoxicity (3) and resistance (4) of ATRA limits its activity against most types of cancer cells. Conversely, fenretinide, a synthetic derivative of ATRA, is particularly promising as an antitumor agent as it has fewer side effects and can induce cell apoptosis even in ATRA-resistant cell lines (5). Thus, modification of the carboxyl end of ATRA with an N-4-hydroxyphenyl group would result in increased efficacy and reduced toxicity compared to other retinoids (6). Furthermore, fenretinide does not induce point mutations or chromosomal aberrations, and is therefore not genotoxic (7). Fenretinide can prevent high-fat diet induced hepatic steatosis, reduce insulin resistance, ameliorate CCl4-induced liver fibrosis, and inhibit angiogenesis (8–10). Conclusively, fenretinide has specific beneficial effects over classical RA and it is important to understand the mechanism that accounts for such differences.
The mechanism by which fenretinide induces apoptosis is not very well understood. The general consensus is that fenretinide is a reactive oxygen species (ROS) generator and it induces apoptosis via lipid second messengers (11, 12) and activated caspases (13). In contrast, ATRA induces differentiation and cytostasis in target cells, but cannot effectively promote apoptosis (14). Whether the function of fenretinide is receptor dependent is another debated issue. Our recent publication shows that fenretinide-induced apoptosis in the Huh7 cell is dependent on the nuclear export of RARβ and Nur77 (15, 16). To gain insight into the molecular mechanisms by which fenretinide functions as an apoptosis-inducing anti-cancer agent with reduced toxicity, the current study compares the differential effects of fenretinide and ATRA using non-biased genome-wide approaches. Chromatin immunoprecipitation followed by next generation sequencing (ChIP-Seq) was performed using anti-retinoid x receptor α (RXRα) antibody because RXRα is highly expressed in the liver and is a master regulator that dimerizes with retinoic acid receptors (RAR) to regulate gene expression (17, 18). In addition, microarray was also performed. This global transcriptome profiling along with RXRα binding allowed us to establish the relationship between gene expression and DNA binding and thus enabled us to differentiate between the direct vs. indirect effects on gene regulation. The data indicated that although fenretinide and ATRA regulate many common target genes, fenretinide is a weaker transcriptional regulator. It uniquely up-regulates pro-apoptosis gene expression and inhibits RAS/RAF/ERK-mediated survival pathways to exert its apoptotic effect.
2. Materials and Methods
2.1 Reagents
All reagents and chemicals used were from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. TRIzol Reagent, Dynase beads and NP40 cell lysis buffer were purchased from Invitrogen (Invitrogen, CA). RNeasy Mini Kit and High Capacity RNA-to-cDNA Kit were purchased from Qiagen (Qiagen, CA) and Applied Biosystems (Applied Biosystems, CA), respectively. Rabbit polyclonal antibodies specific for RXRα and DDIT3, antibodies for IgG, and mouse polyclonal antibodies specific for caspase 4 as well as HRP labeled anti-rabbit and anti-goat IgG were purchased from Santa Cruz (Santa Cruz, CA). Anti-RNA Polymerase II antibody was purchased from Millipore (Millipore, MA). Mouse monoclonal antibody caspase 8, rabbit polyclonal antibody β-actin, and HRP labeled anti-mouse IgG were purchased from Cell Signaling technology (Cell Signaling technology, MA). Rabbit polyclonal antibodies specific for HERPUD1 and HSPA1A/B were purchased from Lifespan Biosciences (Lifespan Biosciences, WA). Protease and phosphatase inhibitors were purchased from Roche Applied Science (Roche Applied Science, IN). Fenretinide (4-(N-hydroxyphenyl) retinamide) and ATRA were dissolved in DMSO at 1 mM as the stock solution and stored at −20°C in the dark.
2.2 Cell culture and treatment
Human hepatocellular carcinoma Huh7 cells were maintained in Dulbecco’s Modification of Eagle’s Medium (DMEM) (Gibco, VA) and supplemented with 10% fetal bovine serum (Atlanta Biologicals, GA). Cells were plated with approximately 1×106 cells per 100-mm dish or 5×104 cells per well in the 6-well plates overnight prior to treatment. Huh7 cells were treated by fenretinide (10 μM) or ATRA (10 μM) in a serum-free medium. After 3 hrs of treatment, cell pellets were fixed in 1% formaldehyde (pH = 7) and then chromatin was extracted. After 12 hrs of treatment, cells were harvested for RNA preparation. The final concentration of DMSO in the culture medium was 0.1% in all treatments.
2.3 RNA Preparation and Microarray
Total RNA was extracted using TRIzol Reagent (Invitrogen, CA) and purified with the RNeasy Mini Kit (Qiagen, CA). The quantity and quality of the total RNA was assessed by Bioanalyzer 2100 (Agilent Technologies, CA). Complementary DNA was made using High Capacity RNA-to-cDNA Kit (Applied Biosystems, CA). The methods used for performing microarray and data processing were described in our publication (19). The Affymetrix Human Genome U133 plus 2.0 Array GeneChip (Affymetrix, CA) was used. Microarray data were annotated using RMA (Robust Multiarray Average) by PARTEK 6.11. 0701Affymetrix Expression Console (MAS5). The probe signal with p values less than 0.05 were used for further analysis. For data validation, total RNA was isolated and reverse transcripted to cDNA (n=3), and then quantified by qRT-PCR on an ABI 7900HT Fast Real time PCR system (Applied Biosystems, CA) using Power SYBR® Green PCR Master Mix (Applied Biosystems, CA). Primers were designed using Primer3 Input Software (v0.4.0) and the primer sequences were available upon request.
2.4 Western blot
Cells were lysed with NP40 cell lysis buffer (Invitrogen, CA) including protease inhibitors (Roche Applied Science, IN). Proteins (30 μg) were ran on a 12% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, CA). The membrane was incubated with 5% non-fat milk in TBST (10 mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween 20) for 1 hour at room temperature to block nonspecific binding. Immunostaining was performed using antibodies for HSPA1A/B, DDIT3, HERPUD1, caspase 4, caspase 8, and β-actin in TBST-milk overnight at 4 °C. After three washes, membranes were incubated with appropriate secondary antibody for 1 h in TBST-milk. The signal was detected using the ECL system SuperSignal West Pico Chemiluminescent Substrates (Pierce, IL).
2.4 Chromatin immunoprecipitation followed by next generation sequencing (ChIP-Seq) and ChIP-qPCR
Cell pellets were fixed in 1% formaldehyde (pH = 7) for 15 minutes before being quenched with 0.125 M glycine. Following cell lysis, the nuclear part was extracted and sonicated to yield 300–500 base-pair (bp) DNA fragments. Genomic DNA was prepared by treating aliquots of chromatin with RNase and proteinase K, heating for de-crosslinking followed by ethanol precipitation. Aliquot chromatin (30 μg) was precleared by Dynase beads (Invitrogen, CA) before incubation with a ChIP-quality anti-RXRα antibody (Santa Cruz, CA). Antibody to IgG (Santa Cruz, CA) and RNA Polymerase II (Millipore, MA) was used as negative and positive control, respectively. Samples were incubated with prepared Dynase beads at 4 °C overnight followed by de-crosslinking and purification. DNA fragment library was size-selected (175–225 bp) on an agarose gel. Amplified DNAs (DNA libraries) were sequenced on the Illumina Genome Analyzer II (High-seq 2000, Illumina, WI). For ChIP-Seq data validation, DNA fragments generated based on the above mentioned method (n=3) were quantified by real-time PCR with Power SYBR® Green PCR Master Mix (Applied Biosystems, CA). Primers were designed using Primer3 Input Software (v0.4.0) and the primer sequences were available if request.
2.5 Alignment, call peak, and annotation of ChIP-Seq data
Primary image and base calling were processed using Genome Analyzer Pipeline Software (Illumina, CA). All sequenced reads were aligned to Human (Homo sapiens) reference genome using bowtie version 0.12.7 (20). Only uniquely mapped reads were included. Regions with reads enrichment were detected using Model-based Analysis of ChIP-Seq (MACS v 1.4.1) method (21). By comparing with IgG background, non-specific peaks with false discovery ratios (FDR) greater than 0.1 were eliminated. The identified peaks were split by Mali Salmon’s PeakSplitter (http://www.ebi.ac.uk/bertone/software.html) and filtered by p values with Poisson distributions lower than 10−5. The peak annotation was based on the UCSC genome NCBI/hg19 database.
2.6 Motif and Pathway Analysis
The sequences that were 100 bp up and downstream from the summit of the top 500 peaks, which have the highest peak score, were subjected to motif analysis by MEME-ChIP (Multiple Em for Motif Elicitation). Furthermore, a Hidden Markov Model (HMM) was established based on the nuclear receptors binding sites from published work (22) and JASPAR CORE (http://jaspar.cgb.ki.se/cgi-bin/jaspar_db.pl) to predict the specific motif information for all of the RXRα binding sites. All biological function and pathway analyses were generated by the Functional Annotation Tool in the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://www.david.niaid.nih.gov). Functional pathways or processes with p < 0.05 and Bonferroni value < 0.1 were accepted.
2.7 Statistical Analysis
For ChIP-qPCR data and microarray data, the differences between the two groups were analyzed with the Student’s t test. p<0.05 was considered statistically significant.
3. Result
3.1 Microarray data and ChIP-Seq data validation
The expressions of 160 genes, which could be regulated by fenretinide and ATRA, were validated by qRT-PCR. The results showed 95% and 92.5% consistency between qRT-PCR and microarray data for the genes regulated by fenretinide and ATRA, respectively. To validate the ChIP-Seq results, RXRα binding sites with various peak scores were randomly selected for confirmation by ChIP-qPCR. Peak score was used to scale the level of RXRα binding. Overall, ChIP-qPCR was performed for 70 RXRα-bound genes, including 10 binding sites with score>100, 25 binding sites with score between 50 and100, and 35 binding sites with score<50. In fenretinide treated cells, 100%, 90%, and 83% of peaks with strong (score>100), medium (50<score<100), and weak (score<50) bindings could be validated by single gene ChIP-qPCR. Similarly, 100%, 95%, and 88% of peaks with strong (score>100), medium (50<score<100), and weak (score<50) bindings could be confirmed in ATRA treated cells.
3.2 Comparison the effect of fenretinide and ATRA in regulating gene expression
After 12 hrs fenretinide treatment, 1 093 genes (594 up- and 499 down-regulated) had a greater than 1.5-fold change at the mRNA level while ATRA changed the expression of 2,811 genes (> 1.5 folds). The numbers of genes that were up- (1 463) or down- (1 348) regulated by ATRA were close (Fig. 1A). The number of fenretinide-regulated genes was only about one-third of those regulated by ATRA. Eighty-six percent of genes (942) regulated by fenretinide were also regulated by ATRA. Thus, fenretinide and ATRA regulate many common genes.
Fig 1. Comparison of differential gene expression, RXRα binding genes, and binding motif between fenretinide- and ATRA-treated Huh7 cells.
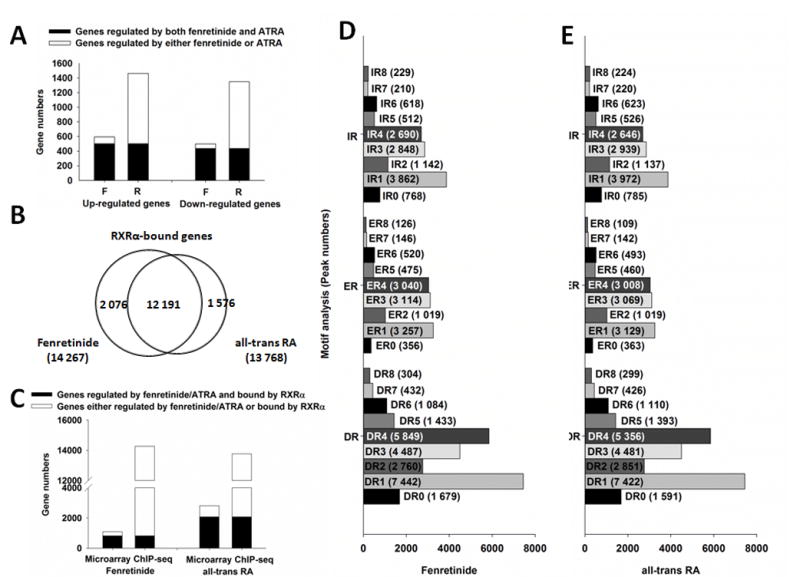
(A) Comparison of fenretinide and all-trans RA based on gene expression. F: fenretinide; R: all-trans RA. (B) Comparison of RXRα binding genes between fenretinide and all-trans RA. (C) Comparison of genes bound by RXRα and regulated by fenretinide and all-trans RA. (D) Motif analysis for genes bound by RXRα and regulated by fenretinide. (E) Motif analysis for genes bound by RXRα and regulated by all-trans RA. For gene expression analysis, total RNA was extracted from Huh7 cells after 12 hrs of fenretinide (10 μM), ATRA (10 μM) or DMSO (control) treatment, followed by microarray analyses. For RXRα binding and motif analysis, Huh7 cells were treated with fenretinide (10 μM) or ATRA (10 μM) for 3 hrs and then subjected to ChIP-Seq assay using IgG (negative control) or anti-RXRα antibody. Global profiling of RXRα binding motifs in Huh-7 cells were predicted by HiddenMarkov Model. DR: direct repeat; ER: everted repeat; IR: inverted repeat.
3.3 Comparison of RXRα binding and binding motif in fenretinide- and ATRA-treated Huh7 cells
In fenretinide- and ATRA-treated Huh7 cells, there were 14 267 and 13 768 RXRα-bound genes, respectively, and most of them (12 191) were the same genes (Fig. 1B). Remarkably, 74.5% (814) of fenretinide-regulated genes and 73.6% (2 069) of ATRA-regulated genes were bound by RXRα (Fig. 1C) suggesting that most of the genes were regulated in an RXRα-dependent manner. The number of RXRα-bound genes was similar in fenretinide- and ATRA-treated Huh7 cells, but the number of the genes regulated by fenretinide was much less, suggesting that binding does not necessarily lead to expression.
Since RXRα dimerizes with multiple nuclear receptors, its binding sites are composed of diverse binding motifs. Motif analysis by Hidden Markov Model confirmed that the RXRα-binding motifs were similar in fenretinide- and ATRA-treated Huh7 cells. Direct repeat with a spacer of one nucleotide (DR1) was the most common motif followed by DR4, DR3, inverted repeat (IR) 1, and everted repeat (ER) 1. Furthermore, spacers 1, 3, and 4 were relatively more prevalent than other spacers (Fig. 1D and E).
3.4 Functional analysis of fenretinide and ATRA based on transcriptome profiling
To study the global role of fenretinide and ATRA, biological function annotation was performed based on the gene expression data. Table 1 shows that proliferation and apoptosis as well as response to wounding, oxidation and reduction etc. were regulated by both compounds. The most striking difference between fenretinide and ATRA was in the lipid/fatty acid metabolic process and the DNA metabolic process. Fenretinide was specifically involved in the biosynthesis of lipid and fatty acid metabolic process; whereas ATRA participated in transcription regulation, DNA metabolic, and response to stress. Among these, an average of 80% and 77% of the genes regulated by fenretinide and ATRA was bound by RXRα, respectively.
Table 1.
Global biological functional comparison of fenretinide and all-trans RA based on gene expression.
| David biological functional annotationa | Fenretinide-regulated genes | RXRα-Bound Genes (%) | p value | All-trans RA- regulated genes | RXRα-Bound Genes (%) | p value |
|---|---|---|---|---|---|---|
|
preferentially regulated by fenretinide
| ||||||
| lipid biosynthetic process | 44 | 30 (68%) | 1.1E-07 | 0 | ||
| fatty acid metabolic process | 26 | 18 (69%) | 1.2E-04 | 0 | ||
| carboxylic acid biosynthetic process (organic acid) | 23 | 18 (78%) | 5.4E-05 | 0 | ||
| carboxylic acid transport (organic acid) | 22 | 18 (82%) | 7.2E-05 | 0 | ||
| amino acid transport | 16 | 14 (88%) | 1.4E-04 | 0 | ||
|
| ||||||
|
preferentially regulated by all-trans RA
| ||||||
| regulation of transcription from RNA polymerase II promoter | 0 | 156 | 130 (83%) | 6.6E-07 | ||
| cellular response to stress | 0 | 126 | 96 (76%) | 1.3E-06 | ||
| DNA metabolic process | 0 | 115 | 85 (74%) | 7.9E-07 | ||
| negative regulation of transcription | 0 | 98 | 89 (91%) | 1.2E-04 | ||
| regulation of locomotion (cell motion) | 0 | 55 | 50 (91%) | 6.2E-06 | ||
| nuclear division (mitosis) | 0 | 54 | 35 (65%) | 8.2E-05 | ||
| response to protein stimulus | 0 | 33 | 25 (76%) | 4.6E-05 | ||
| response to unfolded protein | 0 | 24 | 17 (71%) | 1.4E-04 | ||
|
| ||||||
|
Common processes regulated by fenrentinide and all-trans RA
| ||||||
| cell cycle | 100 | 80 (80%) | 7.2E-08 | 231 | 123 (53%) | 5.7E-13 |
| response to organic substance | 80 | 68 (85%) | 6.1E-09 | 176 | 144 (82%) | 3.2E-12 |
| regulation of cell proliferation | 74 | 60 (81%) | 1.4E-05 | 179 | 159 (89%) | 5.5E-10 |
| regulation of apoptosis | 74 | 67 (91%) | 2.8E-05 | 239 | 190 (80%) | 4.6E-08 |
| oxidation reduction | 62 | 48 (77%) | 3.0E-05 | 132 | 107 (81%) | 3.7E-05 |
| response to wounding | 51 | 44 (86%) | 2.1E-04 | 114 | 100 (88%) | 2.3E-05 |
| response to endogenous stimulus | 47 | 39 (83%) | 4.0E-06 | 100 | 81 (81%) | 1.4E-07 |
| response to extracellular stimulus | 35 | 31 (89%) | 6.9E-08 | 65 | 51 (79%) | 3.1E-08 |
| response to nutrient levels | 34 | 30 (88%) | 1.5E-08 | 62 | 49 (79%) | 5.0E-09 |
| steroid (sterol) metabolic process | 34 | 18 (53%) | 2.7E-08 | 58 | 44 (76%) | 5.3E-07 |
| cholesterol metabolic process | 21 | 16 (76%) | 1.4E-07 | 30 | 21 (70%) | 3.6E-05 |
| steroid (sterol) biosynthetic process | 23 | 15 (65%) | 1.0E-09 | 28 | 16 (57%) | 5.6E-05 |
| cholesterol biosynthetic process | 14 | 10 (71%) | 2.6E-10 | 15 | 8 (53%) | 3.8E-06 |
| response to vitamin | 14 | 12 (86%) | 6.4E-05 | 24 | 21 (88%) | 3.9E-05 |
| glutamine family amino acid metabolic process | 12 | 12 (100%) | 1.2E-04 | 21 | 19 (91%) | 2.4E-05 |
Biological processes were obtained from DAVID function annotation.
Based on the induction or inhibition of the gene expression, the roles of fenretinide and ATRA were compared (Table 2 and 3). Both compounds regulate many common processes. All the sterol and cholesterol metabolic/biosynthetic related genes were down-regulated by both compounds, suggesting that they may play an important role in the metabolism of sterol and cholesterol. Most importantly, many apoptosis related genes were up-regulated and cell cycle related genes were down-regulated by both fenretinide and ATRA. This finding suggests that both fenretinide and ATRA have a role in the control of cell fate and cell metabolism.
Table 2.
Top 10 processes regulated by fenretinide.
| David biological functional annotationa | Fenretinide-regulated genes | RXRα-bound genes (%) | p value |
|---|---|---|---|
|
Up-regulated genes by fenretinide
| |||
| regulation of transcription, DNA-dependent (RNA metabolic process) | 77 | 64 (83%) | 3.5E-04 |
| regulation of apoptosis | 48 | 43 (90%) | 4.3E-06 |
| response to organic substance | 47 | 44 (94%) | 5.1E-07 |
| response to wounding | 33 | 29 (88%) | 8.5E-05 |
| response to nutrient levels(extracelluar stimulus) | 22 | 21 (95%) | 3.2E-07 |
| carboxylic acid transport | 13 | 12 (92%) | 1.3E-03 |
| response to protein stimulus(unfolded protein) | 12 | 11 (92%) | 2.9E-04 |
| cell cycle arrest | 12 | 12 (100%) | 2.1E-04 |
| amino acid transport | 10 | 9 (90%) | 2.9E-04 |
| response to vitamin | 9 | 9 (100%) | 6.5E-04 |
|
| |||
|
Down-regulated genes by fenretinide
| |||
| cell cycle | 60 | 46 (77%) | 3.3E-07 |
| oxidation reduction | 37 | 26 (70%) | 2.1E-05 |
| lipid biosynthetic process | 36 | 25 (69%) | 1.8E-12 |
| DNA metabolic process | 36 | 26 (72%) | 2.8E-07 |
| steroid (sterol) metabolic process | 28 | 21 (75%) | 5.3E-12 |
| steroid (sterol) biosynthetic process | 22 | 15 (68%) | 5.1E-15 |
| DNA replication | 22 | 16 (73%) | 4.0E-08 |
| cholesterol metabolic process | 19 | 14 (74%) | 3.3E-11 |
| fatty acid metabolic process | 17 | 13 (76%) | 8.8E-05 |
| cholesterol biosynthetic process | 14 | 10 (71%) | 2.3E-14 |
Biological processes were obtained from DAVID function annotation.
Table 3.
Top 10 processes regulated by all-trans RA.
| David biological functional annotation.a | All-trans RA-regulated genes | RXRα-bound genes (%) | p value |
|---|---|---|---|
|
Up-regulated genes by all-trans RA
| |||
| regulation of transcription, DNA-dependent (RNA polymerase II promoter) | 175 | 140 (80%) | 1.6E-05 |
| regulation of apoptosis | 142 | 121 (85%) | 3.4E-08 |
| response to organic substance | 88 | 78 (87%) | 1.8E-06 |
| intracellular transport | 82 | 66 (80%) | 1.8E-06 |
| vesicle-mediated transport | 70 | 55 (79%) | 2.7E-05 |
| response to extracellular stimulus | 37 | 28 (76%) | 3.7E-06 |
| response to nutrient levels | 36 | 28 (78%) | 7.2E-07 |
| regulation of locomotion (cell migration) | 34 | 32 (94%) | 1.5E-06 |
| response to unfolded protein | 17 | 14 (82%) | 4.0E-05 |
| response to endoplasmic reticulum stress | 12 | 9 (75%) | 1.8E-05 |
|
| |||
|
Down-regulated genes by all-trans RA
| |||
| cell cycle | 143 | 105 (73%) | 4.8E-14 |
| regulation of cell proliferation | 94 | 82 (87%) | 4.0E-06 |
| response to organic substance | 88 | 65 (74%) | 3.7E-06 |
| oxidation reduction | 83 | 62 (75%) | 6.0E-07 |
| DNA metabolic process | 79 | 52 (66%) | 3.7E-10 |
| cellular response to stress | 70 | 49 (70%) | 2.7E-05 |
| response to endogenous stimulus (hormone stimulus) | 58 | 45 (78%) | 2.0E-06 |
| lipid biosynthetic process | 49 | 28 (57%) | 2.8E-06 |
| cell division | 47 | 29 (62%) | 1.2E-06 |
| mitosis(nuclear division) | 44 | 27 (61%) | 3.5E-09 |
Biological processes were obtained from DAVID function annotation.
3.5 Fenretinide and ATRA regulate genes that involved in apoptosis, proliferation, and cell cycle
To understand the differential effects of fenretinide and ATRA in regulating cell fate, specific genes involved in the apoptosis pathways were analyzed. Fenretinide regulated fewer apoptosis-related genes than ATRA (74 vs. 239 genes). Pro-apoptotic genes DDIT3, NUPR1, JMY, TGFB2, and HERPUD1 were up-regulated by both fenretinide and ATRA after 12 hrs of treatment (Fig. 2A). Fenretinite uniquely induced four pro-apoptotic genes including DEDD2, CASP8, and CASP4, which are involved in the Fas/TNFα-mediate death receptor signaling pathway (Fig. 2B). In contrast, ATRA up-regulated two anti-apoptotic genes (BIRC3 and TNFAIP3), which have negative regulatory effects in the Fas/TNFα-mediate death receptor signaling pathway. In addition, ATRA induced the expression of many anti-apoptotic genes (SOCS2, BIRC3, BRAF, TNFAIP3, and XIAP) and suppressed the expression of pro-apoptotic genes (PCSK9, CITED2, TIMP2, and CASP6) by more than 2-fold (Fig. 2C).
Fig 2. The effect of fenretinide and all-trans RA on apoptosis-related genes.
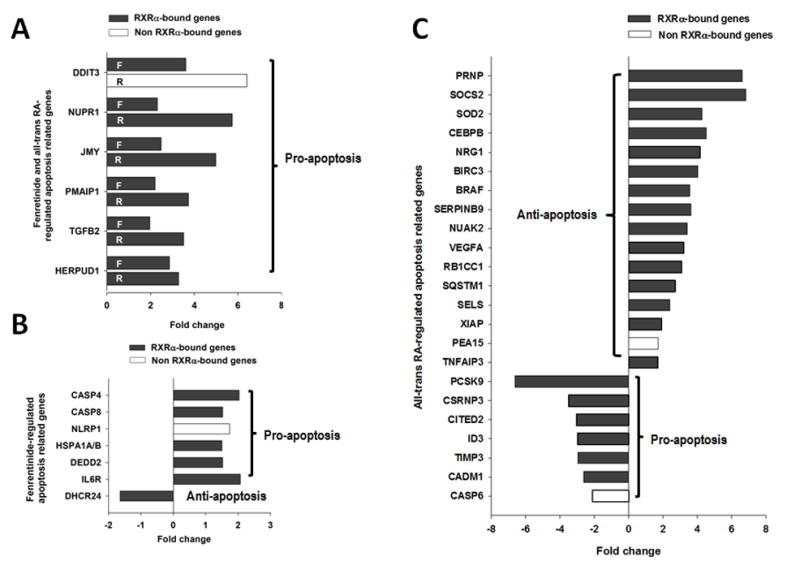
(A) Common apoptosis-related genes regulated by fenretinide and all-trans RA in Huh7 cell. (B) The apoptosis-related genes regulated by fenretinide. (C) The apoptosis-related genes regulated by all-trans RA. Total RNA was extracted from Huh7 cells treated with fenretinide (10 μM), ATRA (10 μM) or DMSO for 12 hrs and subjected to microarray assay.
In cell proliferation pathways, both compounds increased the expression of two anti-proliferation genes (CTH and KLF4) and decreased the expression of six proliferation genes (PLAU, CXCL10, MYCN, VASH2, PGF, and ICMT) (Fig. 3A). However, ATRA had positive effects in regulating the proliferation process. For example, ATRA uniquely increased the expression of proliferation genes ATF3, IL8, EREG, CCL15, HOXA3, HEY2 and TIMP2 and inhibited the expression anti-proliferation genes such as ID4, PROX1, KLF5, CAV2, and TGFBR2 (Fig. 3B). In addition, most of those genes had RXRα binding. These findings suggest the role of ATRA in cell proliferation.
Fig 3. The effect of fenretinide and all-trans RA on proliferation-related genes.
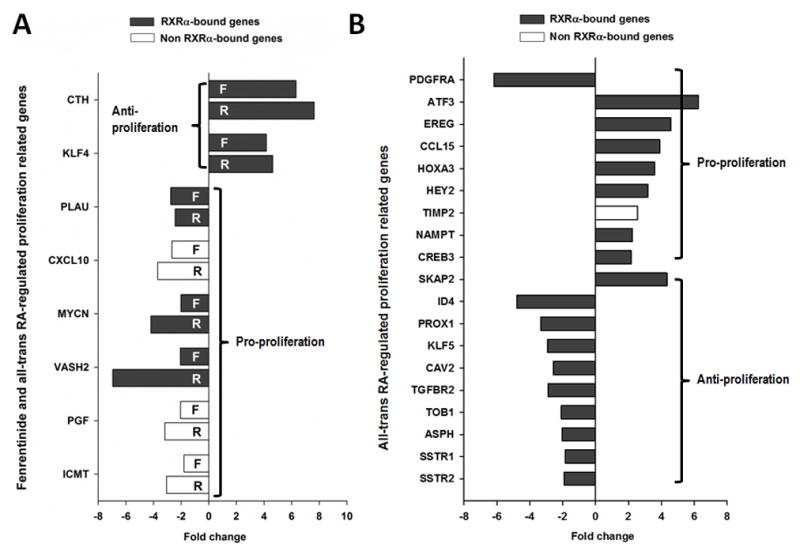
(A) Common proliferation-related genes regulated by both fenretinide and all-trans RA. (B) The proliferation-related genes regulated by all-trans RA. Total RNA was extracted from Huh7 cells treated with fenretinide (10 μM), ATRA (10 μM) or DMSO for 12 hrs and subjected to microarray assay.
As for cell cycle processes, both compounds up-regulated genes that are involved in cell cycle arrest. For example, SESN2, GADD45A, GADD45B, CDKN2B, and IL8 were up-regulated. Both compounds also inhibited the expression of genes that contribute to cell cycle progression (E2F, CCNE1 NEK6, MCM2, MCM3, and MCM6) (Fig. 4A). However, ATRA uniquely up-regulated genes involved in cell cycle stimulation. Those genes include RHOU, HERC5, PDS5A, CDC23, CDC16, and CDC73. In addition, ATRA also uniquely inhibited genes including CDKN2C, HPGD, FAM5C, RGS2, GAS2 and PSMD11that participate in cell cycle arrest (Fig. 4B). Unlike ATRA, fenretinide could not regulate any of those genes. These findings suggest the ATRA’s role in supporting cell cycle progression.
Fig 4. The effect of fenretinide and all-trans RA on cell cycle-related genes.
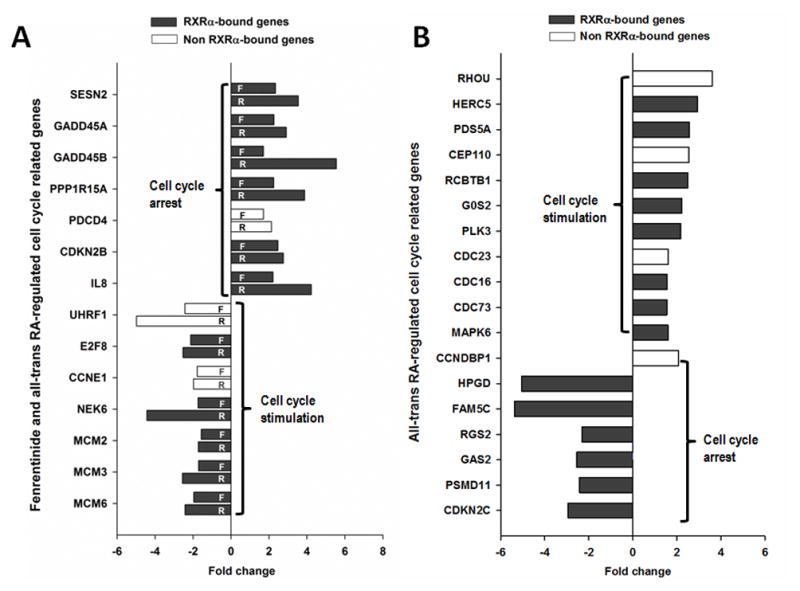
(A) Common cell cycle-related genes regulated by both fenretinide and all-trans RA. (B) The cell cycle-related genes preferentially regulated by all-trans RA. Total RNA was extracted from Huh7 cells treated with fenretinide (10 μM), ATRA (10 μM) or DMSO for 12 hrs and subjected to microarray assay.
3.6 Temporal profiling of the effect of fenretinide and ATRA on the expression of genes in death and survival pathways
Since gene regulation-associated phenotype change is a dynamic process, a time course experiment was performed. The genes studied are those involved in the Fas/TNFα-mediated signaling pathway. Figure 5A shows that many studied genes were up-regulated in the early studied time points and the fold changes were even higher than that at later time points. For example, six and one hr after ATRA treatment, the expression of anti-apoptotic genes BIRC3 and TNFAIP3 was induced, respectively; whereas fenretinide only modestly induced the expression of TNFAIP3 at 12 hrs. ATRA induced survival pathway via the induction of SOCS2, which is a positive regulator for IGF-R1 (23). SOCS2 is also the upstream regulator of MEK1 and ERK2, which were up-regulated by ATRA at 3 hrs (Fig. 5A). In contrast to ATRA, fenretinide had a greater impact on the induction of Fas/TNFα-mediated apoptosis pathway by up-regulating pro-apoptotic genes such as HSPA1A/B, DEDD2, CASP8, and CASP4 (Fig. 5B). Other pro-apoptotic genes including HERPUD1 and DDIT3 were able to be induced by both compounds at multiple studied time points (Fig. 5B). Furthermore, fenretinide inhibited cell survival by decreasing the expression levels of BRAF in the RAS/RAF/ERK mediated survival pathway (Fig. 5A). These findings clearly indicated that ATRA and fenretinide differentially regulate the Fas/TNFα-mediate death receptor signaling pathway and the RAS/RAF/ERK survival pathway.
Fig 5. The effect of fenretinide and all-trans RA on anti-apoptosis genes (A) and pro-apoptosis genes (B) by qRT-PCR.

Total RNA was extracted from Huh7 cells treated with fenretinide (10 μM), ATRA (10 μM), or DMSO for 30 min, 1, 2, 3, 6, and 12 hrs, and the gene expressions were studied by real-time RT-PCR and normalization to that of GAPDH. Data were expressed as mean ± SD from three independent experiments.
3.8 The effects of fenretinide and ATRA on expression levels of pro-apoptotic proteins
A time course experiment was performed to study the effect of fenretinide and ATRA on the expression of pro-apoptotic proteins, HSPA1A/B, caspase 8, caspase 4, HERPUD1, and DDIT3. The data showed that fenretinide and ATRA induced the levels of those five proteins. However, fenretinide had a much greater effect than ATRA in increasing the levels of HSPA1A/B, caspase 8, and capsase 4 as well as HERPUD1. Only fenretinide, and not ATRA, had a time-dependent effect in inducing the protein levels of HSPA1A/B and caspase 8 (Fig. 6). ChIP-qPCR data showed that RXRα bound to HSPA1A/B, caspase 8, caspase 4, HERPUD1, and DDIT3 in fenretinide-treated Huh7 cells (3 hrs). However, RXRα only bound to the caspase 8, caspase 4, and HERPUD1 genes, but not HSPA1A/B and DDIT3, in ATRA-treated cells (data not shown). Thus, binding does not guarantee expression, but expression requires binding.
Fig 6. The effect of fenretinide and all-trans RA on protein levels of HSPA1A/B, caspase 8, caspase 4, HERPUD1, and DDIT3.
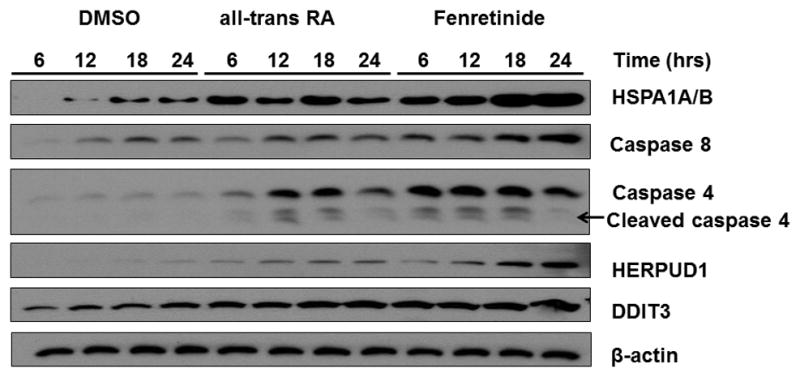
Huh7 cells were treated with fenretinide (10 μM), ATRA (10 μM), or DMSO for 6, 12, 18, and 24 hrs. The protein levels were analyzed by western blot using specific antibodies as described in Section 2. β-actin level was used for loading control. Representative data are shown from three independent experiments. Arrow indicated the position of cleaved caspase 4.
3.8 The effects of fenretinide and ATRA on cytochrome P450 gene expression
The oxidation of RA by CYP26A1 and CYP26B1 is an important mechanism to control the levels of RA in cells and tissues (24, 25). Fig. 7 shows that ATRA highly induced the mRNA levels of CYP26A1 and CYP26B1 at early time points. The induction of mRNA levels of CYP26A1 (19.8-fold) and CYP26B1 (11.8-fold) started at one and two hrs after treatment, respectively. It reached to 1261.4-fold for CYP26A1 and 69.5-fold for CYP26B1 12 hrs after treatment. In contrast, the induction fold of these two genes by fenretinide was much less. The maximal induction was 16.1 and 8.3 folds for CYP26A1 and CYP26B1, respectively, 6 hrs after fenretinide treatment. These findings suggest that fenretinide might be more stable than ATRA in Huh7 cells.
Fig 7. The effect of fenretinide and all-trans RA on CYP26A1 (A) and CYP26B1 (B) gene expression.
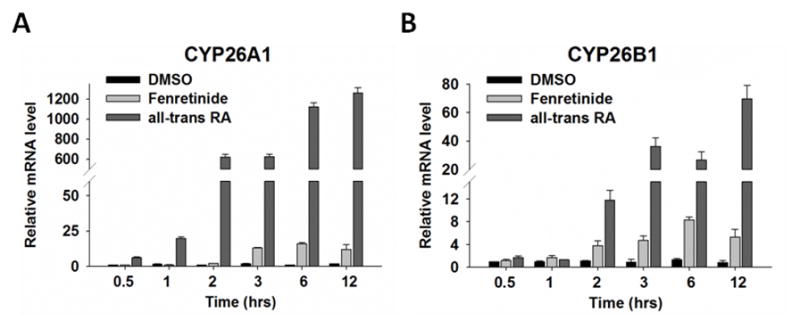
Huh7 cells were treated with fenretinide (10 μM) or ATRA (10 μM) for 30 min, 1, 2, 3, 6, and 12 hrs. Data were expressed as mean ± SD from three independent experiments.
4. Discussion
Although fenretinide and ATRA regulate many common genes, these two compounds have their unique roles. In comparison with ATRA, fenretinide not only has strong apoptosis effects, it also has fewer side effects. The pro-apoptotic effect of fenretinide has been shown in various cancer cell lines including HeLa and breast cancer cell lines, whereas ATRA has shown no growth inhibition effect in these cell lines (26). Fenretinide also induces apoptosis/necrosis in primary Wilm’s tumor cell, but ATRA promotes cell growth (27). The apoptosis effect of fenretinide is cancer cell specific, it does not induce apoptosis in normal hepatocyte (15). In contrast, RA has diverse effects. It is used to treat acute promyelocytic leukemia due to its differentiation effect, but it also has a mild mitogenic effect (3). In vivo study shows that ATRA induces hepatomegaly (28), and compromises liver regeneration in hepatocyte RXRα-deficient livers (29) and in vitamin A-deficient rats (30). These findings indicate the importance of retinoic acid-mediated signaling in liver cell proliferation (31). Despite efforts in identifying fenretinite and ATRA’s role in various cancer cell lines, little is known about the molecular mechanisms by which these two compounds exert their distinct effects. This study might be the first to use a non-biased global approach to determine the differential role. Our findings indicate that fenretinide is a much weaker transcription regulator. By regulating fewer genes, fenretinide has specific effects. Futhermore, although both compounds regulated many common pathways, they have different effects in regulating death and survival genes and thus have distinct roles.
The biochemical pathways leading to fenretinide-induced apoptosis are complex. Whether the action is retinoid receptor-dependent (26, 32) or -independent (5, 33–35) has been a long debated issue. Cells that express mutated RAR are still able to respond to fenretinide suggesting a receptor-independent mechanism (36). However, other studies showed that RARs and RAR antagonists partially inhibit fenretinide-induced apoptosis implying a receptor-dependent mechanism (37). Using a gene knockdown approach, our published data indicated that fenretinide-induced apoptosis of human liver cancer cells is RARβ and Nur77 dependent (16, 32). Furthermore, the sensitivity of the cancer cells to fenretinide-induced apoptosis is positively associated with cytoplasmic enrichment of Nur77 that forms complex with RARβ (15). The current study shows that the majority of the genes regulated by ATRA and fenretinide have the RXRα binding sites implying an RXRα-dependent mechanism. It is likely that both compounds regulate gene expression in an RXRα-dependent manner. Their role in regulating apoptosis and cell survival may require the coordinated effort of multiple nuclear receptors.
It is important to note that HSPA1A/B mRNA was only induced by fenretinide. HSPA1A/B is an endoplasmic reticulum (ER)-stress-induced protein that promotes TNF-mediated apoptosis (38). Thus, ROS generation may also play a role in fenretinide-induced apoptosis. Consistently, fenretinide increased the mRNA and protein level of HSPA1A in human head and neck cancer cells. Hence, chemical inhibition and siRNA-mediated silencing of HSPA1A decrease fenretinide-induced apoptosis (38). In addition to HSPA1A/B, fenretinide also up-regulates other pro-apoptosis genes i.e. DEDD2, CASP8 and CASP4, all of which are involved in the Fas/TNFα-mediated apoptosis pathway. In agreement with this finding, fenretinide-induced apoptosis is also involved in the caspase 8 activation in ovarian cells (13). These data suggest that Fas/TNFα death receptor-mediated signaling is responsible for fenretinide-induced apoptosis in Huh7 cells. In contrast, ATRA induces the expression of BIRC3 and TNFAIP3, which inhibit Fas/TNFα-mediated apoptosis signaling pathway by the interaction with TRAF2, and thus may prevent apoptosis. Furthermore, ATRA inhibits the expression of PCSK9 (7.1 fold reduction), which encodes a novel member of the subtilisin class of proteinase-K-like serine proteinases (39). PCK9 is involved in apoptosis through modulation of the intrinsic apoptotic pathway. Knocking down the expression of PCSK9 inhibits apoptosis by reducing caspase activity (40, 41). Thus, ATRA-reduced PCSK9 may suppress the Fas/TNFα-mediated apoptosis pathway through the reduction of caspase activity. In contrast to the 7.1 fold reduction by ATRA, fenretinide only reduces PCSK9 mRNA levels by 1.7 fold. A schematic of our findings that show the differential mechanism by which fenretinide and ATRA regulate cell death is summarized in Fig. 8A.
Fig 8. Schematic presentations for the differential effects of fenretinide and all-trans RA in regulating apoptosis and survival of Huh7 cells.

(A) Regarding the Fas/TNFα-mediate apoptosis pathway, fenretinide up-regulated the expression of three pro-apoptotic genes including DEDD, CASP8, and CASP4, which subsequently triggered the pathway; Additionally, fenretinide also induced apoptosis by up-regulating pro-apoptotic genes HSPA1A/B, which is activated by ER stress and promotes TNF-mediated apoptosis. In contrast, ATRA induced the expression of two anti-apoptotic genes BIRC3 and TNFAIP3, which negatively regulated the Fas/TNFα-mediate signaling pathway, and eventually inhibited Fas/TNFα-mediated apoptosis. The repressed expression of PCSK9, which play an important role in caspase-3 activation, also decreased ATRA-mediated apoptosis. (B) Regarding the RAS/RAF/ERK-medicated survival pathway, fenretinide repressed the expression of BRAF, and therefore decreased the expression levels of MEK1 and ERK2, which finally inhibited cell survival. Conversely, ATRA up-regulated the expression of SOSC2, successively increased the expression of three key genes: BRAF, MEK1 and ERK2, and therefore induced the RAS/RAF/ERK-mediated survival pathway. Additionally, ATRA promoted the NFkB-dependent survival pathway by repressing CITE2 gene expression. “→” indicates induction and “⊥” indicates repression. The genes marked in green circle indicate pro-apoptosis genes. The genes marked in red circle indicate anti-apoptosis genes. The numbers in bracket represent the fold-change of gene expression changed by fenretinide or ATRA at corresponding time-point.
Fenretinide and ATRA have distinct effects in regulating genes involved in RAS/RAF/ERK mediated survival pathway. Fenretinide inhibits the expression of BRAF, which is upstream of MEK1/ERK2. Consistent with our previous finding, which shows fenretinide induces Huh7 cell death by deactivation of ERK1/2, our published data also showed that the deactivation of ERK1/2 can sensitize fenretinide-resistant liver cancer cells to become sensitive to the apoptotic effects of fenretinide (42). Conversely, ATRA significantly induces the expression of BARF, MEK1, and ERK2. In addition, the expression of SOCS2, downstream of IGF-R1, is also up-regulated by ATRA. The induction of those genes is transient and takes place at relatively early time points (2–3 hrs). The expression of CITED2, a transcriptional repressor of NFkB (43), is reduced by ATRA (5.9-fold). Reduced expression of CITED2 prohibits its ability to repress NFkB, and therefore promotes the NFkB-dependent survival pathway. In comparison, fenretinide does not show any effect on CITED2’s gene expression. A schematic of our current understanding of the differential mechanism by which fenretinide and ATRA-regulates the survival signaling pathway is shown in Fig. 8B. Taken together, ATRA promotes Huh7 cell survival via the RAS/RAF/ERK signaling pathway. These findings not only explain why fenretinide induces apoptosis more effectively than ATRA, they also explain why ATRA has a mitogenic effect.
Among the cell cycle regulated genes, the expressions of CCNE1 and E2F, which facilitate G1/S transition, are down-regulated by fenretinide. Consistently, MCM2, MCM3, and MCM6 are also down-regulated by fenretinide. Loss of MCM causes G1 arrest (44). Conversely, GADD45A (45) and GADD45B (46), which control G2/M arrest, are up-regulated by fenretinide. Essentially, fenretinide-induced growth inhibition is associated with the accumulation of cells in the G1/S and G2/M phases of the cell cycle. In other words, inhibition of Huh7 cell growth by fenretinide is a consequence of blocking the cell cycle at G1/S and G2/M transitions. These results are in agreement with the results demonstrated using PC-3 cells; suppression of PC-3 cell growth by fenretinide is linked to the accumulation of G1 phase cells (47).
In conclusion, the presented data provide a fundamental understanding of how retinoids control cell fate. This study reveals that the distinct roles of the two retinoids lie in their ability in regulating apoptosis and survival. Fenretinide induces apoptosis while ATRA inhibits apoptosis via the death receptor effectors. Similarly, ATRA induces survival while fenretinide reduces survival via RAS/RAF/ERK-mediated pathway. It would be important to further understand how chemical structure changes can result in such drastic alterations in the control of cell fate. The addition of ERK1/2 inhibitor along with fenretinide should have a better effect in inducing apoptosis in cancer cells. We predict that the dose as well as the side effects during cancer treatment can be reduced when a combination of ERK1/2 inhibitor and fenretinide is used.
Acknowledgments
Grant Support
This study is supported by grants funded by National Institutes of Health CA53596 and DK092100.
This study is supported by grants funded by National Institutes of Health CA53596 and DK092100. We thank Dr. Stan Svojanovsky, the associate professor in Molecular & Integrative Physiology of Kansas Medical Center, for processing the microarray data. We also thank Julia Ann Wu and Jessica Tsuei for their assistant in preparation of this manuscript.
Abbreviations
- APL
acute promyelocytic leukemia
- ATRA
all-trans retinoic acid
- ChIP-Seq
chromatin immunoprecipitation followed by next generation sequencing
- DAVID
the Database for Annotation, Visualization and Integrated Discovery
- DR
direct repeat
- ER
everted repeat
- ER stress
endoplasmic reticulum stress
- ERK
extracellular-signal-regulated kinase
- FDR
false discovery ratio
- IR
inverted repeat
- RA
retinoic acid
- RAR
retinoic acid receptor
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- qRT-PCR
real-time quantitative reverse transcription PCR
- qPCR
quantitative PCR
- TNF
tumor necrosis factor
Footnotes
Authors’ Contributions
YH: Performed experiments, analyzed data, generated figures and tables as well as prepared manuscript.
HXL: ChIP-Seq and microarray data validation and assisted in ChIP experiment.
YH: Assisted in statistical analysis of ChIP-Seq.
YF: ChIP-Seq data analyses that include alignment, call peak, annotation, and motif analysis
JF: ChIP-Seq data analyses that include alignment, call peak, annotation, and motif analysis.
YJY W: Generated idea and supervised the all overall performance of the project. Final approval and overall responsibility for the published work.
Disclosure of Potential Conflicts of Interest
The authors declare that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ying Hu, Email: yinghu0821@gmail.com.
Hui-Xin Liu, Email: huixinliu2012@gmail.com.
Yuqi He, Email: hyqjeff@gmail.com.
Yaping Fang, Email: ypfang@ku.edu.
Jianwen Fang, Email: jwfang@ku.edu.
Yu-Jui Yvonne Wan, Email: yjywan@ucdavis.edu.
References
- 1.Choi Y, Kim SY, Kim SH, Yang J, Park K, Byun Y. Inhibition of tumor growth by biodegradable microspheres containing all-trans-retinoic acid in a human head-and-neck cancer xenograft. Int J Cancer. 2003;107:145–8. doi: 10.1002/ijc.11354. [DOI] [PubMed] [Google Scholar]
- 2.Di C, Liao S, Adamson DC, Parrett TJ, Broderick DK, Shi Q, et al. Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all-trans retinoic acid. Cancer Res. 2005;65:919–24. [PubMed] [Google Scholar]
- 3.de-Medeiros BC, Strapasson E, Pasquini R, de-Medeiros CR. Effect of all-trans retinoic acid on newly diagnosed acute promyelocytic leukemia patients: results of a Brazilian center. Braz J Med Biol Res. 1998;31:1537–43. doi: 10.1590/s0100-879x1998001200005. [DOI] [PubMed] [Google Scholar]
- 4.Tari AM, Lim SJ, Hung MC, Esteva FJ, Lopez-Berestein G. Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast cancer cells. Oncogene. 2002;21:5224–32. doi: 10.1038/sj.onc.1205660. [DOI] [PubMed] [Google Scholar]
- 5.Kitareewan S, Spinella MJ, Allopenna J, Reczek PR, Dmitrovsky E. 4HPR triggers apoptosis but not differentiation in retinoid sensitive and resistant human embryonal carcinoma cells through an RARγ independent pathway. Oncogene. 1999;18:5747–55. doi: 10.1038/sj.onc.1202981. [DOI] [PubMed] [Google Scholar]
- 6.Hail N, Jr, Kim HJ, Lotan R. Mechanisms of fenretinide-induced apoptosis. Apoptosis. 2006;11:1677–94. doi: 10.1007/s10495-006-9289-3. [DOI] [PubMed] [Google Scholar]
- 7.Paulson JD, Oldham JW, Preston RF, Newman D. Lack of genotoxicity of the cancer chemopreventive agent N-(4-hydroxyphenyl)retinamide. Fundam Appl Toxicol. 1985;5:144–50. doi: 10.1016/0272-0590(85)90058-2. [DOI] [PubMed] [Google Scholar]
- 8.Qian J, Zhang JS, Wang XQ, Ji JL, Mei S. Fenretinide stimulates the apoptosis of hepatic stellate cells and ameliorates hepatic fibrosis in mice. Hepatol Res. 2009;39:1229–47. doi: 10.1111/j.1872-034X.2009.00562.x. [DOI] [PubMed] [Google Scholar]
- 9.Preitner F, Mody N, Graham TE, Peroni OD, Kahn BB. Long-term Fenretinide treatment prevents high-fat diet-induced obesity, insulin resistance, and hepatic steatosis. Am J Physiol Endocrinol Metab. 2009;297:E1420–9. doi: 10.1152/ajpendo.00362.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sogno I, Vene R, Ferrari N, De Censi A, Imperatori A, Noonan DM, et al. Angioprevention with fenretinide: targeting angiogenesis in prevention and therapeutic strategies. Crit Rev Oncol Hematol. 2010;75:2–14. doi: 10.1016/j.critrevonc.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 11.McCormick DL, Moon RC. Antipromotional activity of dietary N-(4-hydroxyphenyl)retinamide in two-stage skin tumorigenesis in CD-1 and SENCAR mice. Cancer Lett. 1986;31:133–8. doi: 10.1016/0304-3835(86)90003-0. [DOI] [PubMed] [Google Scholar]
- 12.You KR, Wen J, Lee ST, Kim DG. Cytochrome c oxidase subunit III: a molecular marker for N-(4-hydroxyphenyl)retinamise-induced oxidative stress in hepatoma cells. J Biol Chem. 2002;277:3870–7. doi: 10.1074/jbc.M109284200. [DOI] [PubMed] [Google Scholar]
- 13.Kalli KR, Devine KE, Cabot MC, Arnt CR, Heldebrant MP, Svingen PA, et al. Heterogeneous role of caspase-8 in fenretinide-induced apoptosis in epithelial ovarian carcinoma cell lines. Mol Pharmacol. 2003;64:1434–43. doi: 10.1124/mol.64.6.1434. [DOI] [PubMed] [Google Scholar]
- 14.Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–7. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Zhan Q, Wan YJ. Enrichment of Nur77 mediated by retinoic acid receptor beta leads to apoptosis of human hepatocellular carcinoma cells induced by fenretinide and histone deacetylase inhibitors. Hepatology. 2011;53:865–74. doi: 10.1002/hep.24101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Bushue N, Bu P, Wan YJ. Induction and intracellular localization of Nur77 dictate fenretinide-induced apoptosis of human liver cancer cells. Biochem Pharmacol. 2010;79:948–54. doi: 10.1016/j.bcp.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Luca LM. Retinoids and Their Receptors in Differentiation, Embryogenesis, and Neoplasia. FASEB Journal. 1991;5:2924–33. [PubMed] [Google Scholar]
- 18.Shulman AI, Mangelsdorf DJ. Mechanisms of disease: Retinoid X receptor heterodimers in the metabolic syndrome. N Engl J Med. 2005;353:604–15. doi: 10.1056/NEJMra043590. [DOI] [PubMed] [Google Scholar]
- 19.Guo M, Gong L, He L, Lehman-McKeeman L, Wan YJ. Hepatocyte RXRalpha deficiency in matured and aged mice: impact on the expression of cancer-related hepatic genes in a gender-specific manner. BMC genomics. 2008;9:403. doi: 10.1186/1471-2164-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandelin A, Wasserman WW. Prediction of nuclear hormone receptor response elements. Mol Endocrinol. 2005;19:595–606. doi: 10.1210/me.2004-0101. [DOI] [PubMed] [Google Scholar]
- 23.Dey BR, Spence SL, Nissley P, Furlanetto RW. Interaction of human suppressor of cytokine signaling (SOCS)-2 with the insulin-like growth factor-I receptor. J Biol Chem. 1998;273:24095–101. doi: 10.1074/jbc.273.37.24095. [DOI] [PubMed] [Google Scholar]
- 24.Abu-Abed S, MacLean G, Fraulob V, Chambon P, Petkovich M, Dolle P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech Dev. 2002;110:173–7. doi: 10.1016/s0925-4773(01)00572-x. [DOI] [PubMed] [Google Scholar]
- 25.Villani MG, Appierto V, Cavadini E, Valsecchi M, Sonnino S, Curley RW, et al. Identification of the fenretinide metabolite 4-oxo-fenretinide present in human plasma and formed in human ovarian carcinoma cells through induction of cytochrome P450 26A1. Clin Cancer Res. 2004;10:6265–75. doi: 10.1158/1078-0432.CCR-04-0655. [DOI] [PubMed] [Google Scholar]
- 26.Fanjul AN, Delia D, Pierotti MA, Rideout D, Yu JQ, Pfahl M. 4-Hydroxyphenyl retinamide is a highly selective activator of retinoid receptors. J Biol Chem. 1996;271:22441–6. doi: 10.1074/jbc.271.37.22441. [DOI] [PubMed] [Google Scholar]
- 27.Wegert J, Bausenwein S, Kneitz S, Roth S, Graf N, Geissinger E, et al. Retinoic acid pathway activity in Wilms tumors and characterization of biological responses in vitro. Mol cancer. 2011;10:136. doi: 10.1186/1476-4598-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perea G, Salar A, Altes A, Brunet S, Sierra J. Acute hepatomegaly with severe liver toxicity due to all-trans-retinoic acid. Haematologica. 2000;85:551–2. [PubMed] [Google Scholar]
- 29.Yang X, Guo M, Wan YJ. Deregulation of growth factor, circadian clock, and cell cycle signaling in regenerating hepatocyte RXRα-deficient mouse livers. Am J Pathol. 2010;176:733–43. doi: 10.2353/ajpath.2010.090524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu Z, Fujio K, Marsden ER, Thorgeirsson SS, Evarts RP. Hepatic regeneration in vitamin A-deficient rats: changes in the expression of transforming growth factor alpha/epidermal growth factor receptor and retinoic acid receptors!! and ! Cell Growth Differ. 1994;5:503–8. [PubMed] [Google Scholar]
- 31.Bushue N, Wan YJ. Retinoic Acid-mediated Nuclear Receptor Activation and Hepatocyte Proliferation. J Exp Clin Med. 2009;1:23–30. doi: 10.1016/S1878-3317(09)60007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bu P, Wan YJ. Fenretinide-induced apoptosis of Huh-7 hepatocellular carcinoma is retinoic acid receptor beta dependent. BMC Cancer. 2007;7:236. doi: 10.1186/1471-2407-7-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22:7305–15. doi: 10.1038/sj.onc.1206936. [DOI] [PubMed] [Google Scholar]
- 34.Chiantore MV, Giandomenico V, De Luca LM. Carcinoma cell lines resistant for growth inhibition and apoptosis to retinoic acid are responsive to 4-hydroxy-phenyl-retinamide: correlation with tissue transglutaminase. Biochem Biophys Res Commun. 1999;254:636–41. doi: 10.1006/bbrc.1998.9987. [DOI] [PubMed] [Google Scholar]
- 35.Clifford JL, Menter DG, Wang M, Lotan R, Lippman SM. Retinoid receptor-dependent and -independent effects of N-(4-hydroxyphenyl)retinamide in F9 embryonal carcinoma cells. Cancer Res. 1999;59:14–8. [PubMed] [Google Scholar]
- 36.Delia D, Aiello A, Lombardi L, Pelicci PG, Grignani F, Formelli F, et al. N-(4-hydroxyphenyl)retinamide induces apoptosis of malignant hemopoietic cell lines including those unresponsive to retinoic acid. Cancer Res. 1993;53:6036–41. [PubMed] [Google Scholar]
- 37.Sun SY, Li W, Yue P, Lippman SM, Hong WK, Lotan R. Mediation of N-(4-hydoxyphenyl)retinamide-induced apoptosis in human cancer cells by different mechanisms. Cancer Res. 1999;59:2493–8. [PubMed] [Google Scholar]
- 38.Kadara H, Lacroix L, Lotan D, Lotan R. Induction of endoplasmic reticulum stress by the pro-apoptotic retinoid N-(4-hydroxyphenyl)retinamide via a reactive oxygen species-dependent mechanism in human head and neck cancer cells. Cancer Biol Ther. 2007;6:705–11. doi: 10.4161/cbt.6.5.3963. [DOI] [PubMed] [Google Scholar]
- 39.Naureckiene S, Ma L, Sreekumar K, Purandare U, Lo CF, Huang Y, et al. Functional characterization of Narc 1, a novel proteinase related to proteinase K. Arch Biochem Biophys. 2003;420:55–67. doi: 10.1016/j.abb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 40.Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem. 2012;359:347–58. doi: 10.1007/s11010-011-1028-6. [DOI] [PubMed] [Google Scholar]
- 41.Kysenius K, Muggalla P, Matlik K, Arumae U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cell Mol Life Sci. 2012;69:1903–16. doi: 10.1007/s00018-012-0977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ulukaya E, Pirianov G, Kurt MA, Wood EJ, Mehmet H. Fenretinide induces cytochrome c release, caspase 9 activation and apoptosis in the absence of mitochondrial membrane depolarisation. Cell Death Differ. 2003;10:856–9. doi: 10.1038/sj.cdd.4401242. [DOI] [PubMed] [Google Scholar]
- 43.Lou X, Sun S, Chen W, Zhou Y, Huang Y, Liu X, et al. Negative feedback regulation of NF-kB action by CITED2 in the nucleus. J Immunol. 2011;186:539–48. doi: 10.4049/jimmunol.1001650. [DOI] [PubMed] [Google Scholar]
- 44.Borel F, Lacroix FB, Margolis RL. Prolonged arrest of mammalian cells at the G1/S boundary results in permanent S phase stasis. J Cell Sci. 2002;115:2829–38. doi: 10.1242/jcs.115.14.2829. [DOI] [PubMed] [Google Scholar]
- 45.Zhan Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellular response to DNA damage. Mutat Res. 2005;569:133–43. doi: 10.1016/j.mrfmmm.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 46.Vairapandi M, Balliet AG, Hoffman B, Liebermann DA. GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J Cell Physiol. 2002;192:327–38. doi: 10.1002/jcp.10140. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi N, Watanabe Y, Maitani Y, Yamauchi T, Higashiyama K, Ohba T. p-Dodecylaminophenol derived from the synthetic retinoid, fenretinide: antitumor efficacy in vitro and in vivo against human prostate cancer and mechanism of action. Int J Cancer. 2008;122:689–98. doi: 10.1002/ijc.23154. [DOI] [PubMed] [Google Scholar]