

Abstract
A new approach to the synthesis of substituted 5-membered cyclic guanidines is described. Palladium-catalyzed alkene carboamination reactions between acyclic N-allyl guanidines and aryl or alkenyl halides provide these products in good yield. This method allows access to a number of different cyclic guanidine derivatives in only two steps from readily available allylic amines.
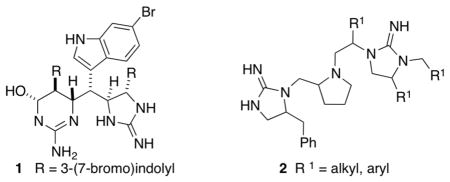
The synthesis of cyclic guanidine derivatives has attracted considerable interest due to the significance and utility of these compounds. Five-membered cyclic guanidine subunits are displayed in a number of natural products such as the araiosamines (e.g., araiosamine A, 1)1 and the plumbagine alkaloids.2,3 These structures are also featured in a number of synthetic molecules with highly interesting biological activities. For example, simple monocyclic guanidine derivatives of general structure 2 have demonstrated potent antimicrobial activity against drug-resistant Gram-positive bacteria including MRSA (methicillin-resistant Staphylococcus aureus).4 Moreover, cyclic guanidines have also been employed as synthetically useful organocatalysts.5
Classical methods for the construction of cyclic guanidines typically involve intramolecular nucleophilic substitution,6 or reactions of 1,2-diamines with phosgene imines or their equivalents.7 While useful, these strategies typically require fairly complex substrates that are prepared with multistep sequences.
In recent years several interesting metal-catalyzed transformations have been employed for the construction of cyclic guanidine derivatives from simple precursors. These strategies include alkene diamination reactions,8,9 C–H functionalization reactions,10 hydroaminations,11 oxidative aminations,12 ring-expansions via carbodiimide insertions,13 and allylic alkylations.14 Although these reactions are highly efficient and powerful methods, none of them effect carbon-carbon bond formation during the ring-closing event.
Over the past ten years we have developed a series of Pd-catalyzed alkene carboamination reactions that afford a broad range of nitrogen heterocycles. These reactions involve the cross-coupling of an aryl/alkenyl halide and an alkene bearing a pendant nitrogen nucleophile to generate a heterocyclic product with formation of both a C–N and a C–C bond.15 We felt the extension of this methodology to the construction of cyclic guanidines could have considerable potential utility, as substrates could be generated in one step from guanylation of readily available allylic amines.16 Moreover, the cyclizations should allow for facile generation of numerous analogs from a single substrate, as many aryl and alkenyl halides can be obtained from commercial sources. However, we felt that this transformation could be challenging to develop since highly basic guanidines are good ligands, and could potentially disrupt key steps in the catalytic cycle by binding to the metal at undesired stages.17 Moreover, the high nucleophilicity of the guanidine unit could also lead to competing N-arylation side reactions.18
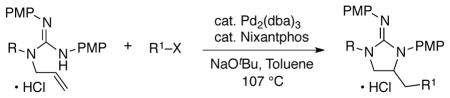
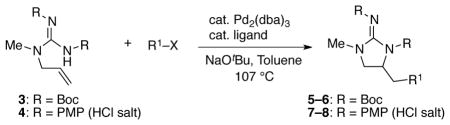
With these concerns in mind we initially elected to examine the Pd-catalyzed carboamination of boc-protected guanidine substrate 3, as the boc-protecting groups should attenuate the basicity of the nucleophile. As shown in Table 1, the coupling of 3 with either 4-bromotoluene or 2-bromonaphthalene was examined using conditions that provided good results in analogous Pd-catalyzed carboaminations of N-allylurea derivatives.19,20 We were pleased to find that use of a catalyst composed of Pd2(dba)3 and Xantphos did afford the desired products 5 and 6 (entries 1–2). However, the yields for these reactions were highly variable (not reproducible), and control experiments revealed that both the boc-protected guanidine starting material and the products decompose under the reaction conditions. To address this issue we replaced the substrate boc-groups with PMP (p-methoxyphenyl) groups, and were gratified to discover that this substrate (4) was transformed to 7 or 8 in good, and reproducible, yields.21 Further examination of reaction conditions indicated that use of the ligand Nixantphos provided improved results as compared to those obtained with Xantphos, whereas a range of other ligands gave lower yields.
Table 1.
Preliminary Experiments and Optimization a
| |||||
|---|---|---|---|---|---|
| entry | substrate | R1 | ligand | product | yield (%)b |
| 1 | 3 | 2-naphthyl | Xantphos | 5 | (20–66) |
| 2 | 3 | 4-tolyl | Xantphos | 6 | (26) |
| 3 | 3 | 2-naphthyl | Nixantphos | 5 | 33 (41) |
| 4 | 3 | 4-tolyl | Nixantphos | 6 | 26 (53) |
| 5 | 3 | 2-naphthyl | Dpe-phos | – | 0 |
| 6 | 3 | 4-tolyl | Dpe-phos | – | 0 |
| 7 | 4 | 2-naphthyl | Xantphos | 7 | (74) |
| 8 | 4 | 4-tolyl | Xantphos | 8 | (65) |
| 9 | 4 | 2-naphthyl | Nixantphos | 7 | 57 (81) |
| 10 | 4 | 4-tolyl | Nixantphos | 8 | 73 (77) |
Conditions: 1.0 equiv of guanidine substrate, 1.5 equiv R–X, 2.4 equiv NaOtBu, 2 mol % Pd2(dba)3, 8 mol % ligand, toluene (0.1 M), 107 °C. Reactions of 4 were quenched with an excess of aqueous HCl (1 M) to ensure complete protonation of the guanidine.
Isolated yields. Numbers in parentheses are NMR yields based on phenanthrene as an internal standard.
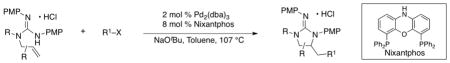
In order to explore the scope of guanidine-forming alkene carboamination reactions, substrate 4 was coupled with a variety of different aryl iodides and bromides. As shown in Table 2, satisfactory results were obtained using electron-rich, electron-neutral, and electron-poor aryl halides (entries 1–7). In addition, the presence of an ortho methyl group on the aryl bromide was also tolerated (entry 8). The main side products formed in these reactions resulted from competing deallylation of the guanidine substrate, although occasionally side products derived from oxidation of the product to the corresponding 2-aminoimidazole were observed.
Table 2.
Pd-Catalyzed Carboamination of N-Allyl Guanidinesa
| |||
|---|---|---|---|
| entry | substrate | product | yield (%)b |
| 1 |
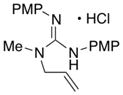 4 |
8
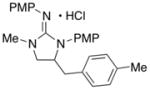
|
76 (X = I) |
| 2 | 73 (X = Br) | ||
| 3 |
12
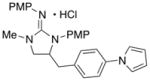
|
62 (X = I) | |
| 4 |
13
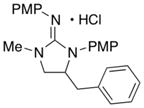
|
72 (X = Br) | |
| 5 |
14
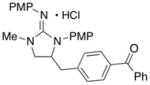
|
65 (X = I) | |
| 6 |
15
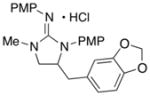
|
78 (X = Br) | |
| 7 |
16
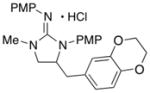
|
83 (X = Br) | |
| 8 |
17
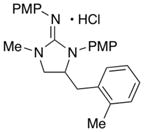
|
75c (X = Br) | |
| 9 |
 9 |
18
|
99 (X = Br) |
| 10 |
19
|
99 (X = Br) | |
| 11 |
20
|
98 (X = Br) | |
| 12 |
21
|
86d (X = Br) | |
| 13 |
|
22a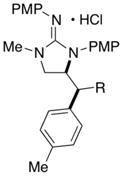
|
21 >20:1 dr (X = Br) |
| 14 | complex mixture | ||
| 15 |
 11 |
 23 |
62 2:1 dr (X = Br) |
Conditions: 1.0 equiv of guanidine substrate, 1.5 equiv R–X, 2.4 equiv NaOtBu, 2 mol % Pd2(dba)3, 8 mol % Nixantphos, toluene (0.1 M), 107 °C. Reactions were quenched with an excess of aqueous HCl (1 M) to ensure complete protonation of the guanidine.
Isolated yields (average of two experiments). Diastereomeric ratios (dr) were determined by 1H NMR analysis of the isolated products; diastereomeric ratios did not change significantly during purifcation.
This product contained ca. 8% of a side product tentatively assigned as the analogous 2-aminoimidazole.
This reaction was conducted using 4.0 equiv of the alkenyl bromide and 4.5 equiv of NaOtBu.
Substrate 10b was employed as a 7:1 mixture of E:Z alkene isomers.
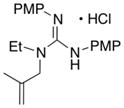
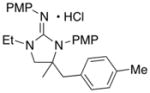
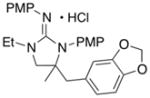
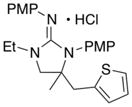
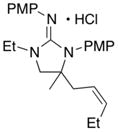
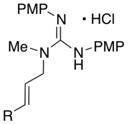
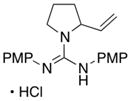
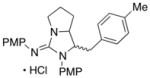
In addition to the simple N-allylguanidine substrate 4, we also examined transformations of substrates bearing additional substituents. Substrate 9, which contains a methyl group on the internal alkene carbon atom, was effectively coupled with aryl bromides using our standard conditions (entries 9–10). The use of the heteroaromatic aryl halide 2-bromothiophene as the electrophile in a reaction with 9 provided 20 in good yield (entry 11). Coupling of 9 with an alkenyl bromide (Z-1-bromobutene) to afford 21 was also achieved. However, use of a large excess of the alkenyl bromide (4 equiv) was required due to a competing homocoupling side reaction of the electrophile (entry 12). The Pd-catalyzed reaction between E-alkene substrate 10a and 4-bromotoluene afforded only a modest 21% yield of the desired product 22a (albeit with >20:1 dr), and efforts to convert related E-alkene substrate 10b provided a complex mixture of products.22 In contrast, conversion of 2-vinylpyrrolidine-derived guanidine 11 to bicyclic product 23 proceeded in good yield, but with low diastereoselectivity. Efforts to transform substrates bearing cyclic alkenes have thus far been unsuccessful.23
The mechanism of these reactions is likely analogous to that of other Pd-catalyzed alkene carboamination reactions.15,19 As shown in Scheme 1, oxidative addition of the aryl (or alkenyl) halide to Pd(0) would generate LnPd(Ar)Br complex 24, which could react with the substrate in the presence of base to afford amido complex 25.18 Migratory insertion of the alkene into the Pd–N bond affords 26,24 which then undergoes reductive elimination to provide the cyclic guanidine products.
Scheme 1.

Mechanism of Guanidine Carboamination Reactions
In order to further explore the potential synthetic utility of these reactions we have conducted preliminary studies on the deprotection of the cyclic guanidine products. Our initial experiments are shown in eq 1, and indicate that removal of one of the two PMP groups is feasible (although yields are modest). However, we have not yet successfully removed the second PMP group. Initial efforts to circumvent this problem via use of unprotected guanidine substrate 28 led to the formation of a complex mixture of undesired products; no cyclic guanidine was obtained.
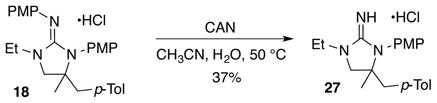 |
(1) |
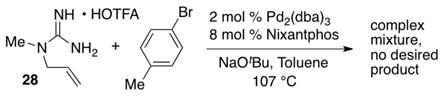 |
(2) |
Given our past success with enantioselective Pd-catalyzed carboamination reactions of related urea substrates, we have also conducted preliminary studies on the use of chiral catalysts for transformations of 9. As shown in Table 3 the monodentate phosphoramidite ligands (S)- and (R)-Siphos-PE, which provided good results in related asymmetric carboamination reactions of N-allylureas25a and N-(pent-4-enyl)carbamates,25b failed to afford significant amounts of the desired product. A preliminary survey of other chiral ligands indicated that the chelating bis-phosphines (S)-Phanephos and (S)-BINAP afforded the desired product in good yield, but with modest enantioselectivity. These studies illustrate the potential feasibility of asymmetric carboamination reactions of N-allylguanidines, but further studies will be needed to develop an efficient catalyst system.
Table 3.
Preliminary Studies on Asymmetric Catalysisa
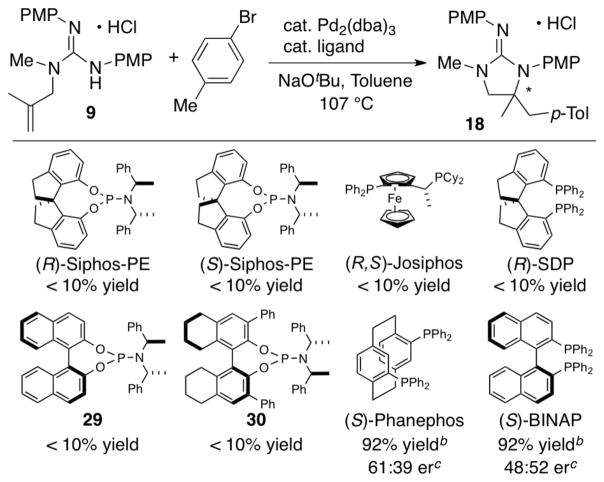
|
Conditions: 1.0 equiv of 9, 1.5 equiv 4-bromotoluene, 2.4 equiv NaOtBu, 2 mol % Pd2(dba)3, 8 mol % ligand (for chelating ligands) or 16 mol % ligand (for monodentate ligands), toluene (0.1 M), 107 °C. Reactions were quenched with an excess of aqueous HCl (1 M) to ensure complete protonation of the guanidine.
Isolated yields.
Enantiomeric ratios were determined by cleavage of one PMP group from the product followed by conversion to the Mosher amide and analysis by 1H NMR.
In conclusion, we have developed a concise approach to the synthesis of substituted 5-membered cyclic guanidines via Pd-catalyzed alkene carboamination reactions. These reactions allow for the preparation of a number of different derivatives in only two steps from readily available allylic amines, and are the first examples of Pd-catalyzed alkene carboamination reactions of guanidine nucleophiles. Further studies on expanding the scope and developing enantioselective variants of these reactions are underway.
Supplementary Material
Acknowledgments
The authors acknowledge the NIH-NIGMS (GM-071650) and the University of Michigan Associate Professor Support Fund for financial support of this work. BZ acknowledges the University of Michigan Department of Chemistry for a Summer Undergraduate Research Fellowship.
Footnotes
Supporting Information Available. Experimental procedures, characterization data for all new compounds, descriptions of stereochemical assignments with supporting crystallographic structural data, and copies of 1H and 13C NMR spectra for all new compounds reported in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wei X, Henriksen NM, Skalicky JJ, Harper MK, Cheatham TE, III, Ireland CM, Van Wagoner RM. J Org Chem. 2011;76:5515–5523. doi: 10.1021/jo200327d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cong H-J, Zhang S-W, Shen Y, Zheng Y, Huang Y-J, Wang W-Q, Leng Y, Xuan L-J. J Nat Prod. 2013;76:1351–1357. doi: 10.1021/np400235s. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews on guandine-containing natural products, see: Berlinck RGS, Trindade-Silva AE, Santos MFC. Nat Prod Rep. 2012;29:1382–1406. doi: 10.1039/c2np20071f.Berlinck RGS, Burtoloso ACB, Trindade-Silva AE, Romminger S, Morais RP, Bandeira K, Mizuno CM. Nat Prod Rep. 2010;27:1871–1907. doi: 10.1039/c0np00016g.Berlinck RGS, Burtoloso ACB, Kossuga MH. Nat Prod Rep. 2008;25:919–954. doi: 10.1039/b507874c.
- 4.(a) Hensler ME, Bernstein G, Nizet V, Nefzi A. Bioorg Med Chem Lett. 2006;16:5073–5079. doi: 10.1016/j.bmcl.2006.07.037. [DOI] [PubMed] [Google Scholar]; (b) Nefzi A, Appel J, Arutyunyan S, Houghten RA. Bioorg Med Chem Lett. 2009;19:5169–5175. doi: 10.1016/j.bmcl.2009.07.010. [DOI] [PubMed] [Google Scholar]; (c) Rideout MC, Boldt JL, Vahi-Ferguson G, Salamon P, Nefzi A, Ostresh JM, Giulianotti M, Pinilla C, Segall AM. Mol Divers. 2011;15:989–1005. doi: 10.1007/s11030-011-9333-2. [DOI] [PubMed] [Google Scholar]
- 5.For a review, see: Selig P. Synthesis. 2013;45:703–718.
- 6.For representative examples, see: O’Donovan DH, Rozas I. Tetrahedron Lett. 2012;53:4532–4535.Kim H-O, Mathew F, Ogbu C. Synlett. 1999:193–194.
- 7.For representative examples, see: Nalli SM, Clarkson GJ, Franklin AS, Bellone G, Shipman M. Synlett. 2008:2339–2341.Huber D, Leclerc G, Ehrhardt JD, Andermann G. Tetrahedron Lett. 1987;28:6453–6456.
- 8.Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem Commun. 2008:2334–2336. doi: 10.1039/b719479j. [DOI] [PubMed] [Google Scholar]
- 9.Zhao B, Du H, Shi Y. Org Lett. 2008;10:1087–1090. doi: 10.1021/ol702974s. [DOI] [PubMed] [Google Scholar]
- 10.Kim M, Mulcahy JV, Espino CG, Du Bois J. Org Lett. 2006;8:1073–1076. doi: 10.1021/ol052920y. [DOI] [PubMed] [Google Scholar]
- 11.(a) Bhonde VR, Looper RE. J Am Chem Soc. 2011;133:20172–20174. doi: 10.1021/ja2098063. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gibbons JB, Gligorich KM, Welm BE, Looper RE. Org Lett. 2012;14:4734–4737. doi: 10.1021/ol3019242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Giles RL, Sullivan JD, Steiner AM, Looper RE. Angew Chem, Int Ed. 2009;48:3116–3120. doi: 10.1002/anie.200900160. [DOI] [PubMed] [Google Scholar]
- 12.Mulcahy JV, Du Bois J. J Am Chem Soc. 2008;130:12630–12631. doi: 10.1021/ja805651g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler DCD, Inman GA, Alper H. J Org Chem. 2000;65:5887–5890. doi: 10.1021/jo000608q. [DOI] [PubMed] [Google Scholar]
- 14.Büchi G, Rodriguez AD, Yakushijin K. J Org Chem. 1989;54:4494–4496. [Google Scholar]
- 15.For reviews, see: Wolfe JP. Eur J Org Chem. 2007:571–582.Wolfe JP. Synlett. 2008:2913–2937.Schultz DM, Wolfe JP. Synthesis. 2012;44:351–361. doi: 10.1055/s-0031-1289668.
- 16.(a) Yong YF, Kowalski JA, Lipton MA. J Org Chem. 1997;62:1540–1542. [Google Scholar]; (b) Li D, Guang J, Zhang W-X, Wang Y, Xi Z. Org Biomol Chem. 2010;8:1816–1820. doi: 10.1039/b923249b. [DOI] [PubMed] [Google Scholar]
- 17.Bailey PJ, Pace S. Coord Chem Rev. 2001;214:91–141. [Google Scholar]
- 18.Rauws TRM, Maes BUW. Chem Soc Rev. 2012;41:2463–2497. doi: 10.1039/c1cs15236j. [DOI] [PubMed] [Google Scholar]
- 19.(a) Fritz JA, Wolfe JP. Tetrahedron. 2008;64:6838–6852. doi: 10.1016/j.tet.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fritz JA, Nakhla JS, Wolfe JP. Org Lett. 2006;8:2531–2534. doi: 10.1021/ol060707b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For Pd-catalyzed carboamination reactions of 2-allylpyrrolidinyl ureas that generate bicyclic products with formation of a six-membered ring, see: Babij NR, Wolfe JP. Angew Chem, Int Ed. 2012;51:4128–4130. doi: 10.1002/anie.201201001.Babij NR, Wolfe JP. Angew Chem, Int Ed. 2013;52:9247–9250. doi: 10.1002/anie.201302720.
- 21.The starting materials for these reactions were generated in one step by guanylation of the corresponding allylic amine. Substrates 4 and 9 were obtained in good yield (60–73%), whereas moderate yields were obtained for 3, 10, and 11 (10–39%). See the Supporting Information for full experimental details.
- 22.The relative stereochemistry of product 22a has been tentatively assigned based on analogy to a closely related urea coupled with the likelihood that these transformations proceed through an alkene syn-aminopalladation mechanism analogous to that of other heterocycle-forming Pd-catalyzed alkene carboamination reactions.
- 23.Substrates bearing a stereocenter at the allylic position were not examined as attempts to prepare these compounds by guanylation reactions of the corresponding allylic amines failed to afford useful quantities of material.
- 24.(a) Neukom JD, Perch NS, Wolfe JP. J Am Chem Soc. 2010;132:6276–6277. doi: 10.1021/ja9102259. [DOI] [PubMed] [Google Scholar]; (b) Hanley PS, Markovic D, Hartwig JF. J Am Chem Soc. 2010;132:6302–6303. doi: 10.1021/ja102172m. [DOI] [PubMed] [Google Scholar]
- 25.(a) Hopkins BA, Wolfe JP. Angew Chem, Int Ed. 2012;51:9886–9890. doi: 10.1002/anie.201205233. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157–12159. doi: 10.1021/ja106989h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.