

Abstract
Portopulmonary hypertension (PPHTN) is a known complication of cirrhosis. Moderate-to-severe PPHTN implies an extremely poor prognosis. It occurs in 5%-10% of patients referred for liver transplantation (LT), and probably with an higher incidence in patients with large portosystemic shunts. Patients with moderate-to-severe pulmonary hypertension have been previously excluded from LT because of the extremely high surgical risk and since the post-transplant outcome reported was poor. Recently, new perspectives in the management of patients with portopulmonary hypertension are emerging. In fact, some pulmonary vasoactive drugs have become routine in the treatment of patients with idiopathic pulmonary hypertension. These drugs, particularly epoprostenol, have been recently introduced in the treatment of patients with PPHTN, and have been shown to be effective in reducing pulmonary artery pressure as well as pulmonary vascular resistances. Furthermore, recent studies seem to demonstrate that treatment with pulmonary vasoactive drugs could allow liver transplantation with acceptable surgical risks and excellent survival. Although there are not large series nor prospective studies addressing this topic, the clinical scenario of patients with PPHTN seems to be positively changing.
Keywords: Portopulmonary hypertension, Cirrhosis, Liver transplantation, Management, Epoprostenol
Core tip: Moderate-to-severe portopulmonary hypertension (PPHTN) implies an extremely poor prognosis and patients are generally excluded from liver transplantation. Recently, some pulmonary vasoactive drugs have become routine in the treatment of patients with idiopathic pulmonary hypertension and have been recently introduced in the treatment of patients with PPHTN. Recent studies seem to demonstrate that treatment with pulmonary vasoactive drugs could allow liver transplantation with acceptable surgical risks and excellent survival. This paper reports a review on management of PPHTN.
INTRODUCTION
Portopulmonary hypertension (PPHTN) is a known and uncommon severe complication of cirrhosis, since moderate-to-severe forms have grave prognostic significance with the majority of patients succumbing within 2 years[1-4].
PPHTN affects 1%-2% of patients with portal hypertension or cirrhosis and 5%-10% of patients being evaluated for liver transplantation[5].
Diagnosis is suggested by either elevation of the right ventricular systolic pressure or right ventricular dysfunction on echocardiography, but requires confirmation by right heart catheterization[6-8].
DEFINITION OF PORTOPULMONARY HYPERTENTION
PPHTN definition has evolved over time, specifically with regard to the cutoff for pulmonary vascular resistances (PVR). Initial studies used PVR > 120 dynes s cm-5 as abnormal. Subsequent studies and the Consensus Report of the European Respiratory Task Force on pulmonary vascular diseases associated with liver diseases have recommended PVR > 240 dynes s cm-5 as the “gold standard”.
Therefore the current consensus diagnostic criteria for PPHTN include mean pulmonary artery pressure (MPAP) > 25 mmHg, PVR > 240 dynes s cm-5, and pulmonary artery occlusion pressure (PAOP) < 15 mmHg[1,6,7].
Since it has been shown that some patients with PAOP > 15 mmHg and PVR > 240 dynes s cm-5 may present a trans-pulmonary gradient (TPG) strongly suggestive of obstruction to pulmonary arterial flow, recently new criteria have been proposed by authors from Mayo Clinic[8]. These are the following: (1) Portal Hypertension and/or liver disease (clinical diagnosis-ascites/varices/splenomegaly); (2) MPAP > 25 mmHg at rest; and (3) PVR > 240 dynes s cm-5; and (4) PAOP < 15 mmHg or TPG > 12 mmHg, where TPG = MPAP - PAOP.
In fact, many patients managed as pulmonary hypertension have elevated left-sided filling pressure, probably due to ventricular interaction. Then, it has been recently proposed that only values of PAOP higher than 18 mmHg exclude the diagnosis of PPHTN[9-12].
PATHOPHYSIOLOGY AND PATHOGENESIS
PPHTN involves endothelial and smooth muscle proliferation, and have the same features of plexogenic arteriopathy of Idiopathic Pulmonary Hypertension (Figure 1). There is not a clear link between portal hypertension and the development of PPHTN. Because only 5%-10% of patients with portal hypertension develop PPHTN, factors other than portal hypertension must be involved in its development. Moreover, the link between liver dysfunction and PPHTN is not obvious, because PPHTN may develop in cases of portal vein thrombosis or idiopathic hypertension, in absence of any liver dysfunction. Data of a recent retrospective study by Talwalkar et al[13], suggest a strong association between large portosystemic shunts, hepatofugal portal blood flow, and PPHTN. These data may support the hypothesis that vasoactive factors from the splanchnic circulation may be pathogenic for PPHTN development. The fact that the most occurring shunt observed in this study was spleno-renal might suggest that the blood flow coming from the spleen, which is involved in the destruction of platelets and prostaglandins delivery, might be of primary importance in the PPHTN pathogenesis. Moreover, presence of large portosystemic shunts, such as those reported in this study, seems to be associated with lack of response to vasoactive treatment.
Figure 1.
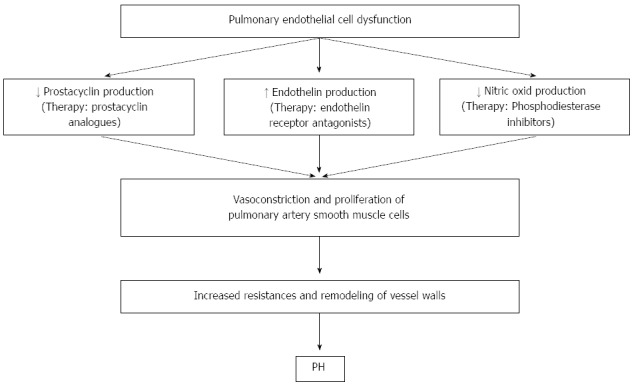
Pathophysiology of pulmonary hypertension and possible drug targets.
PROGNOSIS OF PPHTN AND LIVER TRANSPLANTATION
Moderate-to-severe PPHTN (MPAP ≥ 35 mmHg) that is present in up to 5%-10% of patients referred for liver transplantation (LT)[4,5], excludes patients from LT in most of the centers, since post-transplant outcome is poor and because of the high surgical risk[11-13].
A multicenter transplant database has shown that 36% of PPHTN patients died during the immediate post-transparent period due to progressive right ventricular failure, acute respiratory distress syndrome and cardiovascular collapse[11]. Moreover, Krowka et al[14] reported that only 29% of transplanted patients with untreated PPHTN survived 3 years.
VASODILATION TREATMENT IN PPHTN
Several vasomodulating and vasodilating drugs have been introduced for the treatment of idiopathic pulmonary hypertension. More recently, vasomodulating and vasodilating drugs, available for the treatment of idiopathic pulmonary hypertension, have also been shown to significantly improve pulmonary hemodynamics in some patients with PPHTN[5-16].
Current medications target three pulmonary hypertension pathways. The first agent, the endothelin receptor antagonists, target the vasoconstrictive endothelin pathway. The second agent targets the prostacyclin pathway, resulting in vasodilation, antiplatelet effect, and vascular remodelling. The third one involves nitric oxide-mediated vasodilation through cyclic guanosine monophosphatase and the inhibition of the phosphodiesterase type 5 enzyme. Therefore, some drugs have a vasodilation effect, while others have remodeling and antiplatelet effects.
Since we do not yet know which is the predominant mechanism which determines pulmonary hypertension in patients with portal hypertension[4], different drugs have been used in the treatment of patients with PPHTN.
The drugs that have been mostly used for the treatment of patients with PPHTN are the prostacyclin analogues, such as Epoprostenol, inhaled Iloprost and Treprostinol. Epoprostenol is a potent pulmonary and systemic vasodilator, that also reduces platelet aggregation[16-18]. In PPHTN it has been shown to improve hemodynamics acutely, because of its vasodilator effects[15]. Furthermore, in two studies prolonged use of the drug has shown additional improvement[5,15]. A recent paper by Awdish et al[19] reports the long-term effects of treatment with and without epoprostenol on pulmonary hemodynamics, liver function and survival, in a large retrospective cohort of patients with moderate to severe PPHTN. They showed significant improvements in mean pulmonary artery pressure, pulmonary vascular resistances and cardiac output with epoprostenol after a median of 15.4 mo, and no significant change of liver biochemistry. However, survival seemed not to differ between treatment groups. These studies argue against the hypothesis by Krowka that epoprostenol might worsen portal hypertension by increasing splenic blood flow and portal system congestion[15]. In fact, it is possible to hypothesize that by improving pulmonary hemodynamics and right heart function, epoprostenol could potentially improve liver function[20].
A recent prospective observation study has stressed the utility of early initiation of parenteral prostacyclin therapy in PPHTN patients, so improving 5-year survival, as compared with data of the REVEAL Registry[20].
There are scarce studies concerning the treatment with Treprostenil[21], and inhaled Iloprost[22], sometimes used together with other vasoactive drugs[23,24] in patients with PPHTN.
Concerning the endothelin receptor antagonists, such as Bosentan, there are some recent studies that seem to demonstrate a possible hemodynamic improvement and safety of these drugs[23,25-29]. Moreover, Bosentan has been administered together with different drugs in some studies: inhaled Iloprost[23,24,29], or with Sildenafil and Iloprost[24].
Phosphodiesterase inhibitor Sildenafil has the advantage of being an oral compound with pulmonary vasoselective action but no hepatotoxicity[27]. Few recent retrospective studies, performed in a small number of patients with PPHTN[30-33], seem to show that sildenafil might be effective in monotherapy, and in combination with inhaled prostanoids, leading to hemodynamic improvement. These data have also been confirmed by a recent paper by Krowka[34] reporting favourable follow up in seven patients with moderate or severe PPHTN treated with oral combination therapy.
LIVER TRANSPLANTATION FOLLOWING MEDICAL MANAGEMENT
PPHTN survival in the absence of transplantation has been reported in 38% at 3 years[1] and 28% at 5 years[35]. Furthermore, it has been demonstrated that PPHTN prognosis is worse than that of idiopathic pulmonary hypertension[1].
PPHTN is a serious problem in the context of liver transplantation. Mild PPHTN (MPAP < 35 mmHg) has scarce perioperative risks[36,37], but moderate disease (MPAP 35-45 mmHg, PVR > 250 dyne scm-5) has been associated with a perioperative mortality of 50%-80%[8,38], and a MPAP > 50 mmHg is universally fatal[14,36,37].
Recently numerous studies have demonstrated the possibility of reducing MPAP and PVR, at least in some patients affected by PPHTN, leading to a decrease of perioperative risk due to liver transplantation. Moreover, some studies have shown that PPHTN may resolve following transplantation, presumably by removing the root cause of the problem[39-41].
As a consequence of recent improved knowledge of PPHTN management, a potential therapeutic opportunity arises: PPHTN might initially be controlled with vasodilator therapy and subsequently cured with liver transplantation. Several studies have been performed to examine the feasibility of this hypothesis[31-42]. Moreover, in the last years some retrospective studies have been published reporting patients with PPHTN in whom liver transplantation was successfully performed after treatment with pulmonary vasoactive drugs[22,43,44]. In the study by Ashfaq et al[43], 16 of 20 patients with moderate-to-severe pulmonary hypertension (MPAP ≥ 35 mmHg) were otherwise considered suitable liver transplant candidates and were treated with vasoactive pulmonary drugs (epoprostenol in 13, bosentan + epoprostenol in 1, sequential; bosentan + diltiazem + epoprostenol, sequential in 1, diltiazem in 1). In these patients MPAP fell to less than 35 mmHg in 12 (75%), and 11 of them underwent liver transplantation. One-year survival was 91%, and 5-year survival was 67%. Nine of 11 patients were off vasodilator therapy after a median of 9.2 mo after transplantation. In patients who failed vasodilator therapy median survival was 8 mo. The Authors conclude that effective pharmacologic control of PPHTN before transplantation is associated with posttransplant survival that is similar to patients transplanted for other indications.
In the study by Sussman et al[44], 8 cirrhotic patients with MPAP ≥ 35 mmHg, were treated with continuous intravenous epoprostenol (2-8 ng/kg per minute). In these patients liver transplant was considered if MPAP was lowered to < 35 mmHg. In seven patients the treatment improved hemodynamics within 6.5 mo therapy: mean vascular resistances declined from 410 to 192 dyne s cm-5, and cardiac output increased from 6.6 to 10 L/min. Six of the seven responders were listed for transplantation; two died on the waiting list; four were transplanted and remained alive and well after 9 to 18 mo post LT. Epoprostenol was continued throughout surgery and into the post-transplant period. In two of the patients it was possible to stop the vasodilator therapy, whereas in two patients oral medication with bosentan was continued.
In the study by Swanson et al[4], a retrospective screening-right heart catheterization-survival analysis of patients with PPHTN was performed. Patients were categorized in three subgroups: (1) no vasoactive therapy and no transplantation; (2) therapy for pulmonary hypertension alone; and (3) therapy for pulmonary hypertension followed by liver transplantation. Even though it is a relatively small study, it seems to demonstrate that the survival of untreated patients was poor, whereas subgroups of patients selected to medical treatment with or without liver transplantation had better long-term survival. The best survival was in the subgroup of patients in whom liver transplantation was performed following effective medical therapy for pulmonary hypertension. However, it is to stress that this study is retrospective and reports a small number of patients. Moreover, 4 of 5 deaths in the LT group occurred in patients with PAOP ≤ 10 mmHg, while only two out of seven LT survivors had PAOP ≤ 10 mmHg. The authors hypothesize that lower values of PAOP may be correlated to lower cardiac outputs, and this might contribute to the poor prognosis. In the assessment of patients with PPHTN that are candidate to liver transplantation it is very important to perform an accurate evaluation of right ventricular function, because the success of undertaking liver transplantation will be determined by the ability of the right ventricle to sustain the increase of cardiac output and of pulmonary vascular resistance that acutely occur at the time of reperfusion ant that may cause acute right heart failure[45]. Therefore, pre-transplant evaluation of right ventricular function by means of right heart catheterization and stress echocardiography is essential. Continuous intraoperative trans-oesophageal echocardiography has also been recommended for following right ventricular function[46].
MODEL FOR END-STAGE LIVER DISEASE AND PPHTN
Results of recent studies concerning LT following medical treatment in patients with PPHTN, as discussed above, probably change the clinical scenarios of patients with moderate or even severe pulmonary hypertension and portal hypertension.
Outcome of these patients, traditionally excluded from LT, could benefit from LT if a response to vasoactive therapy is evident. In fact, there is increasing evidence that many patients with PPHTN have important decrease of MPAP and PVR after therapy, so that they have excellent survival following liver transplantation. Furthermore, in some of these patients, vasoactive therapy may be stopped a few months after LT. Moreover, there is not any doubt that many patients who positively respond to medical therapy die on the waiting list for transplantation. Finally, another reason could supports early transplant of these patients. In fact, long-term intravenous epoprostenol, probably the most effective drug, is expensive, labor-intensive, requires hospitalization and is difficult to tolerate. These are the reasons why in some studies patients with PPHTN on intravenous therapy are given a MELD exception of 25 points in Region 4[43].
In a recent paper, Krowka et al[47] discuss this topic suggesting MELD exception (Meld score= 26 points) when the acceptable candidates satisfy the following criteria: (1) POPH exists with severity characterized by MPAP > 35 mmHg; and (2) a minimum of 12 wk of United States Food and Drug Administration-approved pulmonary arterial hypertension therapy results in the following hemodynamic profile: (1) MPAP < 35 mmHg and PVR < 400 dynes scm-5; and (2) satisfactory right ventricular function exists.
LIVER AND LUNG TRANSPLANTATION
Combined lung and liver transplantation is a therapeutic option for selected patients with coexisting lung and liver diseases such as cystic fibrosis and alfa 1 -proteinase inhibitor deficiency, and has also been performed in few cases of patients with PPHTN[48-53]. Recently, a report concerning 13 consecutive patients who underwent combined lung and liver transplantation has been published. In the whole cohort of patients, 5 with PPHTN and 8 with other severe hepatic and pulmonary diseases (sarcoidosis, cystic fibrosis, alfa 1-proteinase inhibitor deficiency), patient and graft survival rates, after combined transplantation, were 69% after 1, 62% after 3, and 49% after 5 years[54]. However, further studies are needed to confirm the efficacy, as well as the indications of this surgical approach in the management of patients with PPHTN.
Footnotes
P-Reviewers: Aghemo A, Ramsay M, Shindoh J S-Editor: Cui XM L-Editor: A E-Editor: Wang CH
References
- 1.Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, Palevsky HI. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005;11:1107–1111. doi: 10.1002/lt.20459. [DOI] [PubMed] [Google Scholar]
- 2.Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17:492–498. doi: 10.1016/s0735-1097(10)80121-4. [DOI] [PubMed] [Google Scholar]
- 3.Hervé P, Lebrec D, Brenot F, Simonneau G, Humbert M, Sitbon O, Duroux P. Pulmonary vascular disorders in portal hypertension. Eur Respir J. 1998;11:1153–1166. doi: 10.1183/09031936.98.11051153. [DOI] [PubMed] [Google Scholar]
- 4.Swanson KL, Wiesner RH, Nyberg SL, Rosen CB, Krowka MJ. Survival in portopulmonary hypertension: Mayo Clinic experience categorized by treatment subgroups. Am J Transplant. 2008;8:2445–2453. doi: 10.1111/j.1600-6143.2008.02384.x. [DOI] [PubMed] [Google Scholar]
- 5.Kuo PC, Plotkin JS, Gaine S, Schroeder RA, Rustgi VK, Rubin LJ, Johnson LB. Portopulmonary hypertension and the liver transplant candidate. Transplantation. 1999;67:1087–1093. doi: 10.1097/00007890-199904270-00001. [DOI] [PubMed] [Google Scholar]
- 6.Colle IO, Moreau R, Godinho E, Belghiti J, Ettori F, Cohen-Solal A, Mal H, Bernuau J, Marty J, Lebrec D, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003;37:401–409. doi: 10.1053/jhep.2003.50060. [DOI] [PubMed] [Google Scholar]
- 7.Benjaminov FS, Prentice M, Sniderman KW, Siu S, Liu P, Wong F. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut. 2003;52:1355–1362. doi: 10.1136/gut.52.9.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krowka MJ, Plevak DJ, Findlay JY, Rosen CB, Wiesner RH, Krom RA. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000;6:443–450. doi: 10.1053/jlts.2000.6356. [DOI] [PubMed] [Google Scholar]
- 9.McGoon MD, Miller DP. REVEAL: a contemporary US pulmonary arterial hypertension registry. Eur Respir Rev. 2012;21:8–18. doi: 10.1183/09059180.00008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodríguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-Hepatic vascular Disorders (PHD) Eur Respir J. 2004;24:861–880. doi: 10.1183/09031936.04.00010904. [DOI] [PubMed] [Google Scholar]
- 11.Hadengue A, Benhayoun MK, Lebrec D, Benhamou JP. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100:520–528. doi: 10.1016/0016-5085(91)90225-a. [DOI] [PubMed] [Google Scholar]
- 12.Krowka MJ, Mandell MS, Ramsay MA, Kawut SM, Fallon MB, Manzarbeitia C, Pardo M, Marotta P, Uemoto S, Stoffel MP, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transpl. 2004;10:174–182. doi: 10.1002/lt.20016. [DOI] [PubMed] [Google Scholar]
- 13.Talwalkar JA, Swanson KL, Krowka MJ, Andrews JC, Kamath PS. Prevalence of spontaneous portosystemic shunts in patients with portopulmonary hypertension and effect on treatment. Gastroenterology. 2011;141:1673–1679. doi: 10.1053/j.gastro.2011.06.053. [DOI] [PubMed] [Google Scholar]
- 14.Krowka MJ, Frantz RP, McGoon MD, Severson C, Plevak DJ, Wiesner RH. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology. 1999;30:641–648. doi: 10.1002/hep.510300307. [DOI] [PubMed] [Google Scholar]
- 15.Halank M, Miehlke S, Hoeffken G, Schmeisser A, Schulze M, Strasser RH. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation. 2004;77:1775–1776. doi: 10.1097/01.tp.0000122420.86904.89. [DOI] [PubMed] [Google Scholar]
- 16.Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Clayton LM, Jöbsis MM, Blackburn SD, Shortino D, Crow JW; Primary Pulmonary Hypertension Study Group. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 17.Barst RJ, Rubin LJ, McGoon MD, Caldwell EJ, Long WA, Levy PS. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med. 1994;121:409–415. doi: 10.7326/0003-4819-121-6-199409150-00003. [DOI] [PubMed] [Google Scholar]
- 18.Fix OK, Bass NM, De Marco T, Merriman RB. Long-term follow-up of portopulmonary hypertension: effect of treatment with epoprostenol. Liver Transpl. 2007;13:875–885. doi: 10.1002/lt.21174. [DOI] [PubMed] [Google Scholar]
- 19.Awdish RL, Cajigas HR. Early Initiation of Prostacyclin in Portopulmonary Hypertension: 10 Years of a Transplant Center’s Experience. Lung. 2013;191:593–600. doi: 10.1007/s00408-013-9501-5. [DOI] [PubMed] [Google Scholar]
- 20.Sakai T, Planinsic RM, Mathier MA, de Vera ME, Venkataramanan R. Initial experience using continuous intravenous treprostinil to manage pulmonary arterial hypertension in patients with end-stage liver disease. Transpl Int. 2009;22:554–561. doi: 10.1111/j.1432-2277.2008.00830.x. [DOI] [PubMed] [Google Scholar]
- 21.Sugimachi K, Soejima Y, Morita K, Ueda S, Fukuhara T, Nagata S, Ikegami T, Taketomi A, Maehara Y. Rapid normalization of portopulmonary hypertension after living donor liver transplantation. Transplant Proc. 2009;41:1976–1978. doi: 10.1016/j.transproceed.2009.02.095. [DOI] [PubMed] [Google Scholar]
- 22.Hoeper MM, Seyfarth HJ, Hoeffken G, Wirtz H, Spiekerkoetter E, Pletz MW, Welte T, Halank M. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J. 2007;30:1096–1102. doi: 10.1183/09031936.00032407. [DOI] [PubMed] [Google Scholar]
- 23.Austin MJ, McDougall NI, Wendon JA, Sizer E, Knisely AS, Rela M, Wilson C, Callender ME, O’Grady JG, Heneghan MA. Safety and efficacy of combined use of sildenafil, bosentan, and iloprost before and after liver transplantation in severe portopulmonary hypertension. Liver Transpl. 2008;14:287–291. doi: 10.1002/lt.21310. [DOI] [PubMed] [Google Scholar]
- 24.Grander W, Eller P, Fuschelberger R, Tilg H. Bosentan treatment of portopulmonary hypertension related to liver cirrhosis owing to hepatitis C. Eur J Clin Invest. 2006;36 Suppl 3:67–70. doi: 10.1111/j.1365-2362.2006.01687.x. [DOI] [PubMed] [Google Scholar]
- 25.Hinterhuber L, Graziadei IW, Kähler CM, Jaschke W, Vogel W. Endothelin-receptor antagonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol. 2004;2:1039–1042. doi: 10.1016/s1542-3565(04)00466-5. [DOI] [PubMed] [Google Scholar]
- 26.Neuhofer W, Gülberg V, Gerbes AL. Endothelin and endothelin receptor antagonism in portopulmonary hypertension. Eur J Clin Invest. 2006;36 Suppl 3:54–61. doi: 10.1111/j.1365-2362.2006.01690.x. [DOI] [PubMed] [Google Scholar]
- 27.Barth F, Gerber PJ, Reichen J, Dufour JF, Nicod LP. Efficiency and safety of bosentan in child C cirrhosis with portopulmonary hypertension and renal insufficiency. Eur J Gastroenterol Hepatol. 2006;18:1117–1119. doi: 10.1097/01.meg.0000231749.60889.f7. [DOI] [PubMed] [Google Scholar]
- 28.Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet. 2004;363:1461–1468. doi: 10.1016/S0140-6736(04)16107-2. [DOI] [PubMed] [Google Scholar]
- 29.Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 30.Makisalo H, Koivusalo A, Vakkuri A, Hockerstedt K. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transpl. 2004;10:945–950. doi: 10.1002/lt.20153. [DOI] [PubMed] [Google Scholar]
- 31.Chua R, Keogh A, Miyashita M. Novel use of sildenafil in the treatment of portopulmonary hypertension. J Heart Lung Transplant. 2005;24:498–500. doi: 10.1016/j.healun.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 32.Reichenberger F, Voswinckel R, Steveling E, Enke B, Kreckel A, Olschewski H, Grimminger F, Seeger W, Ghofrani HA. Sildenafil treatment for portopulmonary hypertension. Eur Respir J. 2006;28:563–567. doi: 10.1183/09031936.06.00030206. [DOI] [PubMed] [Google Scholar]
- 33.Raevens S, De Pauw M, Reyntjens K, Geerts A, Verhelst X, Berrevoet F, Rogiers X, Troisi RI, Van Vlierberghe H, Colle I. Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol. 2013;25:495–502. doi: 10.1097/MEG.0b013e32835c504b. [DOI] [PubMed] [Google Scholar]
- 34.Krowka MJ. Portopulmonary hypertension and the issue of survival. Liver Transpl. 2005;11:1026–1027. doi: 10.1002/lt.20494. [DOI] [PubMed] [Google Scholar]
- 35.Krowka MJ. Editorial: Pulmonary hypertension, (high) risk of orthotopic liver transplantation, and some lessons from “primary” pulmonary hypertension. Liver Transpl. 2002;8:389–390. doi: 10.1053/jlts.2002.33134. [DOI] [PubMed] [Google Scholar]
- 36.Starkel P, Vera A, Gunson B, Mutimer D. Outcome of liver transplantation for patients with pulmonary hypertension. Liver Transpl. 2002;8:382–388. doi: 10.1053/jlts.2002.31343. [DOI] [PubMed] [Google Scholar]
- 37.De Wolf AM, Scott V, Bjerke R, Kang Y, Kramer D, Miro A, Fung JJ, Dodson F, Gayowski T, Marino IR, et al. Hemodynamic effects of inhaled nitric oxide in four patients with severe liver disease and pulmonary hypertension. Liver Transpl Surg. 1997;3:594–597. [PubMed] [Google Scholar]
- 38.Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3:494–500. doi: 10.1002/lt.500030503. [DOI] [PubMed] [Google Scholar]
- 39.Levy MT, Torzillo P, Bookallil M, Sheil AG, McCaughan GW. Case report: delayed resolution of severe pulmonary hypertension after isolated liver transplantation in a patient with cirrhosis. J Gastroenterol Hepatol. 1996;11:734–737. doi: 10.1111/j.1440-1746.1996.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 40.Schott R, Chaouat A, Launoy A, Pottecher T, Weitzenblum E. Improvement of pulmonary hypertension after liver transplantation. Chest. 1999;115:1748–1749. doi: 10.1378/chest.115.6.1748. [DOI] [PubMed] [Google Scholar]
- 41.Losay J, Piot D, Bougaran J, Ozier Y, Devictor D, Houssin D, Bernard O. Early liver transplantation is crucial in children with liver disease and pulmonary artery hypertension. J Hepatol. 1998;28:337–342. doi: 10.1016/0168-8278(88)80022-9. [DOI] [PubMed] [Google Scholar]
- 42.Tan HP, Markowitz JS, Montgomery RA, Merritt WT, Klein AS, Thuluvath PJ, Poordad FF, Maley WR, Winters B, Akinci SB, et al. Liver transplantation in patients with severe portopulmonary hypertension treated with preoperative chronic intravenous epoprostenol. Liver Transpl. 2001;7:745–749. doi: 10.1053/jlts.2001.26057. [DOI] [PubMed] [Google Scholar]
- 43.Ashfaq M, Chinnakotla S, Rogers L, Ausloos K, Saadeh S, Klintmalm GB, Ramsay M, Davis GL. The impact of treatment of portopulmonary hypertension on survival following liver transplantation. Am J Transplant. 2007;7:1258–1264. doi: 10.1111/j.1600-6143.2006.01701.x. [DOI] [PubMed] [Google Scholar]
- 44.Sussman N, Kaza V, Barshes N, Stribling R, Goss J, O’Mahony C, Zhang E, Vierling J, Frost A. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant. 2006;6:2177–2182. doi: 10.1111/j.1600-6143.2006.01432.x. [DOI] [PubMed] [Google Scholar]
- 45.Safdar Z, Bartolome S, Sussman N. Portopulmonary hypertension: an update. Liver Transpl. 2012;18:881–891. doi: 10.1002/lt.23485. [DOI] [PubMed] [Google Scholar]
- 46.Ramsay M. Portopulmonary hypertension and right heart failure in patients with cirrhosis. Curr Opin Anaesthesiol. 2010;23:145–150. doi: 10.1097/ACO.0b013e32833725c4. [DOI] [PubMed] [Google Scholar]
- 47.Krowka MJ, Fallon MB, Mulligan DC, Gish RG. Model for end-stage liver disease (MELD) exception for portopulmonary hypertension. Liver Transpl. 2006;12:S114–S116. doi: 10.1002/lt.20975. [DOI] [PubMed] [Google Scholar]
- 48.Couetil JP, Houssin DP, Soubrane O, Chevalier PG, Dousset BE, Loulmet D, Achkar A, Tolan MJ, Amrein CI, Guinvarch A. Combined lung and liver transplantation in patients with cystic fibrosis. A 4 1/2-year experience. J Thorac Cardiovasc Surg. 1995;110:1415–1422; discussion 1422-1223. doi: 10.1016/s0022-5223(95)70064-1. [DOI] [PubMed] [Google Scholar]
- 49.Couetil JP, Soubrane O, Houssin DP, Dousset BE, Chevalier PG, Guinvarch A, Loulmet D, Achkar A, Carpentier AF. Combined heart-lung-liver, double lung-liver, and isolated liver transplantation for cystic fibrosis in children. Transpl Int. 1997;10:33–39. doi: 10.1007/BF02044339. [DOI] [PubMed] [Google Scholar]
- 50.Dennis CM, McNeil KD, Dunning J, Stewart S, Friend PJ, Alexander G, Higenbottam TW, Calne RY, Wallwork J. Heart-lung-liver transplantation. J Heart Lung Transplant. 1996;15:536–538. [PubMed] [Google Scholar]
- 51.Praseedom RK, McNeil KD, Watson CJ, Alexander GJ, Calne RY, Wallwork J, Friend PJ. Combined transplantation of the heart, lung, and liver. Lancet. 2001;358:812–813. doi: 10.1016/S0140-6736(01)06003-2. [DOI] [PubMed] [Google Scholar]
- 52.Barshes NR, DiBardino DJ, McKenzie ED, Lee TC, Stayer SA, Mallory GB, Karpen SJ, Quiros-Tejeira RE, Carter BA, Fraser CD, et al. Combined lung and liver transplantation: the United States experience. Transplantation. 2005;80:1161–1167. doi: 10.1097/01.tp.0000165717.23652.09. [DOI] [PubMed] [Google Scholar]
- 53.Trulock EP, Edwards LB, Taylor DO, Boucek MM, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-second official adult lung and heart-lung transplant report--2005. J Heart Lung Transplant. 2005;24:956–967. doi: 10.1016/j.healun.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Grannas G, Neipp M, Hoeper MM, Gottlieb J, Lück R, Becker T, Simon A, Strassburg CP, Manns MP, Welte T, et al. Indications for and outcomes after combined lung and liver transplantation: a single-center experience on 13 consecutive cases. Transplantation. 2008;85:524–531. doi: 10.1097/TP.0b013e3181636f3f. [DOI] [PubMed] [Google Scholar]