

Abstract
Catalytic procedures are described for aminedirected borylation of aliphatic and aromatic tertiary amine boranes. Sequential double borylation is observed in cases where two or more C–H bonds are available that allow 5-center or 6-center intramolecular borylation. The HNTf2 catalyzed borylation of benzylamine boranes provides a practical means for the synthesis of ortho-substituted arylboronic acid derivatives, suitable for Suzuki-Miyaura cross-coupling applications.
INTRODUCTION
Recent reports from our laboratory have described intramolecular borylation of aromatic C–H bonds via hydride abstraction from amine borane 1 with Ph3C+ B(C6F5)4− (2; eq. 1).1–4 Nearstoichiometric activation procedures gave the best results using 90 mol% of the trityl salt 2 at room temperature, and borylation was attributed to transient borenium intermediates 3 or equivalent species, although the only observable borocations were the relatively stable hydride-bridged “dimers” 4 (at 50% loading of 2) and the product borenium salts 6 (using 90% of 2).3 Although the mechanistic details remain uncertain, conversion to 64 requires a dehydrogenation step that may correspond to a transition state 5.
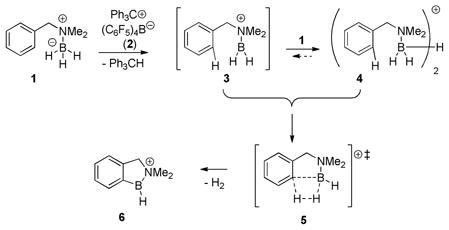 |
(1) |
We have also disclosed related chemistry for the borylation of aliphatic C–H bonds in a preliminary account.5,6 In this case, the stoichiometric process using trityl salt 2 also occurs at room temperature, but cleaner reactions are observed in several examples using the strong acid Tf2NH as catalyst at 160 °C (eq. 2), apparently via the initial conversion of the amine borane 7 to an intermediate 8 and hydrogen gas. Subsequent borylation affords a mixture of the hydrolytically labile 9 and the cyclic amine borane 10 resulting from intermolecular hydride transfer from 7 to 9, and reductive quenching with Bu4NBH4 completes the conversion to the stable product 10. We now describe a more extensive investigation of catalytic and stoichiometric borylations involving aliphatic substrates, and also show that a similar catalytic borylation gives much-improved yields with representative aromatic substrates. In some examples, the stoichiometric and catalytic methods give distinctly different product mixtures.
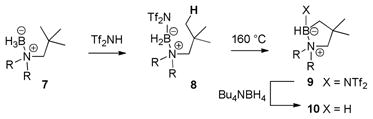 |
(2) |
RESULTS AND DISCUSSION
The first stage of our investigation was designed to clarify the nature of activated intermediates derived from amine boranes and Tf2NH. Thus, treatment of a range of amine borane complexes 11 with 1 equiv of Tf2NH in CD2Cl2 or d8-PhMe generally produced mixtures of two isomeric species (eq 3; Table 1). The reactions proceeded vigorously, and gas liberation ceased within seconds at rt, indicating complete consumption of Tf2NH. The products were identified as N- and O-bound covalent boron bistriflimides 12 and 13 based on multinuclear NMR spectroscopy. Thus, both species could be reliably assigned as tetracoordinate boron complexes based on 11B NMR data. From 19F and 13C NMR data it is also apparent that in one of the isomers both CF3 groups are magnetically equivalent, as expected in the N-bound isomer 12, but they are distinctly different in the O-bound isomer 13. Bistriflimide connectivity isomerism has not previously been observed in boron compounds (very few boron sulfonylimides have been reported so far),7 although similar isomerism is a known phenomenon in Si bistriflimides.8
Table 1.
11B and 19F NMR Data for R3N–BH2NTf2 Complexes 12 and 13a
| entry | R3N | δ 11B, ppm | δ 19F, ppm | 12:13 ratio at equilibriumb | |
|---|---|---|---|---|---|
| 1 | Me3N |
12a 13a |
−3.7 4.0 |
−69.2 −77.6, −79.1 |
7:1 |
| 2 | Et3N |
12b 13b |
−7.4 0.7 |
−68.9 −76.7, −79.1 |
1:4.7 |
| 3 | (i-Pr)2NEt |
12c 13c |
ND 1.2 |
−68.4 −77.0, −79.1 |
<1:25 |
| 4 | p-MeBnNMe2 |
12d 13d |
−3.2 4.0 |
−69.0 −76.6, −79.1 |
4.2:1 |
| 5c |
|
12e 13e |
−4.6 −0.6 |
−69.2 −76.7, −78.7 |
1:2.6 |
Conditions: 1:1 R3N–BH3:Tf2NH, CD2Cl2, rt.
Monitored up to 2–14 days at rt.
In d8-PhMe.
 |
(3) |
In most cases the ratio of 12 to 13 measured immediately following mixing of 11 and Tf2NH was different from that observed after a few hours at room temperature, suggesting a kinetic preference for the formation of one of the isomers. Such kinetic product mixtures initially contained larger amounts of the O-bound isomer 13, which partially turned into 12 over time. Activation of Me3N–BH3 with Tf2NH in d8-PhMe serves as a representative example where equilibration to the thermodynamic product ratio was observed to be particularly slow (hours at rt). To avoid discrepancies caused by slow kinetics, ratios of N- vs. O-bound products (12 vs. 13) summarized in Table 1 were confirmed to remain un- changed for days following the initial equilibration period. The product ratio (12 vs. 13) measured at equilibrium correlates reasonably well with steric properties of the amine fragment. Thus, while 12 is the thermodynamically preferred isomer in the relatively unhindered Me3N series (7:1 12a:13a), the O-bound isomer is clearly the dominant species in the far more hindered i-Pr2NEt derivatives (<1:25 12c:13c). Similar observations in Si bistriflimides have been rationalized based on lower steric demands of the bistriflimide fragment in the O-bound isomer,8 and the same considerations apparently can be extended to boron compounds. The observed equilibration suggests facile interconversion of 12 and 13, although the exact mechanism of this process is unclear.
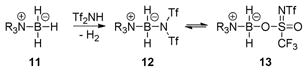
When activations of amine boranes were performed using only a substoichiometric amount of Tf2NH, formation of an additional product was observed by NMR spectroscopy. Thus, when Et3N–BH3 was treated with 0.5 equiv of Tf2NH in CD2Cl2, NMR assay after 30 min at rt indicated the presence of three new compounds aside from unreacted amine borane (eq 4). While two of the products were found to be 12b and 13b in the same 1:4.7 ratio as in the stoichiometric Tf2NH activation experiment (Table 1), the third product was assigned as the unusual H-bridged borocation 14 based on the similarity of the multinuclear NMR spectra to data reported previously for the corresponding [B(C6F5)4]− salt.1 Structurally, the central 3c2e B–H–B bond of borocation 14 can be viewed as being derived from the σ-basic B–H bond of Et3N–BH3 and the formally empty p-orbital of the hypothetical primary borenium cation 15b. The observed equilibrium ratio between Et3N–BH3, 12b, 13b and 14 (ca. 7.2:1.1:5.0:1.0 mol) thus suggests that B–H σ-bonds of Et3N–BH3 are sufficiently nucleophilic to compete with Tf2N− for binding to 15b in the thermodynamic sense.
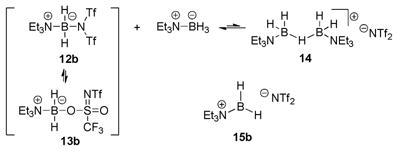 |
(4) |
Despite the structural description of cation 14 as a complex between Et3N–BH3 and 15b, the free primary borenium cation (15b) is not necessarily present at any stage of the reaction shown in eq. 4, since both the forward and the reverse processes can also be envisioned as proceeding by SN2-type displacements at boron for 12b or 13b (forward process) or 14 (reverse process). Presumably, such displacements would involve the weakly nucleophilic Tf2N− anion that is inevitably present as a contaminant in all Tf2NH activations, as suggested by 19F NMR data. On the other hand, reversible dissociation of 12b/13b to 15b, and 14 to Et3N–BH3 and 15b provides a sufficient explanation for our observations in the absence of evidence to the contrary. In terms of reactivity as well as structure, H-bridged cations analogous to 14b occupy an intermediate position between tricoordinate borenium and tetracoordinate boronium species.3 Furthermore, eq. 4 represents the borderline case for the formation of covalent counterion adducts 12/13 in the presence of 14. Any further decrease in the coordinating ability of the anion can be expected to favor conversion of 12/13 to hydride bridged species such as 14 when excess borane complex is present, as observed when the anion is B(C6F5)4−.
Having developed a better understanding of the activation process, we turned attention to broadening the substrate scope for the aliphatic borylations reported in the preliminary study.5 Several amine borane substrates were selected for evaluation under optimum stoichiometric and/or catalytic activation conditions (Scheme 1). In most cases the substrates were chosen such that multiple C–H bonds would be in proximity to the cationic boron center in activated intermediates so that issues of regioselectivity or multiple borylation could be addressed.
Scheme 1.

a Catalytic borylation conditions: 5 mol% Tf2NH, PhMe, sealed tube, 160 °C, 14h, quenched with ca. 10 mol% n-Bu4NBH4. b Stoichiometric borylation conditions: 0.9 equiv Ph3C+ [B(C6F5)4]−, PhF, rt, 4h, quenched with ca. 1.1 equiv n-Bu4NBH4.
First, a substrate 16 was investigated to allow direct assessment of the potential for competition between intramolecular borylation pathways leading to 5- vs. 6-membered rings. Under both catalytic and stoichiometric reaction conditions, borylation of the N-neopentyl group was preferred, resulting in 17 as the dominant product after reductive quenching to convert intermediate B–NTf2 species to B-H. Isomeric 6-membered borylation product 18 or doubly borylated products were not detected in either the catalytic or stoichiometric experiments. In the case of the catalytic reaction using 5 mol% of Tf2NH in toluene at 160 °C (sealed tube), material balance consisted of 17 (72% isolated) together with recovered starting material 16 due to incomplete reaction. The corresponding stoichiometric experiment using 0.90 mol% of Ph3C+ B(C6F5)4 − (2) in fluorobenzene at room temperature gave an 81% yield of 17, but isolation of the product was complicated by substantial amounts of the Ph3CH byproduct.
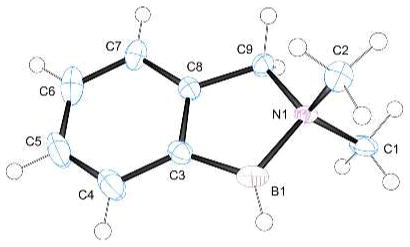
A modified substrate 19 (Scheme 1) was designed to have two reactive C–H’s within 5 atom distance from the boron atom, and formation of the doubly borylated 20 was the result (catalytic conditions; structure of 20 confirmed by a single crystal X-ray diffraction study, Fig. 1). Similar to the experiment starting from 16, no substantial amount of the isomeric product 21 resulting from a 6-center intramolecular borylation was observed. Although the timing of sequential borylation events was not established, formation of 20 indicates that both the aliphatic and aromatic C–H bonds have comparable reactivity in this example.
Figure 1.
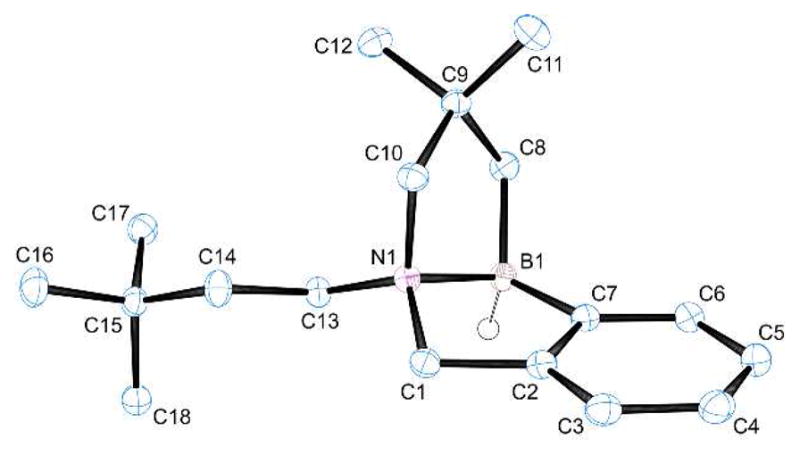
X-Ray structure of amine borane 20 (50% probability ellipsoids, H atoms omitted for clarity except at B). Selected bond lengths (Å) and angles (deg): B1–N1, 1.66; B1–C7, 1.61; B1–C8, 1.63; N1–B1–C7, 97; N1–B1–C8, 101.
The next step was to evaluate the reactivity of C–H bonds in a transannular position, accomplished using 11- and 8-membered ring substrates 22, 24 and 27 (Scheme 2). No borylation of the aliphatic ring was observed in 22 and the spirocyclic structure 23 was the only product isolated under standard catalytic or stoichiometric activation conditions. However, transannular borylation was observed in the 8-membered cyclic amine borane 24 to give a symmetrical bicyclo[3.3.1] product 25. In contrast to the other borylations discussed so far, 25 is the result of a 6-center borylation process. The modest isolated yield (52%) of 25 in the high-temperature catalytic protocol is largely the result of incomplete consumption of the starting material 24, although traces of a second product were detected by NMR spectroscopy. This substance could not be purified, but the limited NMR data are consistent with the tentative assignment of structure 26, having the isomeric bicyclo[4.2.1] skeleton.
Scheme 2.

a Catalytic borylation conditions: 5 mol% Tf2NH, PhMe, sealed tube, 160 °C, 14h, quenched with ca. 10 mol% n-Bu4NBH4. b Stoichiometric borylation conditions: 0.9 equiv Ph3C+ [B(C6F5)4]−, PhF, rt, 4h, quenched with ca. 1.1 equiv n-Bu4NBH4.
We also explored the 8-membered amine borane 27, an analog of 24 having an N-benzyl subunit in place of N-ethyl. The modified substrate 27 showed remarkably rich behavior due to the presence of three reactive C–H bonds (Scheme 2; Table 2). When subjected to the standard catalytic conditions with HNTf2, 27 was converted exclusively to monoborylated products 28 and 29 in a 1:1.7 ratio after the usual reductive quench with n-Bu4NBH4 (1H NMR assay of the crude reaction mixture). Products 28 and 29 were isolated in 30% and 48% yields, respectively. No products of a second borylation event were observed, not even with extended reaction times at 160 °C! A partial explanation for these observations is shown in Scheme 2 by considering a pathway from 27 to the activated intermediate 31 followed by the expected borylation to give 33. Subsequent reductive quenching would then afford the major product 29. On the other hand, stoichiometric activation of 27 with Ph3C+ B(C6F5)4− (2) at rt (4 h) followed by reductive quenching did afford a doubly borylated product (30) in addition to 28 and 29 (Table 2). As established by X-ray crystallography (Fig. 2), structure 30 is noteworthy because it contains a bicyclo[4.2.1] subunit involving the borylated azacene ring. Surprisingly, 30 becomes the dominant product and accumulates at the expense of the symmetrical bicyclo[3.3.1] product 29, as evidenced by the borylation outcome under more forcing stoichiometric conditions at 60 °C (Table 2, entry 3). A convincing explanation for these findings would require a more detailed knowledge of the timing of initial C–H insertion events under stoichiometric conditions, but analogy suggests that a hypothetical 3c2e structure 32 is generated first, and that it undergoes borylation to give 34 or, more likely, the precedented5,6 mono-alkyl borenium cation 35. Either 34 or 35 might serve as the precursor of 29 when the entire stoichiometric reaction sequence, including reductive quenching, is conducted at room temperature. However, if the borylation is conducted at 60 °C, then the activated intermediates apparently can undergo a competing retrohydroboration process. For the sake of simplicity, we assume that the borenium cation 35 undergoes retrohydroboration to generate 36, which rapidly recloses to afford the bicyclo[4.2.1] skeleton followed by borylation of the aromatic ring (not shown). However, it is conceivable that 36 would borylate the aromatic ring faster than it hydroborates the alkene. In either case, reductive quenching would ultimately afford the doubly borylated product 30. Precedents for facile retrohydroboration under similar thermal conditions are well-known,9 including reported cases involving borenium equivalents generated from N-heterocyclic carbene boranes with HNTf2.9e
Table 2.
Borylations of 27
| entry | conditions | yield, % | ||
|---|---|---|---|---|
| 28 | 29 | 30 | ||
| 1 | 5 mol% Tf2NH, 160 °C, 14h | 30a | 48a | 0 |
| 2 | 0.9 equiv Ph3C+ [B(C6F5)4]−, rt, 4h | 29b | 38b | 25b |
| 3 | 0.9 equiv Ph3C+ [B(C6F5)4]−, 60 °C, 20h | 17b | 4b | 70b |
Isolated yields.
NMR yields vs. internal reference.
Figure 2.
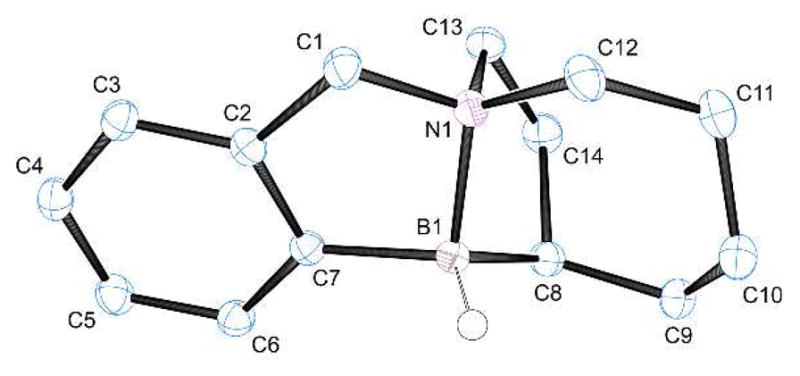
X-Ray structure of amine borane 30 (50% probability ellipsoids, H atoms omitted for clarity except at B). Selected bond lengths (Å) and angles (deg): B1–N1, 1.65; B1–C7, 1.61; B1–C8, 1.62; N1–B1–C7, 97; N1–B1–C8, 101.
It is important to note that 30 was obtained when the borylation of 27 was performed under stoichiometric conditions at room temperature or at 60 °C, but was not detected under catalytic conditions at 160 °C. This observation argues against the involvement of identical activated intermediates in both the catalytic and the stoichiometric experiments. Stated more explicitly, if the borenium cation 35 was responsible for the formation of 30, then it either was not formed, or was not viable in the catalytic process at 160 °C.
As discussed in the preceding sections, the catalytic activation method using HNTf2 allows efficient borylation with optimal substrates such as 22. In other, less hindered aliphatic examples, the catalytic process does not go to completion, suggesting some form of product inhibition of the catalytic cycle, or a dead end decomposition pathway perhaps involving the product borenium cations. The reasons for this behavior remain unexplained in the aliphatic borylations, and the potential for retrohydroboration may be partly responsible for unknown decomposition or disproportionation events that interfere with catalyst turnover. On the other hand, aromatic borylations should be less prone to complications under catalytic conditions. The product borenium cations are somewhat more stabilized due to their “bora-benzylic” nature, involving delocalization between the aromatic π-system and the planar tricoordinate boron. Furthermore, the aromatic borylation products are incapable of undergoing retrohydroboration. Based on these considerations, we tested the catalytic activation method with the simple N,N-dimethylbenzylamine borane 1 (Scheme 3), and were pleased to find that >90% of the known borylation product 37 was formed after reductive quenching. Evidently, an efficient catalytic cycle is possible.
Scheme 3.
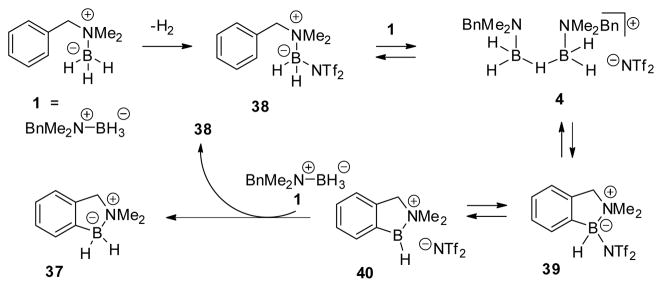
A hypothetical catalytic cycle is illustrated in Scheme 3 so that plausible intermediates can be mentioned. Although several important details are not addressed in this cycle, it does show that the key activated intermediate 38 can be regenerated from the initial borylation product 39 or its corresponding ion pair 40 as required for catalyst turnover. The cycle includes the hydridebridged cation 4 because its presence is predicted from eq. 4 and Table 1. While we have no direct evidence that 4 is the immediate precursor of 39 or 40, our preliminary investigation of internal borylation using stoichiometric activation had found a correlation between effective electrophiles (Ph3C+ salts, Tf2NH, B(C6F5)3, or AlCl3) and the generation of hydride bridged “dimers” similar to 4. Weaker electrophiles such as TfOH converted 1 to a tetracoordinate analogue of 38, gave no 4, and did not induce borylation. Intriguingly, while catalytic amounts of Tf2NH efficiently promoted the borylation of 1 at >120 °C, essentially no cyclization was observed using a 1:1 ratio of Tf2NH and 1. This observation suggests that the crucial borylating agent can be formed from 4, but not by spontaneous B–NTf2 heterolysis from 38 (BnMe2NBH2–NTf2). So far, the observation remains unexplained, and is particularly surprising in view of the comparable affinity of [R3N–BH2]+ for R3N–BH3 and Tf2N− (eq. 4). With a catalytic cycle established for the simple substrate 1 under HNTf2 catalysis, the regioselectivity of aromatic borylations with several m-substituted N,N-dimethylbenzylamine boranes was investigated using similar catalytic conditions (eq. 5; Table 3).10
Table 3.
Aromatic C–H Borylation in m-Substituted Benzylamine Boranes
| entry | substrate | X | R | para:ortho borylation | combined yield,a % |
|---|---|---|---|---|---|
| 1 | 1 | H | Me | 37 | 94 |
| 2 | 41 | Cl | Me | 42:43, 25:1a (1:1.2)b | 93c |
| 3 | 44 | I | Me | 45:46, 40:1a (1:2.4)b | 91c |
| 4 | 47 | Cl | (CH2)4 | 48:49, 10:1a | 95 |
| 5 | 50 | F | Me | 51:52, 13:1a (4:1)b | 95c |
| 6 | 53 | PhO | Me | 54:55, 10:1a | 96 |
Catalytic borylation conditions: 5 mol% Tf2NH, PhMe, sealed tube, 160 °C, 14h, quenched with ca. 10 mol% n-Bu4NBH4.
Stoichiometric borylation conditions: 0.9 equiv Ph3C+ [B(C6F5)4]−, PhBr, rt, 4h, quenched with ca. 1.1 equiv n-Bu4NBH4.
24 h.
 |
(5) |
The most striking difference between the catalytic and stoichiometric borylation processes is in the opposite trends in regioselectivity for the halogenated amine boranes 41, 44, and 50. While in the catalytic process the para:ortho selectivity increases from F or Cl to I, the changes are not systematic in view of the two contrasting chlorine examples (entries 2 and 4). In the stoichiometric trityl-activated borylations at room temperature for the meta halides, selectivity is much lower and decreases from fluoride (4:1, entry 5) to chloride (1:1.2, entry 2), but inverts for iodide (1:2.4, entry 3). It is tempting to assume that the differences in catalytic vs. stoichiometric product ratios arise from thermodynamic equilibration under the high-temperature conditions, but so far all indications suggest that the catalytic borylations are kinetically controlled. Thus, the product regioisomer ratios do not change with % conversion, and the isolated borylation products do not undergo equilibration under the reaction conditions. To support this assertion, each of the purified isomers 42 and 43 was activated with 5 mol% Tf2NH in d8-PhMe, and the resulting solutions were heated at 120 °C.11 No equilibration of pure 42 or 43 to the isomer mixture was observed, although the catalytic activation of 41 under the same conditions gave a ca. 25:1 mixture of 42:43.
Since it can be argued that catalytic activation of the products 42 or 43 did not exactly mimic the borylation conditions because the starting amine borane was not present, a modified set of experiments was also performed. Thus, mixtures of 0.1 equiv of either 42 or 43 with 1 equiv of 1 in toluene were activated with 5 mol% Tf2NH, and then heated in sealed tubes at 160 °C. Under the reaction conditions, full conversion of 1 to 37 was observed after 10 mol% borohydride quench, but 42 and 43 were recovered unchanged. In a second control experiment under similar conditions, 0.1 equiv of 41 and 1 equiv of 1 were used along with 5% HNTf2. This time, a mixture of the cyclized products 37, 42 and 43 was formed, and the ratio of 42 and 43 was found to be the same as in the cyclization of 41 with no 1 present (ca. 25:1 42:43). This suggests that the starting amine borane 41 cyclizes to form a mixture of 42 and 43 under the indicated conditions, while the interconversion of the two isomers does not occur. In this series of experiments, the cyclization of 1 to 37 serves not only to mimic the actual cyclization conditions, but also to act as a probe confirming that the catalytic cycle is viable.
The rates of the catalytic borylation are qualitatively insensitive to the electronic effects in the substrate. In an attempt to estimate the rate effect caused by a phenoxy group positioned para with respect to the C–H bond that undergoes borylation (i.e. the meta-PhO-benzylamine substrate), a 1:1 mixture of 1 and 53 (Table 3) activated with 10% HNTf2 in d8-PhMe was heated at 120 °C. The relative rates of consumption of both amine boranes were monitored in situ using NMR spectroscopy, and were found to be similar (equal, within experimental uncertainty).
In sharp contrast to the high-yielding cyclization of the meta-PhO substrate 53 (96% isolated yield of 54+55), the cyclization of the corresponding para-isomer 56 delivered only 10% of the expected 57, while the major product was found to be p-phenoxytoluene (77% isolated; the only other product detected in the crude mixture was the dimer of Me2NBH2). To explain the net hydrodeamination from 56, we suggest that activation produces 58 as usual, followed by fragmentation to a stabilized benzylic cation 59 that abstracts hydride from the starting 56. Similar hydrodeamination was observed during attempted borylation of α-methylbenzylamine derivative 60 to give ethyltoluene, and, to a lesser extent, for 1-(dimethylaminomethyl)naphthalene borane (61). In the latter case, the expected borylation product 62 was obtained in 52% yield, while 1-methylnaphthalene 63 and (Me2N–BH2)2 constituted the rest of the crude reaction mixture according to NMR and GC-MS assay. Overall, it appears that C–N fragmentation predominates in those cases where a reasonably stable carbocation can be formed.
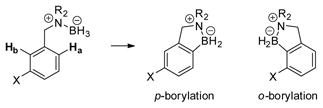
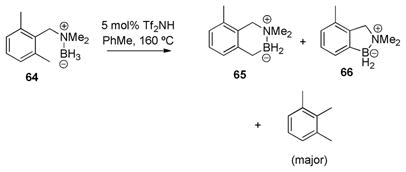
While a substantial degree of C–N cleavage (ca. 70%) was observed in the catalytic borylation of o,o′-disubstituted amine borane 64, this substrate also presented an unusual case of C–C bond reactivity (eq 6). Thus, aside from the 1,2,3-trimethylbenzene and the expected aliphatic borylation product 65, a small amount of the C-demethylation product 66 (ca. 6.3:1 65:66) was isolated and identified by comparison with an authentic sample.2 The mechanism of this process was not investigated, but the observed demethylation can be understood as an insertion of electrophilic tricoordinate boron into a C–C bond, forming CH4 as the byproduct. 12,13
 |
(6) |
The success of small scale catalytic aromatic borylations in the substituted benzylamine borane series prompted experiments to develop a larger scale (3–4 g) reaction protocol featuring a more scalable isolation procedure. Performing the reaction with 5% HNTf2 in dry tetralin at 180 °C obviated the need for sealed glass tubes, and removal of tetralin was accomplished by precipitating the crude product with hexane. Subsequent extraction of the crude solid with either toluene (37) or THF/Et2O (45) separated the products from insoluble Tf2N derivatives, affording pure products 37 (77% after crystallization) and 45 (95%).14
Having demonstrated gram-scale access to representative aromatic borylation products, we briefly tested their conversion to arylboronic acid derivatives that are potentially useful substrates for transition metal catalyzed cross-coupling reactions. Thus, refluxing amine boranes 37 or 45 in aqueous MeOH for 4h followed by concentration and azeotropic removal of water furnished cyclic boroxines 67 and 68 in nearly quantitative yields (Scheme 5). Alternatively, refluxing 37 with catechol in xylenes, afforded the catechol ester 69 in 93% yield after crystallization.
Scheme 5.

Cross-Couplings of 67 and 68
Attempts to use boronic anhydride 67 in Suzuki-Miyaura cross-coupling indicated that its reactivity is somewhat lower compared to that of arylboronic acids (Scheme 5). Thus, 67 could be efficiently coupled with PhI/Pd(PPh3)4 in DME/H2O using Ba(OH)2·8H2O as base (82% of 70 isolated), but no coupling was observed using K3PO4/Pd(OAc)2/SPhos in THF, conditions that are known to effect the cross-coupling of aryl iodides with PhB(OH)2.15 This suggested the possibility of using 68 in a sequence of two distinct Suzuki-Miyaura coupling events. The added base was identified as the crucial variable determining the reactivity of the C–B bond of 67, and a protocol for a selective two-stage cross-coupling of 68 with PhB(OH)2 was developed. First, 68 was reacted with PhB(OH)2 using K3PO4/Pd(OAc)2/SPhos in THF, the same conditions that gave no coupling with 67. Selective coupling was evident from the 1H NMR spectrum of the crude reaction mixture, suggesting that only a single benzylamine derivative (71) was present. In the second stage, Ba(OH)2·8H2O was used to activate the boroxine moiety of 71, and this enabled cross-coupling with added 4-iodoacetophenone. While the unoptimized isolated yield of 72 is modest (40%), the goal of this sequence was to illustrate the potential of the bifunctional coupling partner 68 for selective sequential attachment of two distinct aromatic substituents. The analogy between these results and selective coupling reactions of MIDA-protected boronates16 suggests that the pendant (dimethylamino) methyl moiety is responsible for the decreased boroxine reactivity in 68.
SUMMARY
Catalytic borylation conditions (5% HNTf2, 160 °C) have been developed that allow intramolecular borylation starting from tertiary amine boranes. The reactions of aliphatic amine boranes are shown to have a preference for 5-membered ring formation, although 6-membered rings can be formed with biased substrates. Retrohydroboration appears to be possible under the reaction conditions and is responsible for skeletal isomerization during the borylation step from 29 to 30. Two sequential borylations are demonstrated starting from amine boranes 19 or 27.
The same catalytic conditions are especially effective for amine-directed aromatic borylation and generally give yields well above 90%. The catalytic method is more practical that an earlier variation using stoichiometric Ph3C+ B(C6F5)4− (2) for hydride abstraction from the amine borane.1,2 The catalytic reactions also give considerably higher regioselectivity with meta-substituted benzylamine boranes. Conversion of the borylation products into arylboronic acid derivatives is possible, and is illustrated in selective Suzuki-Miyaura cross-coupling reactions.
EXPERIMENTAL SECTION
General Remarks
All reactions were performed at room temperature (unless otherwise stated), under an atmosphere of dry nitrogen, either in a glove-box, or using standard Schlenk techniques. Nuclear magnetic resonance experiments were performed on Varian Inova 700, Varian Inova 500 and Inova 400 spectrometers at the following frequencies: 1H 700 MHz, 500 MHz or 400 MHz; 11B and 11B{1H} 225 MHz, 160 MHz or 128 MHz; 13C{1H} 176 MHz, 126 MHz or 101 MHz; 19F 471 MHz. All spectra were recorded in CDCl3, d5-PhBr, or CD2Cl2 and referenced to the 1H signal of internal Me4Si according to IUPAC recommendations,17 using a Ξ (referencing parameter) of 32.083974 for BF3–OEt2 (11B), a Ξ of 25.145020 for Me4Si (13C), and a Ξ of 94.094011 for CCl3F (19F). When the internal Me4Si reference could not be used, residual solvent peaks in 1H NMR spectra were referenced instead. Hexanes, CH2Cl2, and THF were dried by passing through a column of activated alumina. Hexanes and CH2Cl2 were further dried by storing over activated 3Å molecular sieves in the glovebox. Commercially available NMR grade deuterated solvents (Cambridge Isotope Laboratories), as well as benzene and fluorobenzene were not distilled; instead they were simply dried over a large amount of activated 3Å molecular sieves in the glovebox. All other reagents were used as received from commercial suppliers, or prepared according to published procedures.
Amine Borane Activations with Tf2NH. In Situ NMR Study
Every possible effort was made to protect the reaction mixtures from exposure to air and moisture. The reactions were set up in dry J. Young NMR tubes under N2 atmosphere in a glovebox. The NMR tubes were dried in a heating oven at ca. 200 °C overnight, and the fitted Teflon valves were dried in a desiccator over Drierite. Commercial grade Tf2NH and amine boranes (Me3N–BH3 (11a), Et3N–BH3 (11b) and (iPr)2EtN–BH3 (11c)) were used without further purification. Benzylic amine borane p-MeC6H4CH2NMe2–BH3 (11d)2 and 1-neopentylpyrrolidine borane (11e)5 were prepared as reported previously. Commercial grade CD2Cl2 and d8-PhMe (Cambridge Isotope Laboratories) were not distilled, but simply dried with freshly activated molecular sieves in the glovebox.
When solid amine boranes were used (Me3N–BH3 (11a), p-MeC6H4CH2NMe2–BH3 (11d), 1-neopentylpyrrolidine borane (11e)), the reaction tube was charged with a mixture of solid Tf2NH and the corresponding amine borane. The solid mixture was dissolved by adding the solvent (either 0.6 mL CD2Cl2 or 0.8 mL d8-PhMe) to the tube in one portion at rt. Gas liberation was observed, although no substantial exotherm was noted, potentially due to the small scale of the reaction. The tube was sealed with the fitted Teflon valve, and then shaken vigorously for ca. 1 min. The amounts of the reagents used in each particular case are listed below.
When liquid amine boranes were used (Et3N–BH3 (11b) and (iPr)2EtN–BH3 (11c)), the reaction tube was charged with a solution of Tf2NH in 0.6 mL of CD2Cl2. Neat amine borane was then added to the solution via a microsyringe at rt, causing intense gas liberation, although no substantial exotherm was noted, potentially due to the small scale of the reaction. The tube was sealed with the fitted Teflon valve, and then shaken vigorously for ca. 1 min. The amounts of the reagents used in each particular case are listed below.
The ratios of N-/O-bound isomers of the products were measured by NMR after the initial equilibration had completed, and were confirmed to remain stable for 2–14 days at rt.
12a:13a, 7:1 ratio after equilibration. The following reagents were used: trimethylamine borane (11a) (8.0 mg, 0.109 mmol), Tf2NH (30.7 mg, 0.109 mmol), CD2Cl2 (0.6 mL). 12a: 1H NMR (500 MHz, CD2Cl2): δ 3.2-1.9 (br m, 2H), 2.65 ppm (s, 9H). 11B NMR (160 MHz, CD2Cl2): δ −3.7 ppm (t, J = 115 Hz). 13C NMR (126 MHz, CD2Cl2): δ 119.6 (q, JC–F = 326 Hz), 51.8 ppm. 19F NMR (471 MHz, CD2Cl2): δ −69.2 ppm (s). 13a: 1H NMR (500 MHz, CD2Cl2): δ 3.2-1.9 (br m, 2H), 2.69 ppm (s, 9H). 11B NMR (160 MHz, CD2Cl2): δ 4.0 ppm (t, J = 120 Hz). 13C NMR (126 MHz, CD2Cl2): δ 119.1 (q, JC–F = 320 Hz), 118.7 (q, JC–F = 321 Hz), 49.7 ppm. 19F NMR (471 MHz, CD2Cl2): δ −76.6 (s), −79.1 ppm (s).
12b:13b, 1:4.7 ratio after equilibration. The following reagents were used: triethylamine borane (12b) (13.3 μL, 90.7 μmol), Tf2NH (25.5 mg, 90.7 μmol), CD2Cl2 (0.6 mL). 12b: 1H NMR (500 MHz, CD2Cl2): δ 3.4- 1.9 (br m, 2H), 2.88 (q, J = 7.2 Hz, 6H), 1.21 ppm (t, J = 7.2 Hz, 9H). 11B NMR (160 MHz, CD2Cl2): δ −7.4 ppm (unres t). 13C NMR (126 MHz, CD2Cl2): δ 119.6 (q, JC–F = 327 Hz), 49.8, 8.2 ppm. 19F NMR (471 MHz, CD2Cl2): δ −68.9 ppm (s). 13b: 1H NMR (500 MHz, CD2Cl2): δ 3.4-1.9 (br m, 2H), 2.90 (q, J = 7.2 Hz, 6H), 1.21 ppm (t, J = 7.2 Hz, 9H). 11B NMR (160 MHz, CD2Cl2): δ 0.7 ppm (unres t). 13C NMR (126 MHz, CD2Cl2): δ 119.2 (q, JC–F = 320 Hz), 118.7 (q, JC–F = 321 Hz), 49.2, 7.5 ppm. 19F NMR (471 MHz, CD2Cl2): δ −76.7 (s), −79.1 ppm (s).
12c:13c, <1:25 ratio after equilibration. The following reagents were used: (iPr)2EtN–BH3 (11c) (26.7 μL, 0.153 mmol), Tf2NH (43.0 mg, 0.153 mmol), CD2Cl2 (0.6 mL). Due to the low concentration of the N-bound isomer 12c in solution, only 19F signals were assigned. 12c: 19F NMR (471 MHz, CD2Cl2): δ −68.4 ppm (s). 13c: 1H NMR (500 MHz, CD2Cl2): δ 3.70-3.60 (m, 2H), 3.4-2.1 (br m, 2H), 3.01 (q, J = 7.3 Hz, 2H), 1.37-1.33 (m, 12H), 1.26 ppm (t, J = 7.3 Hz, 3H). 11B NMR (160 MHz, CD2Cl2): δ 1.2 ppm (unres t). 13C NMR (126 MHz, CD2Cl2): δ 119.2 (q, JC–F = 321 Hz), 118.7 (q, JC–F = 321 Hz), 56.7, 56.6, 45.3, 18.3 (overlapping s), 9.5 ppm. 19F NMR (471 MHz, CD2Cl2): δ −77.0 (s), −79.1 ppm (s).
12d:13d, 4.2:1 ratio after equilibration. The following reagents were used: p-MeC6H4CH2NMe2–BH3 (11d) (23.0 mg, 0.141 mmol), Tf2NH (39.6 mg, 0.141 mmol), CD2Cl2 (0.6 mL). 12d: 1H NMR (500 MHz, CD2Cl2): δ 7.30-7.17 (m, 4H), 3.96 (s, 2H), 3.3-2.0 (br m, 2H), 2.48 (s, 6H), 2.39 ppm (s, 3H). 11B NMR (160 MHz, CD2Cl2): δ −3.2 ppm (unres t). 13C NMR (126 MHz, CD2Cl2): δ 140.0, 132.5, 129.4, 125.5, 119.7 (q, JC–F = 326 Hz), 64.9, 46.8, 20.9 ppm. 19F NMR (471 MHz, CD2Cl2): δ −69.0 ppm (s). 13d: 1H NMR (500 MHz, CD2Cl2): δ 7.30-7.17 (m, 4H), 3.99-3.97 (m, 2H), 3.3-2.0 (br m, 2H), 2.54 (s, 3H), 2.53 (s, 3H), 2.39 ppm (s, 3H). 11B NMR (160 MHz, CD2Cl2): δ 4.0 ppm (unres t). 13C NMR (126 MHz, CD2Cl2): δ 140.3, 132.3, 129.5, 125.1, 119.2 (q, JC–F = 321 Hz), 118.8 (q, JC–F = 321 Hz), 63.5, 45.5, 45.4, 20.9 ppm. 19F NMR (471 MHz, CD2Cl2): δ −76.6 (s), −79.1 ppm (s).
12e:13e, 1:2.6 ratio after equilibration. The following reagents were used: 1-neopentylpyrrolidine borane (11e) (10.9 mg, 70.3 μmol), Tf2NH (19.8 mg, 70.3 μmol), d8-PhMe (0.8 mL). 12e: 1H NMR (700 MHz, d8-PhMe): δ 3.2-2.3 (br m, 2H), 3.01-2.94 (m, 2H), 2.60-2.55 (m, 2H), 2.44 (s, 2H), 1.57-1.42 (m, 2H), 1.22-1.11 (m, 2H), 0.73 ppm (s, 9H). 11B NMR (225 MHz, d8-PhMe): δ −4.6 ppm (unres t). 13C NMR (176 MHz, d8-PhMe): δ 120.5 (q, JC–F = 327 Hz), 68.8, 57.8, 33.0, 30.3, 22.0 ppm. 19F NMR (471 MHz, d8-PhMe): δ −69.2 ppm (s). 13e: 1H NMR (700 MHz, d8-PhMe): δ 3.2-2.3 (br m, 2H), 3.01-2.94 (m, 1H), 2.79-2.74 (m, 1H), 2.33 (d, J = 13.7 Hz, 1H), 2.08 (d, J = 13.7 Hz, 1H), 1.98-1.93 (m, 1H), 1.93-1.86 (m, 1H), 1.57-1.42 (m, 2H), 1.22-1.11 (m, 2H), 0.78 ppm (s, 9H). 11B NMR (225 MHz, d8-PhMe): δ 0.6 ppm (unres t). 13C NMR (176 MHz, d8-PhMe): δ 120.2 (q, JC–F = 320 Hz), 119.6 (q, JC–F = 320 Hz), 72.2, 60.4, 59.4, 33.1, 29.7, 22.6, 22.1 ppm. 19F NMR (471 MHz, d8-PhMe): δ −76.7 (s), −78.7 ppm (s).
Preparation of Borylation Substrates
Preparation of 16
N-(3,3-Dimethylbutyl)pivalamide
Pivaloyl chloride (2.68 mL, 2.62 g, 21.7 mmol) in 20.0 mL of CH2Cl2 was slowly added to a stirred solution of 3,3-dimethylbutan-1-amine (2.00 g, 19.8 mmol) and Et3N (3.0 mL, 2.20 g, 21.7 mmol) in 20.0 mL of CH2Cl2. The reaction mixture was stirred at room temperature for 6h, following which it was acidified with 1N HCl, and extracted with CH2Cl2. The organic extracts were washed twice with saturated NaHCO3 solution, then dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting amide (2.49 g, 68%) was used in subsequent transformations without further purification. The expected structure was confirmed by 1H NMR spectroscopy. 1H NMR (500 MHz, CDCl3): δ 5.52 (s, 1H), 3.30-3.18 (m, 2H), 1.43-1.37 (m, 2H), 1.18 (s, 9H), 0.93 ppm (s, 9H).
Amine borane 16
The commercially available KH suspension in mineral oil (30 wt%, 0.78 g, 5.82 mmol) was shaken vigorously, and the resulting slurry was quickly transferred to a flask with a pipette and weighed, then immediately sealed with a septum and purged with dry nitrogen. Anhydrous THF (10.0 mL) was added to the flask, and a solution of N-(3,3-dimethylbutyl)pivalamide (980 mg, 5.29 mmol) in 10.0 mL THF was then added dropwise. The reaction mixture was stirred for 1h at room temperature. Ethyl iodide (0.47 mL, 907 mg, 5.82 mmol) was added dropwise, and the reaction mixture was stirred for an additional 5h at room temperature, following which it was carefully quenched with isopropanol and diluted with water. The reaction mixture was extracted 3 times with EtOAc, and the combined organic extracts were washed with brine, then dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting yellow oil was dissolved in 10.0 mL of anhydrous THF, and treated with Me2S·BH3 (0.95 mL, 9.52 mmol). The addition of the borane complex resulted in an exothermic reaction after a short induction period. After the exothermic reaction had ceased, the reaction mixture was refluxed for 1h, following which it was filtered through a short (2–3 cm) plug of silica, while eluting with CH2Cl2, to decompose the residual Me2S·BH3. Crystallization from hexanes afforded 0.691 g (61%) of 16 as a white crystalline solid, mp 49 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 3.02-2.90 (m, 2H), 2.89-2.79 (m, 2H), 2.70 (AB q, J = 14.3 Hz, 2H), 1.9-1.1 (br m, 3H), 1.71-1.55 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H), 1.15 (s, 9H), 0.93 ppm (s, 9H). 13C NMR (126 MHz, CDCl3): δ 69.4, 57.0, 55.0, 36.2, 33.6, 31.0, 29.9, 29.4, 9.4 ppm. 11B NMR (128 MHz, CDCl3): δ −11.79 ppm (q, J = 90 Hz). HRMS (EI): m/z calculated for C13H30N [M−BH3+H]+ 200.2373, found 200.2376 (+1 ppm). IR (CDCl3, NaCl): 3258, 2954, 2865, 1465, 1364, 1311, 1215, 1027 cm−1.
Preparation of 19
Prepared following the procedure listed above for the preparation of 16, using BnBr (0.549 mL, 789 mg, 4.61 mmol) instead of EtI. Yield: 0.685 g (59%) of a white crystalline solid, mp 107 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.48-7.43 (m, 2H), 7.38-7.33 (m, 3H), 4.11 (d, J = 12.9 Hz, 1H), 3.96 (d, J = 12.9 Hz, 1H), 2.98-2.88 (m, 1H), 2.79 (d, J = 13.9 Hz, 1H), 2.73 (td, J = 12.1, 5.7 Hz, 1H), 2.61 (d, J = 13.9 Hz, 1H), 2.0-1.3 (br m, 3H), 1.84-1.72 (m, 2H), 1.19 (s, 9H), 0.90 ppm (s, 9H). 13C NMR (126 MHz, CDCl3): δ 132.8, 132.1, 128.7, 127.9, 70.6, 66.0, 56.4, 36.6, 33.7, 31.2, 30.1, 29.5 ppm. 11B NMR (128 MHz, CDCl3): δ −11.23 ppm (q, J = 70 Hz). HRMS (EI): m/z calculated for C18H32N [M−BH3+H]+ 262.2529, found 262.2534 (+2 ppm). IR (CDCl3, NaCl): 2958, 2867, 2383, 2335, 2283, 2242, 1455, 1364, 1172 cm−1.
Preparation of 22
1-Benzylazacycloundecan-2-one
The commercially available KH suspension in mineral oil (30 wt%, 1.54 g, 11.5 mmol) was shaken vigorously, and the resulting slurry was quickly transferred to a flask with a pipette and weighed, then immediately sealed with a septum and purged with dry nitrogen. Anhydrous THF (10.0 mL) was added to the flask, and a solution of undecan-2-one (1.77 g, 10.5 mmol) in 20.0 mL THF was then added dropwise. The reaction mixture was stirred for 1h at room temperature. Benzyl bromide (1.50 mL, 2.15 g, 12.6 mmol) was then added dropwise, and the reaction mixture was stirred for an additional 8h at room temperature, and then carefully quenched with isopropanol and diluted with water. The reaction mixture was extracted 3 times with EtOAc, and the combined organic extracts were washed with brine, then dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by column chromatography (10/1 Hexanes/EtOAc) afforded 2.45 g (90%) of the amide as a white powder. The product was obtained as a mixture of E/Z amide isomers at room temperature, which prevented accurate assignment of the peaks and their integral values. 1H NMR (500 MHz, CDCl3): δ 7.36-7.22 (m), 7.16 (d, J = 7.3 Hz), 5.00 (d, J = 16.7 Hz), 4.63 (s), 4.46-4.39 (m), 4.36 (d, J = 16.7 Hz), 3.43 (s), 2.65-2.45 (m), 2.22-2.11 (m), 1.94-1.63 (m), 1.64-1.19 ppm (m). 13C NMR (126 MHz, CDCl3): δ 175.5, 173.8, 137.8, 137.2, 128.8, 128.5, 128.3, 127.5, 127.2, 126.4, 51.7, 46.5, 46.3, 44.9, 34.0, 29.3, 27.3, 26.3, 25.7, 25.14, 25.07, 25.0, 24.9, 24.7, 24.3, 24.1, 23.8, 23.6, 22.6 ppm. HRMS (ES+): m/z calculated for C17H26NO [M+H]+ 260.2009, found 260.2011 (+1 ppm).
Amine borane 22
Dry 1-benzylazacycloundecan-2-one (2.41 g, 9.3 mmol) was dissolved in 9.3 mL of anhydrous THF, and then treated with Me2S·BH3 (1.7 mL, 17 mmol). The addition of the borane complex resulted in an exothermic reaction after a short induction period. After the exothermic reaction had ceased, the reaction mixture was refluxed for 1h, following which it was filtered through a short (2–3 cm) plug of silica, while eluting with CH2Cl2, to decompose the residual Me2S·BH3. Crystallization of the crude product from hexanes afforded 1.71 g (71%) of pure 22 as a white crystalline solid, mp 103 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.54-7.46 (m, 2H), 7.36-7.31 (m, 3H), 3.81 (s, 2H), 3.03-2.91 (m, 2H), 2.89-2.75 (m, 2H), 1.9-1.1 (br m, 3H), 1.79-1.66 (m, 4H), 1.56-1.33 ppm (m, 12H). 13C NMR (126 MHz, CDCl3): δ 133.0, 131.9, 128.7, 127.7, 65.8, 56.5, 25.3, 24.9, 24.5, 20.3 ppm. 11B NMR (128 MHz, CDCl3): δ −12.0 ppm (q, J = 75 Hz). HRMS (EI): m/z calculated for C17H28N [M-BH3+H]+ 246.2216, found 246.2221 (+2 ppm). IR (CDCl3, NaCl): 2927, 2894, 2845, 2408, 2343, 2287, 1481, 1453, 1172, 742, 696 cm−1.
N-Ethylheptamethyleneimine borane (24)
Acetic anhydride (0.86 mL, 0.93 g, 9.14 mmol) was added dropwise to a solution of heptamethyleneimine (0.941 g, 8.31 mmol) in 5 mL of anhydrous CH2Cl2. Triethylamine (2 mL) was added to the mixture, and the resulting solution was stirred overnight at rt. Following concentration under reduced pressure the residue was partitioned between CH2Cl2 and 10% NaOH solution (10 mL), and the aqueous layer was additionally extracted with 2×10 mL CH2Cl2. The combined organic extracts were dried with MgSO4, filtered and concentrated. The residual oil was dissolved in anhydrous THF (10 mL), and Me2S·BH3 (1.4 mL, 14 mmol) was then added. The addition of the borane complex resulted in an exothermic reaction after a short induction period. After the exothermic reaction had ceased, the reaction mixture was refluxed for 1h, following which it was quenched with water (frothing!) and extracted with CH2Cl2. The combined extracts were dried with MgSO4, filtered, and concentrated under reduced pressure. Column chromatography (~150 mL silica gel, PhMe) afforded 0.591 g (49%) of a colorless oil. 1H NMR (700 MHz, CDCl3): δ 3.14-3.09 (m, 2H), 2.82-2.78 (m, 2H), 2.78 (q, J = 7.2 Hz, 2H), 1.91-1.83 (m, 2H), 1.8-1.2(br m, 3H), 1.78-1.65 (m, 5H, integral intensity somewhat uncertain due to overlap with B–H signal), 1.59-1.45 (m, 3H, integral intensity uncertain due to overlap with B–H signal), 1.29 ppm (t, J = 7.2 Hz). 13C NMR (101 MHz, CDCl3): δ 56.0, 55.3, 27.4, 25.2, 22.9, 9.6 ppm. 11B NMR (128 MHz, CDCl3): δ −12.3 ppm (q, J = 100 Hz). IR (CDCl3, NaCl): 2926, 2378, 2278, 1482, 1448, 1175, 1038 cm−1.
N-Benzylheptamethyleneimine borane (27)
Benzoyl chloride (1.44 mL, 1.73 g, 12.3 mmol) in 12.0 mL of CH2Cl2 was slowly added to a solution of heptamethyleneimine (1.27 g, 11.2 mmol) and Et3N (2.35 mL, 1.70 g, 16.8 mmol) in 12.0 mL of CH2Cl2 at rt. After 18 h at rt the reaction mixture was acidified with 1N HCl, and extracted with CH2Cl2. The organic phase was washed with saturated Na-HCO3 solution, then dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting dark yellow oil was dissolved in 11 mL of anhydrous THF, and then treated with Me2S·BH3 (2.1 mL, 21 mmol). The addition of the borane complex resulted in an exothermic reaction after a short induction period. After the exothermic reaction had ceased, the reaction mixture was refluxed for 1h, following which it was filtered through a short (2–3 cm) plug of silica, while eluting with CH2Cl2, to decompose the residual Me2S·BH3. Crystallization of the crude product from hexanes afforded 2.20 g (90%) of the pure product as a white crystalline solid, mp 76 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.51-7.42 (m, 2H), 7.40-7.31 (m, 3H), 3.87 (s, 2H), 3.10-2.92 (m, 4H), 2.01 (m, 2H), 1.9-1.2 (br m, 3H), 1.77-1.65 (m, 1H), 1.66-1.43 ppm (m, 7H). 13C NMR (126 MHz, CDCl3): δ 132.8, 132.0, 128.7, 127.8, 67.0, 55.5, 27.2, 25.3, 23.5 ppm. 11B NMR (128 MHz, CDCl3): δ −11.7 ppm (q, J = 93 Hz). HRMS (EI): m/z calculated for C14H22N [M-BH3+H]+ 204.1747, found 204.1751 (+2 ppm). IR (CDCl3, NaCl): 2993, 2952, 2916, 2871, 2860, 2398, 2334, 2275, 1482, 1454, 1177, 1168, 748, 695 cm−1.
N,N-Dimethyl-3-iodobenzylamine borane (44)
Prepared according to the previously published procedure for substituted N,N-dimethylbenzylamine borane complexes from m-iodobenzyl bromide. 2 1H NMR (500 MHz, CDCl3): δ 7.76 (dt, J = 8.0, 1.5 Hz, 1H), 7.69 (t, J = 1.5 Hz, 1H), 7.34-7.31 (m, 1H), 7.15 (t, J = 8.0 Hz, 1H), 3.91 (s, 2H), 2.52 (s, 6H), 2.2-1.4 ppm (br m, 3H). 13C NMR (101 MHz, CDCl3): δ 140.9, 138.2, 133.5, 131.5, 130.1, 94.1, 66.8, 49.9 ppm. 11B NMR (128 MHz, CDCl3): δ −8.2 ppm (q, J = 90 Hz). HRMS (EI): m/z calculated for [M-3H]+ 272.0108, found 272.0108 (0 ppm).
N,N-Dimethyl-3-chlorobenzylamine borane (47)
Neat pyrrolidine (20 mL, 17.4 g, 0.245 mol) was added slowly to a stirred solution of 3-chlorobenzyl bromide (1.96 g, 9.54 mmol) in MeOH (10 mL) at 0 °C. Upon full consumption of the bromide the reaction mixture was concentrated in vacuum, and the residue was dissolved in 10 mL of 6M HCl. The solution was washed with 3×10 mL Et2O (washes discarded), and then carefully made strongly basic by adding NaOH. The reaction mixture was then extracted with Et2O, the combined extracts were dried with MgSO4, filtered and concentrated under reduced pressure. Treatment of the resulting oil with Me2S·BH3 (0.96 mL, 9.5 mmol) in CH2Cl2 afforded a clear solution, which was concentrated under reduced pressure. Dissolving the crude product in CHCl3 followed by filtration through a fine frit afforded a white solid after concentration. Crystallization of the solid from cyclohexane/CHCl3 produced 1.57 g (79%) of the desired amine borane. 1H NMR (500 MHz, CDCl3): δ 7.41-7.39 (m, 1H), 7.39-7.35 (m, 1H), 7.34-7.29 (m, 2H), 3.99 (s, 2H), 3.16-3.05 (m, 2H), 2.88-2.78 (m, 2H), 2.28-2.12 (m, 2H), 2.1-1.2 (br m, 3H), 1.89-1.74 ppm (m, 2H). 13C NMR (101 MHz, CDCl3): δ 134.1, 133.9, 132.4, 130.8, 129.4, 129.1, 64.9, 59.2, 22.5 ppm. 11B NMR (128 MHz, CDCl3): δ −11.1 ppm (q, J = 95 Hz). HRMS (ES+): m/z calculated for [M+Na]+ 232.1040, found 232.1042 (+1 ppm).
N,N-dimethyl-3-phenoxybenzylamine borane (53)
A solution of 3-phenoxybenzoic acid (2.14 g, 10 mmol) in 20 mL SOCl2 was refluxed for 3.5 h. Unreacted SOCl2 was distilled off, and the residual oil was dissolved in 20 mL CH2Cl2. The acid chloride solution was then added dropwise to 40% aqueous Me2NH (40 mL) at 0 °C, and the resulting mixture was stirred at rt for several hours. The reaction mixture was then carefully acidified with 10% aqueous HCl, and extracted with 3×40 mL CH2Cl2. The combined extracts were washed with 10% NaOH, dried with MgSO4, concentrated, and dried in vacuum. The crude amide was dissolved in 25 mL THF, and then treated with BH3·SMe2 (3.0 mL). After a brief induction period an exothermic reaction followed. The reaction mixture was then refluxed for 1h, diluted with hexanes (~20 mL) and left in the freezer overnight. The slurry was filtered through a glass frit, and the clear solution was then passed through a short plug of silica, while eluting with CH2Cl2. Concentration of the solution afforded a clear oil, which was crystallized from CHCl3/hexanes, affording 1.90 g (79%) of a colorless solid. 1H NMR (500 MHz, CDCl3): δ 7.39-7.32 (m, 3H), 7.16-7.11 (t, J = 7.3 Hz, 1H), 7.08-6.99 (m, 4H), 6.97 (s, 1H), 3.94 (s, 2H), 2.51 (s, 6H), 2.2-1.2 ppm (br m, 3H). 13C NMR (101 MHz, CDCl3): δ 157.4, 156.7, 133.1, 129.9, 129.7, 126.9, 123.8, 122.4, 119.2, 119.0, 67.2, 49.8 ppm. 11B NMR (128 MHz, CDCl3): δ −8.2 ppm (q, J = 80 Hz). HRMS (ES): m/z calculated for C15H20BNONa [M+Na]+ 264.1530, found 264.1532. mp 68 °C (CHCl3/hexanes).
N,N-Dimethyl-4-phenoxybenzylamine borane (56)
Prepared following the procedure listed above for the preparation of 53, using 4-phenoxybenzoic acid (2.14 g, 10 mmol) instead of 3-phenoxybenzoic acid. Crystallized by dissolving the crude product oil in cyclohexane, and allowing to stand at rt overnight. Yield: 1.36 g (56%) of 56, mp 144 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.40-7.33 (m, 2H), 7.27 (m, 2H), 7.18-7.13 (m, 1H), 7.06-7.02 (m, 2H), 7.02-6.97 (m, 2H), 3.95 (s, 2H), 2.51 (s, 6H), 2.1-1.5 ppm (br m, 3H). 13C NMR (126 MHz, CDCl3): δ 158.4, 156.3, 133.7, 129.9, 125.7, 124.0, 119.5, 118.0, 66.9, 49.7 ppm. 11B NMR (128 MHz, CDCl3): δ −8.5 ppm (q, J = 70 Hz). HRMS (EI): m/z calculated for C15H18NO [M-BH3+H]+ 228.1383, found 228.1382. IR (CDCl3, NaCl): 3003, 2949, 2373, 2324, 2272, 2248, 1589, 1508, 1489, 1243, 1168, 1017 cm−1.
N,N-Dimethyl-1-naphthylmethylamine borane (61)
Prepared according to the previously published procedure for substituted N,N-dimethylbenzylamine borane complexes from 1-chloromethyl-naphthalene. 2 1H NMR (500 MHz, CDCl3): 8.31 (d, J = 8.5 Hz, 1H), 7.95-7.88 (m, 2H), 7.59 (ddd, J = 8.2, 6.8, 1.4 Hz, 1H), 7.55-7.48 (m, 3H), 4.55 (s, 2H), 2.53 (s, 6H), 2.6-1.6 ppm (br m, 3H). 11B NMR (128 MHz, CDCl3): δ −7.7 ppm (q, J = 95 Hz).
N,N-Dimethyl-2,6-dimethylbenzylamine borane (64)
Prepared according to the previously published procedure for substituted N,N-dimethylbenzylamine borane complexes from 2,6-dimethylbenzylchloride. 2 1H NMR (500 MHz, CDCl3): δ 7.19 (dd, J = 8.2, 6.9 Hz, 1H), 7.11 (d, J = 7.5 Hz, 2H), 4.26 (s, 2H), 2.52 (s, 6H), 2.44 (s, 6H), 2.3-1.5 ppm (br m, 3H). 13C NMR (101 MHz, CDCl3): δ 139.8, 129.2, 128.9, 128.7, 59.4, 50.1, 21.6 ppm. 11B NMR (128 MHz, CDCl3): δ −7.7 ppm (q, J = 95 Hz). HRMS (ES+): m/z calculated for C11H18N [M-BH3+H]+ 164.1434, found 164.1431 (−2 ppm).
Intramolecular C–H Borylation. General Procedures
Small Scale Activation
Stoichiometric borylations using 0.9 equiv Ph3C[B(C6F5)4] were performed as described in the previously published procedure.2 Catalytic activation used the previously published procedure as follows.5 A dry 12 mL thick-walled Schlenk tube fitted with a teflon stopper was charged with a mixture of solid amine borane (1.32 mmol) and Tf2NH (18.6 mg, 66.2 μmol). Solvent (3 mL) was then added, and some minor frothing due to gas formation was observed. The gas formed during the initial activation stage was identified as H2 in an in situ NMR study. After H2 liberation ceased, and the gas was allowed to escape the reaction vessel, the tube was sealed and heated at 160 °C (bath) for the indicated time. When liquid amine borane complexes were used, the substrate was first dissolved in 1 mL of the solvent, and then Tf2NH and the additional solvent were added. The reaction mixture was quenched by adding solid n-Bu4NBH4 (~30 mg) under N2 atmosphere. The mixture was then diluted with CH2Cl2, and filtered through a short plug of silica, eluting with CH2Cl2 or CHCl3. The products were isolated by concentrating the solution, followed by crystallization or chromatography to isolate pure isomers.
Large Scale Catalytic Borylation of 1
A dry 50 mL flask was charged with a mixture of solid BnNMe2–BH3 (1) (4.00 g, 26.8 mmol) and Tf2NH (0.377 g, 1.34 mmol). To the solid mixture, 10 mL of dry tetralin was added, and the resulting suspension was heated at 180 °C for 17 h. Upon cooling the reaction mixture to rt, 20 mL of hexanes was added, and the resulting suspension was left in the freezer overnight. The solid was filtered out and thoroughly washed with 2×10 mL of cold hexanes. The crude product was extracted on the filter with 20 mL + 2×10 mL of PhMe, and the combined toluene extracts were concentrated in vacuum. Recrystallization from 10 mL of cyclohexane, followed by washing the product with 2×4 mL of cyclohexane and drying in vacuum afforded 3.05 g (77%) of 37 as a white solid, identical by NMR assay to material prepared on small scale using the stoichiometric method.5
Large Scale Catalytic Borylation of 44
A dry 50 mL flask was charged with a mixture of solid 44 (4.00 g, 14.5 mmol) and Tf2NH (204 mg, 0.727 mmol). To the solid mixture 10.0 mL of dry tetralin was added, and the resulting suspension was heated at 180 °C for 17 h. Upon cooling the reaction mixture to room temperature, 20 mL of hexanes were added, and the resulting suspension was left in the freezer overnight. The solid was collected by filtration and thoroughly washed with 2×10 mL of cold hexanes. The crude product was extracted on the filter with 20 mL + 2×15 mL of a 2:1 THF/Et2O mixture, the organic extracts were combined, and solvents were evaporated under reduced pressure. The product 45 (3.77 g, 95%) was found to be sufficiently pure to be used further without recrystallization.
Intramolecular C–H Borylation Products
17: clear oil. 1H NMR (400 MHz, CDCl3): δ 2.99-2.67 (m, 4H), 2.64-2.51 (unres AB q, J = 12.6 Hz, 2H), 2.3-1.7 (br m, 2H), 1.58 (td, J = 12.6, 4.3 Hz, 1H), 1.30 (ddd, J = 12.9, 10.5, 4.1 Hz, 1H), 1.13 (m, 9H), 0.93 (s, 9H), 0.68 ppm (t, J = 5.5 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 73.0, 53.7, 52.1, 37.6, 36.6, 32.5, 32.4, 30.3, 29.7, 29.3, 9.2 ppm. 11B NMR (128 MHz, CDCl3): δ −4.4 ppm (t, J = 80 Hz). HRMS (ESI+): m/z calculated for C13H29BN [M−H]+ 210.2388, found 210.2388. IR (CDCl3, NaCl): 2955, 2864, 2348, 1467, 1366, 1235, 1197, 1171, 1133 cm−1.
20: white crystalline solid, mp 78 °C (hexanes). 1H NMR (400 MHz, CDCl3): δ 7.34 (d, J = 7.2 Hz, 1H), 7.18 (t, J = 7.2 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 4.21 (d, J = 14.6 Hz, 1H), 4.08 (d, J = 14.6 Hz, 1H), 3.57-2.60 (br m, 1H), 3.09 (ddt, J = 17.2, 13.5, 8.3 Hz, 2H), 2.88 (d, J = 12.4 Hz, 1H), 2.68 (d, J = 12.4 Hz, 1H), 1.56 (dd, J = 17.2, 9.1 Hz, 2H), 1.11 (d, J = 9.9 Hz, 3H), 1.05 (dd, J = 13.4, 7.1 Hz, 1H), 0.94 (s, 9H), 0.83-0.78 ppm (m, 4H). 13C NMR (126 MHz, CDCl3): δ 157.0-155.7 (br m), 138.1, 128.9, 127.1, 124.8, 121.3, 72.1, 66.2, 58.1, 39.6, 37.5, 32.9, 31.3, 29.8, 29.3 ppm. The additional 13Caliph–B signal is likely to be located between 33.6-32.0 ppm as suggested by peak shape analysis, but precise assignment is complicated by the overlapping sharp signal at 32.9 ppm. 11B NMR (128 MHz, CDCl3): δ 6.0 ppm (unres. d). HRMS (ESI+): m/z calculated for C18H29BN [M−H]+ 270.2388, found 270.2396 (+3 ppm). IR (CDCl3, NaCl): 3056, 3000, 2956, 2898, 2863, 2833, 2351, 2330, 1456, 1446, 1365, 1172, 1070, 1018, 844 cm−1.
23: white crystalline solid, mp 73 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.41 (d, J = 7.1 Hz, 1H), 7.17 (td, J = 7.1, 1.8 Hz, 1H), 7.10 – 7.03 (m, 2H), 3.97 (s, 2H), 3.11 – 2.97 (m, 4H), 2.96 – 2.34 (br m, 2H), 1.91 – 1.78 (m, 2H), 1.78 – 1.66 (m, 2H), 1.53 (m, 4H), 1.49 – 1.35 ppm (m, 8H). 13C NMR (126 MHz, CDCl3): δ 154.0-152.2 (br m), 138.5, 129.6, 127.0, 124.7, 121.5, 65.0, 54.7, 25.7, 25.3, 24.8, 21.4 ppm. 11B NMR (128 MHz, CDCl3): δ −2.9 ppm (t, J = 90 Hz). HRMS (ESI+): m/z calculated for C17H27BN [M−H]+ 256.2231, found 256.2233 (+1 ppm). IR (CDCl3, NaCl): 2999, 2960, 2915, 2859, 2386, 2343, 2299, 1480, 1448, 1173, 1072 cm−1.
25: white crystalline solid, mp 56 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 2.94-2.87 (m, J = 11.7 Hz, 2H), 2.87-2.74 (m, 2H), 2.78 (q, J = 7.4 Hz, 2H), 2.22-2.07 (m, 2H), 2.07-1.36 (br m, 2H), 1.89 (ddd, J = 17.7, 12.1, 5.6 Hz, 2H), 1.76-1.64 (m, 4H), 1.18 (t, J = 7.4 Hz, 3H), 0.93 ppm (br s, 1H). 13C NMR (126 MHz, CDCl3): δ 61.5, 57.4, 30.8, 24.8, 21.1-20.0 (br m), 7.1 ppm. 11B NMR (128 MHz, CDCl3): δ −4.8 ppm (t, J = 95 Hz). HRMS (ESI+): m/z calculated for C9H19BN [M−H]+ 152.1605, found 152.1604 (−1 ppm). IR (CDCl3, NaCl): 2989, 2893, 2834, 2315, 1468, 1450, 1184, 1150, 794 cm−1.
Formation of isomers 28, 29 and 30 by borylation of 27
The isomer mixture was separated by preparative TLC on silica gel (1:1 CH2Cl2:hexanes); 28: white crystalline solid, mp 85 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.40 (d, J = 7.2 Hz, 1H), 7.17 (td, J = 7.1, 1.9 Hz, 1H), 7.09-7.03 (m, 2H), 4.03 (s, 2H), 3.24 (tt, J = 30.6, 12.0 Hz, 2H), 3.08 (ddd, J = 14.0, 8.3, 2.0 Hz, 2H), 2.94-2.41 (br m, 2H), 1.97-1.87 (m, 2H), 1.86-1.75 (m, 2H), 1.74-1.59 ppm (m, 8H). 13C NMR (126 MHz, CDCl3): δ 138.5, 129. 6, 127.0, 124.7, 121.6, 66.1, 55.0, 27.4, 24.7, 23.5 ppm; aromatic 13C–B signal not detected. 11B NMR (128 MHz, CDCl3): δ −2.3 ppm (t, J = 91 Hz). HRMS (ESI+): m/z calculated for C14H21BN [M−H]+ 214.1762, found 214.1764 (+1 ppm). IR (CDCl3, NaCl): 2920, 2855, 2342, 2283, 1476, 1446, 1340, 1177, 1068, 1021 cm−1. 29: white crystalline solid, mp 105 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.39-7.35 (m, 3H), 7.31 (dt, J = 7.8, 3.8 Hz, 2H), 3.89 (s, 2H), 3.04-2.90 (m, 2H), 2.85 (dd, J = 11.9, 6.3 Hz, 2H), 2.04 (dtd, J = 19.3, 13.0, 6.2 Hz, 2H), 1.97-1.86 (m, 2H), 1.84-1.59 (br m, 2H), 1.75-1.64 (m, 4H), 0.96 ppm (s, 1H). 13C NMR (126 MHz, CDCl3): δ 132.6, 130.4, 128.8, 128.2, 71.4, 57.7, 30.7, 25.0, 21.9-20.2 ppm (br m). 11B NMR (128 MHz, CDCl3): δ −3.3 ppm (t, J = 85 Hz). HRMS (ESI+): m/z calculated for C14H21BN [M−H]+ 214.1762, found 214.1763. IR (CDCl3, NaCl): 2895, 2839, 2319, 1452, 1150, 1097, 1021 cm−1. 30: white crystalline solid, mp 94 °C (hexanes). 1H NMR (500 MHz, CDCl3): δ 7.32 (d, J = 7.1 Hz, 1H), 7.19-7.15 (m, 1H), 7.10-7.04 (m, 2H), 4.26 (d, J = 13.2 Hz, 1H), 4.07 (d, J = 13.2 Hz, 1H), 3.46-2.79 (br m, 1H), 3.12 (dd, J = 9.3, 3.6 Hz, 2H), 2.99 (td, J = 12.4, 6.1 Hz, 1H), 2.87 (ddd, J = 12.4, 9.3, 5.5 Hz, 1H), 2.16-2.06 (m, 1H), 2.05-1.93 (m, 2H), 1.91-1.83 (m, 1H), 1.82-1.72 (m, 2H), 1.71-1.61 (m, 1H), 1.36 ppm (ddd, J = 14.3, 12.1, 5.1 Hz, 2H). 13C NMR (126 MHz, CDCl3): δ 138.8, 128.6, 126.9, 124.7, 121.7, 68.4, 61.9, 59.2, 37.1, 35.8, 29.5-27.4 (br m), 26.3, 25.6 ppm; aromatic 13C–B signal not detected. 11B NMR (128 MHz, CDCl3): δ 2.25 ppm (d, J = 95 Hz). HRMS (ESI+): m/z calculated for C14H19BN [M−H]+ 212.1605, found 212.1610 (+2 ppm). IR (CDCl3, NaCl): 3057, 2999, 2923, 2833, 2325, 1459, 1448, 1334, 1177, 1177, 1117, 1055, 990 cm−1.
Borylation of 47 to give 48 and 49
The major product 48 was isolated by crystallizing the crude isomer mixture from hexanes. The minor product 49 was recovered from the concentrated mother liquor by preparative TLC on silica gel (4:1 hexanes: EtOAc). 48: white crystalline solid. 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 7.8 Hz, 1H), 7.14 (dd, J = 7.8, 1.9 Hz, 1H), 7.05 (d, J = 1.9 Hz, 1H), 4.07 (s, 2H), 3.36-3.25 (m, 2H), 3.1-2.3 (br m, 2H), 2.29-2.82 (m, 2H), 2.23-2.11 (m, 2H), 2.05-1.94 ppm (m, 2H). 13C NMR (101 MHz, CDCl3): δ 153.0-150.3 (br m), 140.9, 130.7, 130.4, 127.0, 121.5, 66.3, 60.3, 22.6 ppm. 11B NMR (128 MHz, CDCl3): δ −3.3 ppm (t, J = 95 Hz). HRMS (EI): m/z calculated [M−H]+ 206.0908, found 206.0916 (+4 ppm). 49: colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.15 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.7, 1H), 6.95 (d, J = 7.5 Hz, 1H), 4.15 (s, 2H), 3.41-3.32 (m, 2H), 3.2-2.3 (br m, 2H), 2.94-2.83 (m, 2H), 2.27-2.15 (m, 2H), 2.06-1.95 ppm (m, 2H). 13C NMR (101 MHz, CDCl3): δ 152.7-150.5 (br m), 140.7, 136.1, 127.3, 126.7, 119.5, 66.9, 60.7, 22.5 ppm. 11B NMR (128 MHz, CDCl3): δ −3.6 ppm (t, J = 95 Hz). HRMS (EI): m/z calculated [M−H]+ 206.0908, found 206.0899 (−4 ppm).
Borylation of 53 to form 54 and 55
The major product 54 was isolated by crystallizing the crude isomer mixture from hexanes/CHCl3. The minor product 55 was recovered from the concentrated mother liquor by preparative TLC on silica gel (eluted with 2:1 hexanes:EtOAc to collect fraction with Rf 0.38, which was then additionally purified by preparative TLC using PhMe eluent). 54: white solid, mp 109 °C (hexanes), Rf 0.53 (2:1 hexanes:EtOAc). 1H NMR (700 MHz, CDCl3) δ 7.37 (d, J = 7.8, 1H), 7.31-7.28 (m, 2H), 7.04 (tt, J = 7.4, 0.9 Hz, 1H), 6.99-6.96 (m, 2H), 6.89 (dd, J = 7.8, 2.2 Hz, 1H), 6.77 (d, J = 1.7 Hz, 1H), 4.00 (s, 2H), 3.1-2.4 (br m, 2H), 2.77 (s, 6H). 13C NMR (176 MHz, CDCl3): δ 158.3, 154.8, 148.4-147.3 (br m), 140.2, 130.7, 129.5, 122.4, 118.6, 118.1, 113.2, 69.4, 51.0 ppm. 11B NMR (128 MHz, CDCl3): δ −1.5 ppm (unres. t). HRMS (EI): m/z calculated for C15H17BNO [M−H]+ 238.1403, found 238.1412 (+4 ppm). 55: white solid. 1H NMR (700 MHz, CDCl3) δ 7.27-7.24 (m, 2H), 7.10 (t, J =7.6 Hz, 1H), 6.99 (tt, J = 7.4, 1.1 Hz, 1H), 6.96-6.93 (m, 2H), 6.90 (d, J = 7.4 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 4.05 (s, 2H), 3.0-2.3 (br m, 2H), 2.73 (s, 6H). 13C NMR (176 MHz, CDCl3): δ 158.4, 157.3, 144.4-143.0 (br m), 141.4, 129.1, 127.0, 121.7, 118.7, 118.0, 117.8, 69.4, 50.9 ppm. 11B NMR (225 MHz, CDCl3): δ −2.6 ppm (unres. t). HRMS (ES+): m/z calculated for C15H17BNO [M−H]+ 238.1398, found 238.1401 (+1 ppm).
57: white colorless solid, mp 88 °C (hexanes). 1H NMR (700 MHz, CDCl3) δ 7.30 (dt, J = 8.3, 4.8 Hz, 2H), 7.07-6.99 (m, 5H), 6.74 (dd, J = 8.0, 2.3 Hz, 1H), 4.02 (s, 2H), 3.0-2.4 (br m, 2H), 2.76 (s, 6H). 13C NMR (176 MHz, CDCl3): δ 157.9, 156.6, 156.4-155.5 (br m), 133.6, 129.5, 122.7, 122.5, 120.1, 118.6, 115.7, 69.2, 50.9 ppm. 11B NMR (225 MHz, CDCl3): δ −1.5 ppm (unres. t). HRMS (ESI+): m/z calculated for C15H17BNO [M−H]+ 238.1398, found 238.1400 (+1 ppm).
62: 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 6.6 Hz, 1H), 7.45 (dd, J = 8.0, 6.8 Hz, 1H), 7.32 (dd, J = 8.2, 6.9 Hz, 1H), 7.12 (m, 1H), 4.22 (s, 2H), 3.3-2.4 (br m, 2H), 2.72 ppm (s, 6H). 11B NMR (128 MHz, CDCl3): δ −4.0 ppm (t, J = 95Hz).
Borylation of 64; isolation of 65
Isolation of 64 and 65 was accomplished by repeated preparative TLC on silica gel (4:1 hexanes:EtOAc). The identity of C–C bond insertion product 64 was established by 1H and 13C NMR analysis.2 65: colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.13 (d, J = 7.6 Hz, 1H), 7.08 (t, J = 7.6 Hz, 1H), 6.91 (d, J = 7.3 Hz, 1H), 3.82 (s, 2H), 2.67 (s, 6H), 2.4-1.6 (br m, 2H), 2.19 (s, 3H), 1.97 ppm (unres t, 2H). 13C NMR (101 MHz, CDCl3): δ 140.9, 134.4, 130.1, 129.6, 126.8, 126.3, 62.4, 51.2, 20.1-18.4 (br m), 19.5 ppm. 11B NMR (128 MHz, CDCl3): δ −6.0 ppm (t, J = 90Hz). HRMS (EI): m/z calculated [M]+ 175.1532, found 175.1525 (−4 ppm).
Hydrolysis of 45 and preparation of 72
A 250 mL round-bottom flask was charged with 45 (3.50 g, 12.8 mmol), 90 mL of MeOH and 18 mL of H2O, and the resulting mixture was refluxed for 4 h, following which the contents were concentrated under reduced pressure. The residue was then azeotropically dried by evaporating with toluene (5–7 times) under reduced pressure, affording 3.60 g (98%) of the boroxine 68 as a pale yellow powder after drying under vacuum, sufficiently pure for use in the next stage. Under N2 atmosphere, in a pre-heated 20 mL vial Pd(OAc)2 (13 mg, 58 μmol) and SPhos (48 mg, 116 μmol) were dissolved in 10.0 mL of degassed, anhydrous THF, and the resulting solution was stirred at rt for 20 min. A pre-heated 100 mL round-bottom flask was charged with a portion of the boroxine 68 from above (0.50 g, 0.58 mmol), phenylboronic acid (425 mg, 3.49 mmol), dry K3PO4 (740 mg, 3.49 mmol), and 20.0 mL of degassed, anhydrous THF. Under N2 atmosphere, the catalyst solution was then added to this mixture, which was then stirred for 22 h at 65 °C. A solution of p-iodoacetophenone (1.14 g, 4.63 mmol) in 5.0 mL of degassed, anhydrous THF and solid Ba(OH)2·8H2O (1.10 g, 3.49 mmol) were then added under N2 atmosphere, and the reaction mixture was stirred for additional 20 h at 65 °C. The reaction was filtered through celite, concentrated under reduced pressure and purified by column chromatography (95/5/2 CH2Cl2/MeOH/NH4OH). The purified compound still contained minor impurities, so it was acidified with 1N HCl and washed with Et2O (x3). The aqueous layer was then made basic by adding 10% NaOH solution, and extracted with Et2O (x3). Organic layers were combined, dried over MgSO4, and concentrated to afford 228 mg (40%) of 72 as a pale yellow powder. 72: 1H NMR (500 MHz, CDCl3): δ 8.06 – 7.99 (m, 2H), 7.81 (t, J = 7.4 Hz, 1H), 7.69 – 7.63 (m, 2H), 7.58 – 7.52 (m, 3H), 7.45 (dd, J = 10.6, 4.7 Hz, 2H), 7.38 – 7.35 (m, 1H), 7.31 (t, J = 8.0 Hz, 1H), 3.38 (s, 2H), 2.68 – 2.60 (m, 3H), 2.18 ppm (s, 6H). 13C NMR (126 MHz, CDCl3): δ 197.9, 146.3, 140.69, 140.63, 140.3, 136.8, 135.7, 130.3, 129.9, 128.82, 128.77, 128.1, 127.4, 127.2, 125.6, 61.2, 45.3, 26.7 ppm. HRMS (ES+): m/z calculated [M+H]+ 330.1852, found 330.1854 (+1 ppm).
Supplementary Material
Scheme 4.

Benzylic Amine Borane Hydrodeamination
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the NIH (GM067146).
Footnotes
The authors declare no competing financial interests.
X-ray crystallography data and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.De Vries TS, Vedejs E. Organometallics. 2007;26:3079. doi: 10.1021/om070228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Vries TS, Prokofjevs A, Harvey JN, Vedejs E. J Am Chem Soc. 2009;131:14679. doi: 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) De Vries TS, Prokofjevs A, Vedejs E. Chem Rev. 2012;112:4246. doi: 10.1021/cr200133c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piers WE, Bourke SC, Conroy KD. Angew Chem Int Ed. 2005;44:5016. doi: 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]; (c) Kölle P, Nöth H. Chem Rev. 1985;85:399. [Google Scholar]
- 4.Recent examples of electrophilic C–H borylation by amine-based borenium cations (also see refs. 2 and 5): Bagutski V, Del Grosso A, Carrillo JA, Cade IA, Helm MD, Lawson JR, Singleton PJ, Solomon SA, Marcelli T, Ingleson MJ. J Am Chem Soc. 2013;135:474. doi: 10.1021/ja3100963.Solomon SA, Del Grosso A, Clark ER, Bagutski V, McDouall JJW, Ingleson MJ. Organometallics. 2012;31:1908.Del Grosso A, Singleton PJ, Muryn CA, Ingleson MJ. Angew Chem Int Ed. 2011;50:2102. doi: 10.1002/anie.201006196.Del Grosso A, Helm MD, Solomon SA, Caras-Quinteroa D, Ingleson MJ. Chem Commun. 2011;47:12459. doi: 10.1039/c1cc14226g.Prokofjevs A, Kampf JW, Vedejs E. Angew Chem Int Ed. 2011;50:2098. doi: 10.1002/anie.201005663.Ishida N, Moriya T, Goya T, Murakami M. J Org Chem. 2010;75:8709. doi: 10.1021/jo101920p.
- 5.Prokofjevs A, Vedejs E. J Am Chem Soc. 2011;133:20056. doi: 10.1021/ja208093c. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
6.To strengthen the structural assignment of the product cation 6 previously deduced based on NMR data, X-ray quality crystals of the [HCB11Cl11]− salt were obtained from CH2Cl2/hexanes. The structure is fully consistent with the proposed connectivity, but extraction of exact structural parameters was prevented by disorder; the X-Ray structure is shown for the borenium salt corresponding to 6 with anion = [HCB11Cl11]− (50% probability ellipsoids; the counterion omitted for clarity).
- 7.Wirth A, Moers O, Blaschette A, Jones PG. Z Anorg Allg Chem. 1998;624:991. [Google Scholar]
- 8.Simchen G, Jonas S. J Prakt Chem. 1998;340:506. [Google Scholar]
- 9.(a) Brown HC, Knights EF, Scouten CG. J Am Chem Soc. 1974;96:7765. [Google Scholar]; (b) Brown HC, Bhatt MV. J Am Chem Soc. 1966;88:1440. [Google Scholar]; (c) Rickborn B, Wood SE. J Am Chem Soc. 1971;93:3940. [Google Scholar]; (d) Parks DJ, Piers WE, Yap GPA. Organometallics. 1998;17:5492. [Google Scholar]; (e) Prokofjevs A, Boussonnière A, Li L, Bonin H, Lacôte E, Curran DP, Vedejs E. J Am Chem Soc. 2012;134:12281. doi: 10.1021/ja305061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preliminary results on the catalytic borylations of 1 and 41 were reported in refs. 2 and 5, respectively.
- 11.The NMR study to probe 42/43 interconversion was performed at 120 °C for reasons of experimental convenience. The temperature decrease from 160 °C to 120 °C was confirmed to have no effect on the ratio of 42/43 (ca. 25:1) formed in the cyclization of 41.
- 12.Electrophilic borane insertions in C–C bonds are rare. For selected examples, see: Xu BH, Kehr G, Fröhlich R, Erker G. Chem Eur J. 2010;16:12538. doi: 10.1002/chem.201002047.Xu BH, Kehr G, Fröhlich R, Grimme S, Erker G. J Am Chem Soc. 2011;133:3480. doi: 10.1021/ja1092369.
- 13.The reductive C–N bond cleavage is made possible by the combination of strongly electrophilic and reducing conditions, and is somewhat reminiscent of the recently studied silylium ion-promoted hydrodefluorination (Douvris C, Ozerov O. Science. 2008;321:1188. doi: 10.1126/science.1159979.). In view of this analogy, the reactivity of PhCF3 with 1 equiv Me3N–BH3 activated by 5 mol% Tf2NH was briefly explored. Indeed, after 18h at 120 °C ca. 30% conversion to PhCH3 was observed, although neither Tf2N−, nor n-C12F26 were affected.
- 14.Due to differences in solubility among benzylamine boranes with different substituents, the solvent mixture used for product extraction from the crude mixture needs to be optimized in each individual case.
- 15.Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 16.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 17.Harris RK, Becker ED, Cabral de Menezes SM, Goodfellow R, Granger P. Pure Appl Chem. 2001;73:1795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.