

Abstract

Herein we report the site-selective silylation of the ribonucelosides. The method enables a simple and efficient procedure for accessing suitably protected monomers for automated RNA synthesis. Switching to the opposite enantiomer of catalyst allows for the selective silylation of the 3′-hydroxyl, which could be used in the synthesis of unnatural RNA or for the analoging of ribonucelosides. Lastly, the procedure was extended to ribavirin a potent anti-viral therapeutic.
Site-selective catalysis1 is the ability to differentiate a functional group, when multiple similar groups are present in the molecule. Over the past several years site-selective catalysis has emerged as a powerful and efficient means of derivatizing and analoging natural products.2,3 To achieve site selectivity in the absence of a catalyst, synthetic chemists either rely on a significant bias in innate reactivity within the substrate (e.g. functionalizing a primary over a tertiary hydroxyl) or protecting group strategies, which mask other potential reactive sites. Beyond late stage functionalization, site-selective catalysis can also be employed in more traditional synthetic streamlining; in particular, it has been employed in the selective functionalization of monosaccharides.4,5
A significant challenge for site-selective catalysis is the manipulation of sites that are less or similarly reactive to other positions within the molecule. To this end, we have been pursuing catalysts that react with specific functional group motifs within complex molecules. To achieve this goal we have synthesized catalysts that have a catalytic residue appended to a substrate-binding site (Figure 1). The substrate-binding event occurs through a reversible covalent bond,6 minimizing the molecular interactions necessary for substrate localization. Through this design, selectivity and rate acceleration are achieved through proximity effects.7 This is an ideal mechanism for site-selective catalysis, because acceleration is directly linked to the structural arrangement of the functional groups. Because proximity effects can result in orders of magnitude of rate acceleration, differentiation of groups that have similar reactivity and even the functionalization of inherently less reactive sites are possible.8
Figure 1.

Design of Scaffolding Catalysts
Most recently we applied scaffolding catalysts to the derivatization of monosaccharides and natural products.9 In this article we apply the catalyst to the selective functionalization of ribonucleosides.10 Ribonucleosides are the core building blocks for RNA and also serve as templates for the development of therapeutics, in particular antiviral agents.11 Beyond RNA’s traditional role in protein expression (i.e. tRNA and mRNA), RNA has been found to be critical in gene regulation through riboswitches12 and RNAi.13 These discoveries have led to an explosion in the use of synthetic RNA as potential therapeutics14 as well as tools for the biological sciences. Although the expectation would be that RNA and DNA synthesis would be similar, the additional 2′-hydroxyl adds a layer of complexity to the synthesis of RNA and derivatives, because this hydroxyl has to be differentiated from the 3′-position. Current automated methods for RNA synthesis require that the 2′-hydroxyl be protected. One of the most popular protecting groups used with the RNA monomers is a tert-butyldimethylsilyl (TBS) group due to its chemical orthogonality. The current synthesis of these monomers either requires an unselective silylation of the 2′ and 3′ positions followed by separation of the isomers15 or a multistep protecting group sequence.16 Given the increasing importance of RNA synthesis, we have devised a simple one-step procedure that provides the desired monomers in high yield and selectivity for all the natural ribonucleosides. Moreover, switching to the opposite enantiomer of catalyst enables the selective protection of the 3′-hydroxyl, which can be potentially be used in the synthesis of unnatural ribonucleosides.
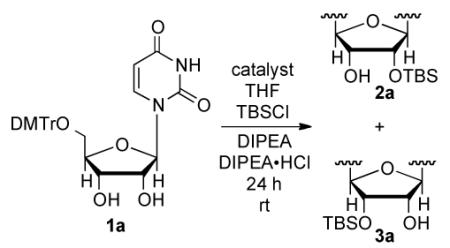
We initially investigated site-selective functionalization of uridine using TBSCl as the electrophile.17 As a control reaction, we employed N-methylimidazole (NMI) as the catalyst, and similar to previous reports15c the product forms in 71:29 ratio of the silylated C2′-OH:C3′-OH (2a:3a, Table 1 entry 1). Using scaffolding catalyst (−)-4a or (−)-4b18 results in a dramatic improvement in selectivity to 98:2 with more moderate conversion (~70%, Table 1, entries 2 and 3). To test for the necessecity of covalent bonding to (−)-4b, an additional control reaction was performed with (−)-5, which lacks a substrate binding site; both the reactivity and selelctivity (2a:3a = 45:55, Table 1, entry 4) decreased as expected. By both increasing the number of equivalents of TBSCl and the concentration of the reaction (1.0 M in substrate), complete conversion was achieved without loss of selectivity (Table 1, entry 5). Moreover, the catalyst loading could be lowered to 10% 4b affording an isolated yield of 93% and a site selectivity of 98:2 (2a:3a, Table 1, entry 6). Lowering the catalyst loading to 5% results in a modest decrease in yield (84%) with no change in selectivity (98:2, Table 1, entry 7).
Table 1.
Optimization of D-uridine silylationa
| entry | catalyst | equiv TBSCl/DIPEA |
2a:3a d | conversion (%)c |
|---|---|---|---|---|
| 1 | 20% NMI | 1.5/2.0 | 71:29 | 100 |
| 2 | 20% (−)-4a | 1.5/2.0 | 98:2 | 70 |
| 3 | 20% (−)-4b | 1.5/2.0 | 98:2 | 72 |
| 4 | 20% (−)-5 | 1.5/2.0 | 45:55 | 45 |
| 5b | 20% (−)-4b | 2.0/1.5 | 98:2 | 100 |
| 6b | 10% (−)-4b | 2.0/1.5 | 98:2 | 100 (93)e |
| 7b | 5% (−)-4b | 2.0/1.5 | 98:2 | 88 (84)e |
Reactions performed at 0.2 M of 1a with 3 mol % DIPEA-HCl.
Reactions run at 1.0 M of 1a.
Conversion determined by 1H NMR using trimethoxybenzene as an internal standard.
Ratio was determined by 1H NMR.
Isolated yield.
With the optimal conditions in hand, we surveyed the remaining naturally-occurring ribonucleosides. To our delight all the ribonucleosides yielded the desired TBS protected product in excellent selectivity and yield (Table 2). The most problematic substate was guanosine which required an increase in the catalyst loading (20 mol %) and a decrease in substrate concentration (0.5 M) to obtain the optimal selectivity (2d:3d = 97:3) and yield (75%, Table 2, entry 3).
Table 2.
Silylation of natural ribonucleosides
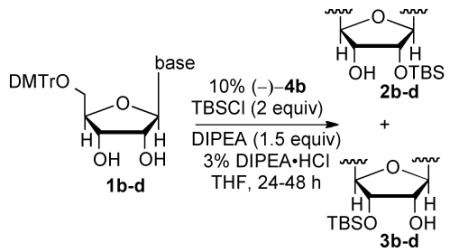
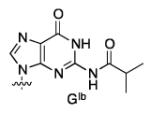
Reaction condition 0.5 M concentration in substrate
Reaction condition 1.0 M concentration in substrate
Reaction condition: 20 mol % (−)-4b, 0.5 M concentration in substrate
Ratio was determined by 1H NMR.
isolated yield
A way to view the current silylation reaction is as a pseudo-desymmetrization of a cis-1,2-diol. This would imply that switching to the opposite enantiomer of catalyst would result in silylation of the C3′-hydroxyl. Initial efforts to TBS protect the 3′ position resulted in poor yield and low selectivity in the reaction. We have previously reported that for inherently less reactive positions within molecules it is often necessary to use more reactive electrophiles. Using chlorotriethylsilane (TESCl) in place of TBSCl allowed for the highly selective protection of the 3′-hydroxyl of uridine (2′:3′ = 7:93, Table 3, entry 1). With the more reactive electrophile the catalyst loading can be reduced to 5 mol % with a reaction time of 4 hours. Moreover, switching to (−)-4b affords the 2′-protected product in excellent yield and selectivity (Table 3, entry 2). Application of the scaffolding catalysts to the other natural ribonucleosides allows for switching of the site selectivity through simply changing the enantiomer of the catalyst. In general either isomer of product can be isolated in >95:5 selectivity and >80% yield using 5% catalyst (Table 3). Guanosine proved to be the most challenging substrate, but by increasing the catalyst loading to 20 mol % the desired product was isolated in 78% yield (5:6 = 14:86, Table 3, entry 7).
Table 3.
Protection of the C3-hydroxyl of ribonucleosides
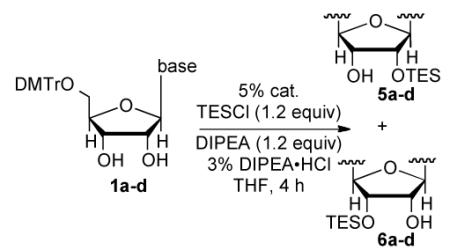
| nucleobase | entry | catalyst | 5:6 a | yield (%)b |
|---|---|---|---|---|
| U (1a) | 1 | (+)-4a | 7:93 | 80 |
| 2 | (−)-4b | >98:<2 | 86 | |
| ABz(1b) | 3 | (+)-4a | 4:96 | 85 |
| 4 | (−)-4b | 98:2 | 88 | |
| CBz(1c) | 5 | (+)-4b | 2:98 | 86 |
| 6 | (−)-4b | 98:2 | 86 | |
| GIb(1d) | 7c | (+)-4a | 14:86 | 78 |
| 8d | (−)-4b | 92:8 | 71 |
Ratio was determined by 1H NMR.
isolated yield.
20 mol % (+)-4a.
10 mol % (−)-4b.
Unnatural ribonucleosides have been found to be effective anti-viral agents, and so the selective modification of these compounds is critical to the development of novel therapeutics. We investigated whether the scaffolding catalysts would also be effective in the selective protection of ribavirin, which is currently used to treat Hepatitis C virus (HCV). Using unprotected ribavirin, catalyst (−)-4b silylates the 5′ and 2′ positions with high selectivity (7:8 = 94:6, Figure 2), leaving the 3′ hydroxyl available for further manipulation. Switching to catalyst (+)-4a affords the product with the 5′ and 3′ hydroxyls protected, allowing access to the 2′-hydroxyl (Figure 2).
Figure 2.

Functionalization of ribavirin
Using a scaffolding catalyst, site-selective functionalization of ribonucleosides was achieved enabling an efficient synthesis of appropirately protected monomers for automated RNA synthesis. Moreover, simply switching the antipode of the catalyst allows for toggling of the site selectivity to favor protection of the 3′-hydroxyl. We believe that developing catalysts that recognize specific functional group motifs will provide a practical and predictable method for manipulating polyfunctional molecules.
Supplementary Material
Acknowledgment
We thank the Alfred P. Sloan foundation (KLT), NSF (CHE-1150393) and NIGMS (RO1GM087581) for funding of this project. Mass spectrometry instrumentation at Boston College is supported by funding from the NSF (DBI-0619576).
Footnotes
Supporting Information Available Experimental procedures, characterization data, and spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mahatthananchai J, Dumas AM, Bode JW. Angew. Chem., Int. Ed. 2012;51:10954–10990. doi: 10.1002/anie.201201787. [DOI] [PubMed] [Google Scholar]
- 2 (a).Lewis CA, Miller SJ. Angew. Chem., Int. Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (c) Lewis CA, Longcore KE, Miller SJ, Wender PA. J. Nat. Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yoshida K, Furuta T, Kawabata T. Tetrahedron Lett. 2010;51:4830–4832. [Google Scholar]; (e) Fowler BS, Laemmerhold KM, Miller SJ. J. Am. Chem. Soc. 2012;134:9755–9761. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pathak TP, Miller SJ. J. Am. Chem. Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Bruckl T, Baxter RD, Ishihara Y, Baran PS. Acc. Chem. Res. 2012;45:826–839. doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Beale TM, Taylor MS. Org. Lett. 2013;15:1358–1361. doi: 10.1021/ol4003042. [DOI] [PubMed] [Google Scholar]; (i) Pathak TP, Miller SJ. J. Am. Chem. Soc. 2013;135:8415–8422. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For examples of directed and reagent controled functionalization of natural products see: Breslow R, Baldwin S, Flechtne T, Kalicky P, Liu S, Washburn W. J. Am. Chem. Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. Breslow R, Corcoran RJ, Snider BB, Doll RJ, Khanna PL, Kaleya R. J. Am. Chem. Soc. 1977;99:905–915. doi: 10.1021/ja00445a038. Breslow R, Heyer D. J. Am. Chem. Soc. 1982;104:2045–2046. Wilcock BC, Uno BE, Bromann GL, Clark MJ, Anderson TM, Burke MD. Nat. Chem. 2012;4:996–1003. doi: 10.1038/nchem.1495.
- 4.For a review on sugar functionalization see: Lee D, Taylor MS. Synthesis. 2012;44:3421–3431.
- 5 (a).Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869–8875. [Google Scholar]; (b) Kawabata T, Muramatsu W, Nishio T, Shibata T, Schedel H. J. Am. Chem. Soc. 2007;129:12890–12895. doi: 10.1021/ja074882e. [DOI] [PubMed] [Google Scholar]; (c) Kawabata T, Furuta T. Chem. Lett. 2009;38:640–647. [Google Scholar]; (d) Chan L, Taylor MS. Org. Lett. 2011;13:3090–3093. doi: 10.1021/ol200990e. [DOI] [PubMed] [Google Scholar]; (e) Gouliaras C, Lee D, Chan L, Taylor MS. J. Am. Chem. Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; (f) Lee D, Taylor MS. J. Am. Chem. Soc. 2011;133:3724–3727. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]; (g) Lee D, Williamson CL, Chan L, Taylor MS. J. Am. Chem. Soc. 2012;134:8260–8267. doi: 10.1021/ja302549c. [DOI] [PubMed] [Google Scholar]
- 6 (a).Park YJ, Park JW, Jun CH. Acc. Chem. Res. 2008;41:222–234. doi: 10.1021/ar700133y. [DOI] [PubMed] [Google Scholar]; (b) Rousseau G, Breit B. Angew. Chem., Int. Ed. 2011;50:2450–2494. doi: 10.1002/anie.201006139. [DOI] [PubMed] [Google Scholar]; (c) Tan KL. ACS Catal. 2011;1:877–886. [Google Scholar]
- 7 (a).Page MI, Jencks WP. Proc. Natl. Acad. Sci. USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pascal R. Eur. J. Org. Chem. 2003:1813–1824. [Google Scholar]
- 8.Worthy AD, Sun X, Tan KL. J. Am. Chem. Soc. 2012;134:7321–7324. doi: 10.1021/ja3027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun X, Lee H, Lee S, Tan KL. Nat. Chem. 2013;5:790–795. doi: 10.1038/nchem.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 (a).Somoza A. Chem. Soc. Rev. 2008;37:2668–2675. doi: 10.1039/b809851d. [DOI] [PubMed] [Google Scholar]; (b) Zewge D, Gosselin F, Sidler R, DiMichele L, Cvetovich RJ. J. Org. Chem. 2010;75:5305–5307. doi: 10.1021/jo100648e. [DOI] [PubMed] [Google Scholar]
- 11 (a).Butler MS. Nat. Prod. Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]; (b) Leyssen P, De Clercq E, Neyts J. Antivir. Res. 2008;78:9–25. doi: 10.1016/j.antiviral.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serganov A, Nudler E. Cell. 2013;152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurreck J. Angew. Chem., Int. Ed. 2009;48:1378–1398. doi: 10.1002/anie.200802092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 (a).Kim DH, Rossi JJ. Nat. Rev. Genet. 2007;8:173–184. doi: 10.1038/nrg2006. [DOI] [PubMed] [Google Scholar]; (b) Keefe AD, Pai S, Ellington A. Nat. Rev. Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Davidson BL, McCray PB. Nat. Rev. Genet. 2011;12:329–340. doi: 10.1038/nrg2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15 (a).Hakimelahi GH, Proba ZA, Ogilvie KK. Tetrahedron Lett. 1981;22:4775–4778. [Google Scholar]; (b) Hakimelahi GH, Proba ZA, Ogilvie KK. Tetrahedron Lett. 1981;22:5243–5246. [Google Scholar]; (c) Hakimelahi GH, Proba ZA, Ogilvie KK. Can. J. Chem. 1982;60:1106–1113. [Google Scholar]; (d) MatulicAdamic J, Beigelman L, Portmann S, Egli R, Usman N. J. Org. Chem. 1996;61:3909–3911. doi: 10.1021/jo960091b. [DOI] [PubMed] [Google Scholar]; (e) Song Q, Wang W, Fischer A, Zhang X, Gaffney BL, Jones RA. Tetrahedron Lett. 1999;40:4153–4156. [Google Scholar]
- 16.Erlacher MD, Lang K, Wotzel B, Rieder R, Micura R, Polacek N. J. Am. Chem. Soc. 2006;128:4453–4459. doi: 10.1021/ja0588454. [DOI] [PubMed] [Google Scholar]
- 17.For examples of catalyzed silyl transfer see: Isobe T, Fukuda K, Araki Y, Ishikawa T. Chem. Commun. 2001:243–244. Rendler S, Auer G, Oestreich M. Angew. Chem., Int. Ed. 2005;44:7620–7624. doi: 10.1002/anie.200502631. Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67–70. doi: 10.1038/nature05102. Zhao Y, Mitra AW, Hoveyda AH, Snapper ML. Angew. Chem., Int. Ed. 2007;46:8471–8474. doi: 10.1002/anie.200703650. You Z, Hoveyda AH, Snapper ML. Angew. Chem., Int. Ed. 2009;48:547–550. doi: 10.1002/anie.200805338. Weickgenannt A, Mewald M, Muesmann TWT, Oestreich M. Angew. Chem., Int. Ed. 2010;49:2223–2226. doi: 10.1002/anie.200905561. Weickgenannt A, Mewald M, Oestreich M. Org. Biomol. Chem. 2010;8:1497–1504. doi: 10.1039/b925722e. Rodrigo JM, Zhao Y, Hoveyda AH, Snapper ML. Org. Lett. 2011;13:3778–3781. doi: 10.1021/ol2010819. Sheppard CI, Taylor JL, Wiskur SL. Org. Lett. 2011;13:3794–3797. doi: 10.1021/ol2012617. Sun X, Worthy AD, Tan KL. Angew. Chem., Int. Ed. 2011;50:8167–8171. doi: 10.1002/anie.201103470. Giustra ZX, Tan KL. Chem. Commun. 2013;49:4370–4372. doi: 10.1039/c2cc33633b.
- 18.Catalysts 4a and 4b are available at Strem Chemicals.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.