

Multiple cis-elements surrounding the β-globin gene locus combine to target this locus to the nuclear periphery through at least two different epigenetic marks.
Abstract
Increasing evidence points to nuclear compartmentalization as a contributing mechanism for gene regulation, yet mechanisms for compartmentalization remain unclear. In this paper, we use autonomous targeting of bacterial artificial chromosome (BAC) transgenes to reveal cis requirements for peripheral targeting. Three peripheral targeting regions (PTRs) within an HBB BAC bias a competition between pericentric versus peripheral heterochromatin targeting toward the nuclear periphery, which correlates with increased H3K9me3 across the β-globin gene cluster and locus control region. Targeting to both heterochromatin compartments is dependent on Suv39H-mediated H3K9me3 methylation. In different chromosomal contexts, PTRs confer no targeting, targeting to pericentric heterochromatin, or targeting to the periphery. A combination of fluorescent in situ hybridization, BAC transgenesis, and knockdown experiments reveals that peripheral tethering of the endogenous HBB locus depends both on Suv39H-mediated H3K9me3 methylation over hundreds of kilobases surrounding HBB and on G9a-mediated H3K9me2 methylation over flanking sequences in an adjacent lamin-associated domain. Our results demonstrate that multiple cis-elements regulate the overall balance of specific epigenetic marks and peripheral gene targeting.
Introduction
Spatial compartmentalization of chromatin may contribute to regulation of genome function (Zhao et al., 2009; Cope et al., 2010; Geyer et al., 2011; Meldi and Brickner, 2011). In many higher metazoans, transcriptionally silent genes are preferentially located toward the nuclear periphery, with more active genes preferentially located in the nuclear interior (Peric-Hupkes and van Steensel, 2010; Shevelyov and Nurminsky, 2012). Recent genome-wide studies using the DNA adenine methyltransferase identification (DamID) method have mapped preferred genome–lamin interactions in Drosophila melanogaster and cultured mammalian cells, suggesting increased interaction of transcriptionally inactive regions with the nuclear lamina (Peric-Hupkes and van Steensel, 2010). In human fibroblasts, >1,300 sharply defined domains with sizes of 0.1–10 Mb were shown to preferentially interact with the nuclear lamina (Guelen et al., 2008). These lamina-associated domains (LADs) are enriched in repressive chromatin marks and genes with low expression levels. Similarly, some inactive genes localize to pericentromeric heterochromatin (PCH). In cycling primary B lymphocytes or developing T cells, PCH association correlated with heritable gene silencing (Brown et al., 1997; Hahm et al., 1998).
Many developmentally regulated genes locate at the nuclear periphery in their silent state but reposition to the nuclear interior upon gene activation, suggesting that peripheral gene localization may help establish and/or maintain developmental gene repression (Kosak et al., 2002; Ragoczy et al., 2006; Williams et al., 2006; Yao et al., 2011; Kohwi et al., 2013). In yeast, tethering to the nuclear periphery restored gene repression to a defective silencer (Andrulis et al., 1998). In mammalian cells, similar tethering experiments (Finlan et al., 2008; Kumaran and Spector, 2008; Reddy et al., 2008) have suggested that gene repression associated with tethering was promoter specific and quantitative, modulating transcription rather than turning it from on to off.
Little is known about how endogenous gene loci are targeted to the nuclear periphery. Different models could explain targeting of single copy gene loci to the nuclear periphery. Peripheral targeting of transcriptionally inactive genomic regions could be the default, with transcriptionally active genome regions actively targeted to the nuclear interior (model 1). Alternatively, specific DNA sequences, and/or proteins binding to these sequences, might target chromatin to the nuclear periphery either through direct molecular interactions with nuclear envelope proteins (model 2) or through establishment of a distinct, epigenetically marked chromatin domain, with peripheral targeting downstream of this chromatin domain establishment (model 3).
Two very recent studies have begun to address these possible molecular mechanisms. Supporting model 2, an autonomous bacterial artificial chromosome (BAC)–targeting approach identified lamin-associated sequences (LAS) conferring peripheral targeting from the IgH and Cyp3a multigene loci (Zullo et al., 2012). These LAS contained GA motifs binding the cKrox GAGA transcription factor, which was proposed to peripherally tether these sites through interactions with the inner nuclear membrane protein Lap2-β and HDAC3. Supporting model 3, tethering of repetitive gene arrays to the nuclear periphery in Caenorhabditis elegans was dependent on H3K9 methyltransferases, whereas chromosome arm regions with high levels of H3K9 methylation showed reduced interactions with the nuclear lamina after H3K9 methylation knockdown (Towbin et al., 2012).
The high compaction of large-scale chromatin folding complicates identification of cis- and trans-elements that target chromosome regions to particular nuclear compartments. Active targeting via a single sequence region may lead to apparent targeting of 100–1,000 s of kilobases of the adjacent chromosomal sequence, depending on the resolution of the imaging modality used to map intranuclear localization. Conversely, redundant targeting mechanisms operating over adjacent chromosome regions may make identification of individual targeting mechanisms difficult.
Here, we use the mammalian β-globin gene (HBB) locus to identify cis requirements for chromosome targeting to the nuclear periphery. HBB targets either to the nuclear periphery (Ragoczy et al., 2006; Hepperger et al., 2008) or to PCH (Brown et al., 2001) in several cell types in which it is inactive. By analyzing the autonomous nuclear periphery targeting of BAC transgenes, we identify a Suv39H, H3K9me3-dependent pathway involved in tethering the HBB locus to the nuclear periphery, in competition with targeting to PCH, which acts separately from an independent G9a, H3K9me2-dependent pathway, which tethers adjacent LAD sequences to the periphery.
Results
A BAC containing the human β-globin locus autonomously targets to the nuclear periphery in mouse fibroblasts
Multiple, cointegrated copies of a 207-kb BAC (CTD-2643I7) containing the human β-globin locus target to the nuclear periphery in mouse embryonic stem cells largely independent of their chromosomal insertion sites (Sinclair et al., 2010). This autonomous targeting mirrors the peripheral targeting of the endogenous mouse β-globin locus in mouse embryonic stem cells (Hepperger et al., 2008).
We applied this autonomous targeting assay to dissect cis-elements conferring targeting to the nuclear periphery (Fig. S1). Inserting a lac operator (LacO) 256 mer into the BAC and expressing GFP-lac repressor (GFP-LacI) provided direct visualizzation of integrated BAC transgene location. To further facilitate this assay, we used mouse NIH 3T3 cells, an immortalized, fibroblast cell line with high transfection and subcloning efficiency.
FISH on human BJ–human telomerase reverse transcriptase (hTERT) cells revealed >50% of endogenous β-globin loci within 0.5 µm from the nuclear lamina (Fig. 1, a and g). Measurements were made from single optical sections from the nuclear midsection. Peripheral localization of the endogenous β-globin locus also was observed in mouse NIH 3T3 fibroblasts (Fig. 1, b and g). No significant peripheral localization was observed for the endogenous human α-globin locus, flanked by multiple housekeeping genes (Fig. 1, c and g).
Figure 1.

200-kb human β-globin BAC transgene targets to nuclear periphery similarly to endogenous β-globin locus. (a–c) FISH shows peripheral localization of endogenous β-globin locus in human BJ-hTERT (a) and mouse NIH 3T3 (b) fibroblasts but interior localization for the α-globin locus in mouse NIH 3T3 cells (c; DAPI [blue], FISH [green], and lamin A immunostaining [red]). Arrowheads show DNA FISH signals. (d–f) Peripheral localization of HBB BAC identified by EGFP-LacI binding (green) in HBB C3 NIH 3T3 cell clone using DAPI staining (blue; d), lamin A immunostaining (red; e), or nuclear pore staining (red; f) to define the nuclear periphery. Arrowheads show the HBB transgenes. (g) Fraction of cells with peripheral localization in each NIH 3T3 subclone for HBB (black) or DHFR (green) BAC transgenes as compared with endogenous β-globin loci (HBB) in human BJ-hTERT (dark gray) or mouse NIH 3T3 (red) cells or α-globin loci (HBA) in human BJ-hTERT cells. HBB BAC (no LacO; yellow) refers to FISH measurements from a mixed population of stable NIH 3T3 clones with HBB BAC with just a selectable marker but no LacO repeat inserted. Random shows a fraction of DAPI staining within 0.5 µm from the periphery. At least 50 cells from each BAC transgene NIH 3T3 cell clone and ≥45 cells for each endogenous gene FISH experiment were analyzed. Bars, 2 µm.
The peripheral localization of human β-globin (HBB) BACs in mouse NIH 3T3 fibroblasts similarly mirrored the peripheral localization of endogenous mouse and human HBB loci in fibroblasts. In 4/5 randomly selected, stable clones, BAC transgenes were located within 0.5 µm from the nuclear periphery in 30–64% of the cells (Fig. 1, d–f), with the transgene radial positioning distribution recapitulating the endogenous β-globin locus distribution (Fig. S2). Peripheral localization in all clones was significantly higher than the 14.8% predicted geometrically for an average-sized, 2D elliptical nucleus (16- and 11-µm diameters; Fig. 1 g). The LacO repeat does not contribute to this peripheral localization; we observed by FISH a 50% peripheral localization of HBB BAC transgenes lacking an inserted LacO repeat in a mixed population of stable clones (Fig. 1 g). In contrast, dihydrofolate reductase (DHFR) BAC transgene arrays showed a 5–16% peripheral localization (Fig. 1 g). Previously, we observed interior nuclear localization for LacO-tagged BAC transgene arrays carrying metallothionein, Hsp70, DHFR, or α-globin gene loci (Hu et al., 2009; Sinclair et al., 2010).
β-Globin genes and regulatory regions are not required for peripheral targeting
Movement of the β-globin genes from the nuclear periphery to interior accompanies their transcriptional activation during erythrocyte maturation (Ragoczy et al., 2006). The human β-globin gene cluster consists of five globin genes (HBE1, HBG1, HBG2, HBD, and HBB). The 39.5-kb “Hispanic” deletion upstream of the β-globin gene cluster causes profound alteration of β-globin expression (Driscoll et al., 1989; Bender et al., 2006). The most important regulatory region overlapping with the Hispanic deletion is the locus control region (LCR) containing six DNase I hypersensitive sites (HSs), located 6–22 kb upstream of the HBE1 gene and required for high expression level of all β-globin genes (Bender et al., 2006). The 27-kb region upstream of the LCR (upstream Hispanic region [UHR]) may also contain functional elements for β-globin gene regulation. Two erythroid-specific DNase I HSs, the 3′ HS1 located between the HBB and olfactory receptor OR51V1 genes and HS-110 located 110 kb upstream of HBE1 between olfactory receptor genes OR51B6 and OR51M1, have also been implicated in β-globin regulation (Palstra et al., 2003; Fang et al., 2007).
We used λ red BAC recombineering to remove specific sequences from the HBB BAC (Fig. 2 and Fig. S3; Warming et al., 2005). Cycles of galK insertion and removal using positive and negative selection allowed repeated rounds of deletions. Deleting the LCR (D1) and the β-globin gene cluster (D2) as well as the entire β-globin locus (D5) including the UHR and 3′ HS1 sequences (D5) all failed to eliminate peripheral targeting (Fig. 2, a and b). HS-110 also is not required (D9 and double deletion D8D11; Fig. 2, f and g).
Figure 2.

Sequence dissection of β-globin BAC reveals three peripheral PTRs. (a) Map of ∼200-kb human β-globin BAC aligned with the Lamin B1 DamID (human fibroblast Tig3 cells), DNase1 hypersensitivity (normal human lung fibroblasts), and CTCF binding (normal human lung fibroblasts) maps from the University of California, Santa Cruz, Genome Browser. Red lines indicate specific deletions made. (b–d, f, and g) Statistics for BAC transgene peripheral localization in independent NIH 3T3 subclones. (b) The LCR (D1), β-globin gene cluster (D2), and entire 116-kb region (D5) encompassing the β-globin locus, including the UHR and 3′ HS1; all are dispensable (D5) for peripheral localization. (c) Loss of peripheral targeting by the D5D7 double deletion, a control containing mostly BAC vector backbone and Tn5 transposon sequence, and the D4 deletion suggests that specific cis-elements outside the β-globin locus are required for targeting. (d) At least two functionally redundant regions within the D4 region are sufficient for peripheral targeting because D8, D9, D10, and double deletions D5D8 and D5D10 BACs display similar peripheral targeting. (e–g) Further sequence dissection reveals at least three PTRs sufficient for peripheral targeting. (e) Additional deletions relative to PTR locations (red bars) as shown for ∼80 kb of the 3′ end of BAC. kan/neo, kanamycin/neomycin. (f) Nested set of deletions reveals 6.3-kb PTR1 by loss of peripheral targeting in D8D14 deletion and persistence of peripheral targeting in D8D13D22 triple deletion, PTR2 by persistence of peripheral targeting in D8D17 double deletion, and PTR3 by D8 deletion. (g) Summary of sequence dissection showing median peripheral targeting levels for five cell clones analyzed for each BAC shown. Distances between BAC transgenes and nuclear periphery were measured in ≥50 cells for each cell clone.
Three cis-regions are each sufficient for HBB BAC peripheral targeting
Instead, deleting the entire 80-kb region (D7) upstream of the β-globin gene cluster and LCR eliminated peripheral targeting (Fig. 2, a, c, and f). This peripheral targeting capability was further narrowed to a 56-kb region by the D4 deletion (Fig. 3, a, c, and f). As a negative control, a double deletion (HBBD5D7; Fig. 2 a) localized to the nuclear interior (Fig. 2, c and f). This BAC double deletion removes nearly the entire human DNA insert but retains the LacO repeat, selectable marker, and vector backbone.
Figure 3.

Competition between nuclear peripheral versus chromocenter targeting. (a) D4 and D8D14 BAC transgenes exhibit significantly higher percentages of chromocenter association compared with intact (HBB) or D5 deletion (HBBD5) β-globin BAC transgenes. (b) Representative cells from three independent cell clones showing association of HBBD4 transgenes (green) with chromocenters. Blue, DNA DAPI staining. Bars, 2 µm. The presented images were collected in several different experiments and using different exposure times, as appropriate to each particular cell. (c) Stacked bar plots show a near constant sum of peripheral or chromocenter transgene targeting for multiple, independent cell clones carrying HBB, HBBD4, or HBBD5 transgenes or a mixed population of stably selected cell clones for HBB BAC transgenes not containing LacO repeats, suggesting competition between targeting to the nuclear periphery versus chromocenter. (a and c) Localization of BAC transgenes to either the nuclear periphery or chromocenter was measured in ≥50 cells for each cell clone. (d and e) Quantitative real-time PCR analysis of relative mRNA expression levels, normalized by copy number, for β-globin (HBB), olfactory receptor (OR), and selectable marker (kanamycin/neomycin [KN]) genes in cell clones containing either HBB BAC or HBBD4 BAC. Expression per gene copy was normalized relative to the expression in the cell clone containing HBB BAC (d) or the expression of the endogenous mouse HBB gene (e). Data show means ± SEM from three independent experiments.
Previous studies in human lung fibroblasts mapped a 32-kb LAD region at the 3′ end of the HBB BAC (Fig. 2 a; Guelen et al., 2008). Neither the LAD (D8) nor the LAD plus boundary region containing HS-110 and a CTCF binding site cluster (D9) was required for peripheral targeting. This means either the LAD region does not contain a peripheral targeting sequence or functionally redundant targeting sequences exist outside the LAD region.
Further dissection revealed three, functionally redundant peripheral targeting regions (PTRs; Fig. 2 e). The 6.3-kb PTR1 was identified using nested deletions D11 through D14 combined with the D8 deletion (Fig. 2, e–g). The triple deletion D8D13D22 confirmed that additional sequences flanking PTR1 were not required for targeting (Fig. 2, e and f). The 23-kb PTR2 was revealed by double deletion D8D17, which removed PTR1 but preserved peripheral targeting (Fig. 2, e and f). PTR3 was mapped to the 32 kb corresponding to the intersection of the D8 and D4 deletions, based on the peripheral targeting of the D10 deletion, the loss of peripheral targeting with the D4 deletion, and the peripheral targeting of the combined D5 and D10 deletions (Fig. 2, c and d). Only PTR3 is contained within the LAD (Fig. 2 e).
Competition between PCH and peripheral targeting
Association of inactive genes with PCH occurs in several cell types (Brown et al., 1997, 1999; Francastel et al., 2001). This includes association of the inactive β-globin locus with PCH in cycling human lymphocytes (Brown et al., 2001) and localization of the inactive β-globin locus to both centromeres and the nuclear periphery in mouse erythroleukemia cells (Francastel et al., 2001).
Interestingly, our deletion analysis revealed competition between HBB BAC targeting to the nuclear periphery versus PCH. In mouse cells, PCH clusters into DNA-dense bodies called chromocenters. BAC deletions that removed all three PTRs (D4 and D8D14) lost peripheral targeting activity while showing a commensurate increase in chromocenter association (Fig. 3, a and b).
These observations prompted reexamination of HBB BAC localization. Cell clones with different chromosome integration sites and different HBB peripheral targeting frequencies showed a near constant sum of chromocenter or peripheral targeting (Fig. 3 c). A similar, near constant sum for peripheral and chromocenter targeting was observed for HBB BAC deletions preferentially targeting to the periphery (D5) or to the chromocenter (D4; Fig. 3 c).
RT–quantitative PCR (qPCR) measurements of β-globin and olfactory gene expression, normalized by copy number, revealed similar expression for BAC transgenes localizing either to the periphery or the PCH (Fig. 3 d). HBB transgene expression levels were very close to copy number–normalized expression levels for the endogenous mouse Hbb-b1 gene (mouse HBB; Fig. 3 e), homologous to the human HBB gene. An approximately threefold expression increase was observed for the selectable marker in the BAC targeted to the PCH versus the nuclear periphery (Fig. 3 d, kanamycin).
PTR intranuclear targeting is context specific
The 6.3-kb PTR had no effect on plasmid transgene positioning (Fig. 4 a), suggesting a position effect for PTR-targeting activity. Multicopy plasmid arrays are known to show strong transgene silencing; this likely is related to their abnormally condensed chromatin conformation (Belmont et al., 2010; Bian and Belmont, 2010).
Figure 4.

Peripheral targeting activity of PTRs is context specific. Histograms showing targeting frequency to nuclear periphery versus chromocenter for five independent clones containing different plasmid or BAC constructs. (a) PTR1 does not confer targeting to the nuclear periphery when placed in a plasmid containing a 256 mer lac operator (LacO) repeat. (top) Plasmid maps with vector backbone (gray), PTR1 (red), and kanamycin/neomycin (Kan/Neo)-selectable marker (yellow). (b) Cointegrated DHFR and β-globin BACs (DHFR_HBB) target with high frequency to chromocenter rather than the peripheral targeting seen for HBB BACs alone. Map of ∼180-kb DHFR BAC. Green, LacO; red, Zeocin-selectable marker. (c) Examples of chromocenter association for several independent clones with varying size; cointegrated DHFR/HBB BAC transgene arrays LacO staining (green) and DAPI DNA staining (blue). Bars, 2 µm. The presented images were collected in several different experiments and using different exposure times, as appropriate to each particular cell. (d) Insertion of the 6.3-kb PTR1 to HBBD4 BAC by transposon restores peripheral targeting. (top) Map showing PTR containing transposon (red arrowhead). (e and f) Whereas insertion of PTR1 into BAC CTD-2207K13 changes neither peripheral nor chromocenter targeting (e), PTR1 insertion into BAC RP11-2I1 significantly increases chromocenter, but not peripheral, targeting. Localization of BAC transgenes to either the nuclear periphery or chromocenter was measured in ≥50 cells for each cell clone.
Position effect modulation of PTR activity also is implied by the clonal variation in peripheral targeting of HBB BACs inserted at different chromosome sites. This position effect could arise either from a dominant targeting activity of endogenous sequences flanking the BAC transgenes and/or to a long-distance, antagonistic activity of these flanking sequences on the cis-targeting elements within the PTR.
To produce an experimentally reproducible position effect, we cotransfected DHFR and HBB BAC transgenes and isolated clones in which these BACs cointegrated. DHFR BAC transgenes reconstitute an open large-scale chromatin conformation and confer position-independent, copy number–dependent expression of a reporter gene independent of chromosome insertion site (Bian and Belmont, 2010).
Surprisingly, clones carrying cointegrated DHFR and HBB BACs transgenes showed preferential targeting not to the periphery, where HBB BACs localized, and not to the nuclear interior, where DHFR BACs localized, but instead to the PCH (Fig. 4, b and c). PCH association frequencies were similar to that observed for the HBBD4 BAC lacking all PTRs, suggesting that flanking DHFR BAC sequences inhibit PTR peripheral targeting activity.
These results suggested an epigenetic component to PTR targeting. We next tested PTR1 within the context of BACs containing large, gene-free DNA regions. First, as a positive control, we showed that random insertion of PTR1 into HBBD4 by Tn5 transposition restored peripheral targeting (Fig. 4 d). Therefore, positioning of the β-globin locus by PTR1 does not require a fixed position of PTR1 within the HBB BAC.
We then inserted PTR1 via Tn5 transposition into BACs containing two different, ∼200-kb human sequences. CTD-2207K13 contains an insert from a large gene desert region, whereas RP11-2I1 contains an intergenic sequence from a gene-rich RIDGE (Goetze et al., 2007) region. Both “neutral” BACs with control transposons containing only the LacO repeat and selectable marker showed interior nuclear localization. Adding PTR1 to the CTD-2207K13 BAC produced no change in interior localization (Fig. 4 e). However, inserting PTR1 into the RP11-2I1 BAC retargeted these BAC transgenes from the nuclear interior to the PCH (Fig. 4 f). Thus, PTR1 confers peripheral targeting to the HBBD4 BAC, which would otherwise target to the PCH, but PCH targeting to the RP11-2I1 BAC, which would otherwise target to the nuclear interior, strongly suggests an epigenetic mechanism for PTR targeting.
Targeting to the nuclear periphery correlates with BAC colocalization with H3K9me3 foci
Repressed genes are known to associate with both the nuclear periphery and the PCH. We therefore used immunofluorescence to examine colocalization of BAC transgenes with specific heterochromatin marks. HBB BACs did not obviously associate with H3K27me3 foci but did show elevated H3K9me3 immunostaining (Fig. 5, a–c). The shape and size of the elevated H3K9me3 immunostaining corresponded closely to the shape and size of the GFP signal, suggesting an increased H3K9me3 modification over the BAC transgene array itself (∼1 Mbp in size for the approximately five BAC copies in the HBB-C3 clone). Approximately 80% of HBB transgenes showed “strong” H3K9me3 immunostaining independent of peripheral versus interior localization. All full-length HBB BAC and HBB BAC deletions targeting to the periphery showed elevated H3K9me3 immunostaining, whereas all HBB BAC deletions that did not target to the periphery showed significantly lower levels of H3K9me3 (Fig. 5, d–g).
Figure 5.

Differential targeting to the nuclear periphery versus chromocenter correlates with H3K9me3 levels over BAC transgenes. (a–f) BAC transgene locations (EGFP-LacI staining), H3K9me3 immunostaining, DAPI (blue). Enlarged insets show regions of magnification with red arrowheads in red channel (middle) pointing to location of transgenes in green (top) channel. (a–c) HBB BAC transgene locations in clone HBB-C3 overlap with H3K9me3 immunostaining foci regardless of whether transgenes are located at nuclear periphery (a), chromocenter (b), or interior (c). (d–f) Examples from clone HBB C3 showing strong (d), weak (e), or no (f) H3K9me3 signals over the BAC transgenes. (g) Strength of H3K9me3 immunostaining over different BAC transgenes (left) correlates with differential targeting of BAC transgenes (right) to nuclear periphery versus chromocenter. Bars, 2 µm. Data shown for each BAC are pooled from at least two independent experiments.
Inserting PTR1 to a new location within the HBBD4 BAC (D4-PTR1) restored peripheral localization and elevated H3K9me3 staining (Fig. 5 g). Cointegration of HBB BAC transgenes with the active housekeeping DHFR BAC transgenes led to reduced H3K9me3 over the transgenes (Fig. 5 g).
H3K9me3 chromatin immunoprecipitation (ChIP) over PTR1 and the HBB locus and its correlation with H3K9m3 immunofluorescence and nuclear localization
To provide an independent, biochemical measure of the H3K9me3 modification, we used qPCR to quantitate H3K9me3 ChIP. Primer pairs were spaced every ∼5 kb over the HBB BAC insert except for PTR1, in which we used six primer pairs spaced over 6.3 kb (Fig. 6 a). Three biological replicates of ChIP over the HBB BAC transgene in cell clone HBBC3 (approximately five BAC copies estimated by qPCR) showed consistently elevated levels over PTR1 at both 5′ and 3′ PTR1 ends, with two primer pairs (28–29) showing peak values over the entire BAC (Fig. 6 b). To normalize ChIP data between different experiments, we linearly mapped measurements on a 0–1 scale: 0 corresponded to the percent input values for the GAPDH promoter negative control, whereas 1 corresponded to percent input values measured for the intracisternal A-particle (IAP) transposon-positive control. The GAPDH promoter H3K9me3 ChIP modification level is among the lowest in the genome, whereas the IAP transposon-positive control H3K9me3 ChIP modification level, higher than that observed over major and minor satellite and LINE-1 repeats (unpublished data), represents the high end for H3K9me3 genomic modification.
Figure 6.

ChIP-qPCR measurements of context-specific H3K9me3 modifications over PTR1 and HBB BAC transgenes. (a) Map of HBB BAC with location of primers used for ChIP-qPCR; primer spacing was ∼1 kb over PTR1 but ∼5 kb elsewhere. (b–d) ChIP-qPCR measurements of H3K9me3 levels with the percent input values normalized by scaling linearly between 0 (GAPDH promoter) and 1 (IAP transposon; see Results H3K9me3 ChIP over PTR1 and the HBB locus and its correlation with H3K9m3 immunofluorescence and nuclear localization section). (b) Three biological replicates (A, B, and C) show reproducibility of H3K9me3 ChIP over HBB BAC with consistent peak over PTR1. (c) H3K9me3 ChIP values of full-length HBB BAC (green) versus HBBD4 BAC deleted of all PTRs (red) or HBBD4 BAC with PTR1 reinserted at arrow location (blue; inset shows PTR1 values with actual orientation). (d) H3K9me3 ChIP values for HBB BAC by itself or intact HBB BAC flanked by transcriptionally active DHFR BAC transgenes. (e) Mean H3K9me3 ChIP levels are lower for HBBD4 relative to HBB or HBBD4 + PTR1 BACs. (f) Mean H3K9me3 immunofluorescence (IF) levels are similarly reduced for HBBD4 versus HBB transgenes using a normalized, linear scaling of immunofluorescence values between DAPI regions with low immunofluorescence (0) and chromocenter immunofluorescence (1) (see Results section). (g) Normalized mean H3K9me3 ChIP values are decreased over PTR1 in plasmid transgenes relative to HBB, HBBD4 + PTR1, or HBB flanked by DHFR BAC transgenes. (h) Reduced H3K9me3 mean, normalized values for an ∼100-kb HBB region (primer pairs 2–18) without PTRs in HBBD4 BAC lacking PTRs but also in full-length HBB BAC transgenes flanked by DHFR BACs. (c–h) Error bars show SEM. (e–h) Statistical significance: *, P < 0.05; **, P < 0.01.
Normalized ChIP values showed an improved reproducibility across the HBB BAC transgene. Because PTR1 was oversampled relative to other HBB BAC sequences, we may have missed localized H3K9me3 peaks in PTR2 and PTR3. Elevated H3K9me3 levels were also seen over PTR1 inserted into the HBBD4 BAC with all PTRs removed (HBBD4 + PTR1; clone A3 containing approximately three BAC copies; Fig. 6 c).
An observable increase in H3K9me3 immunostaining over BAC transgenes would require increased H3K9me3 levels over large regions of the HBB BAC. To compare ChIP with immunofluorescence data, we estimated mean H3K9me3 levels (weighted by probe separation distances) over the HBB BAC. Mean ChIP values were nearly twofold higher over the intact HBB BAC versus the HBBD4 BAC with all PTRs deleted; adding PTR1 back to the HBBD4 BAC increased mean ChIP levels close to that of the full-length HBB BAC (Fig. 6, c and e).
To compare ChIP and immunofluorescence results, we normalized immunostaining intensity values using a similar two point linear interpolation procedure. H3K9me3 immunostaining intensities over DAPI-stained regions devoid of obvious H3K9me3 foci were mapped to 0, whereas immunostaining values over chromocenters were mapped to 1. H3K9me3 immunostaining intensities directly over the GFP-LacI peak intensity were renormalized to this 0–1 scale. Given the resolution of light microscopy, we expected spreading of the high H3K9me3 signal from the intensely stained chromocenters would artificially elevate intensity values over transgenes at the chromocenter periphery. We therefore made measurements over BAC transgenes not associated with chromocenters.
Normalized immunostaining H3K9me3 levels were similar to normalized ChIP H3K9me3 levels (Fig. 6 f). ChIP and immunofluorescence H3K9me3 values were significantly higher for the full-length HBB BAC compared with the HBBD4 BAC deleted of PTRs. No significant differences in mean ChIP or immunofluorescence values were observed between the full-length HBB BAC and the HBB BAC deleted of PTRs but with PTR1 added back (D4 + PTR1).
PTR1-containing transgenes that target to the nuclear interior show significantly lower PTR1 mean H3K9me3 levels compared with PTR1 levels within HBB or HBBD4 + PTR1 BACs that target to the nuclear periphery (Fig. 6 g). PTR1 H3K9me3 levels in cointegrated HBB and DHFR BACs were similar to levels in full-length HBB BAC transgenes despite their different nuclear targeting (Fig. 6, d and g). However, over the non-PTR regions (primer pairs 2–18), mean H3K9me3 ChIP levels were significantly lower in the cointegrated DHFR/HBB BAC arrays than peripherally targeted HBB or HBBD4 + PTR1 BAC arrays but similar to levels in the HBBD4 BAC that also targeted to the PCH (Fig. 6 h).
H3K9 methylation is required for peripheral and PCH HBB BAC targeting
Cells were infected with pooled lentiviruses expressing shRNA directed against both Suv39H1 and Suv39H2 and selected using puromycin. A significant fraction of cells surviving drug selection showed weaker and more diffuse H3K9me3 staining than control cells. In cell clone C3 containing the HBB BAC, infected cells were classified into three categories based on H3K9me3 staining levels (Fig. 7, a–c). A dose-dependent reduction of peripheral targeting was observed with a drop from 61 to 23% peripheral association between the “normal” and “none” categories (Fig. 7 d). A similar dose-dependent reduction in PCH targeting was observed in cells (clone C40.10) carrying the HBBD4 BAC with all PTRs deleted (Fig. 7, e–h).
Figure 7.

Reducing H3K9 methylation inhibits β-globin BAC targeting to periphery and chromocenter. (a–d) Reduced peripheral targeting of HBB clone C3 cells with increased reduction of H3K9me3. (a–c, left) HBB C3 cells after combined Suv39H1 and Suv39H2 shRNA knockdown showing normal level (a), reduced level (b), or no (c) H3K9me3 staining. (d) Statistics for peripheral targeting. (e–h) Reduced association with chromocenter of HBBD4 BAC transgenes with increased reduction of H3K9me3. (e–g) HBBD4 clone C40.10 cells after combined Suv39H1 and Suv39H2 shRNAs knockdown showing normal (e), reduced (f), and no (g) H3K9me3 immunostaining. (h) Statistics for chromocenter association. (a–c and e–g) Green, EGFP-LacI; arrowheads indicate BAC transgenes. (middle) DAPI staining. (right) H3K9me3 staining. Bars, 2 µm. Data shown are pooled from at least three independent experiments.
Combined H3K9me3, H3K9me2 knockdown detaches HBB locus from the periphery—H3K9me2- or H3K9me3-dependent peripheral interactions over spatially separate chromosome regions
We observed no change in peripheral localization of the endogenous β-globin locus in mouse NIH 3T3 cells (not depicted) or human Tig3ET fibroblasts (Fig. 8, f and g) after Suv39H1/H2 RNAi knockdown. Previously, we estimated compaction levels ∼1–3 Mbp/µm for large-scale chromatin fibers (Tumbar et al., 1999; Hu et al., 2009), whereas our criterion for peripheral localization is a signal <0.5 µm from the nuclear. We hypothesized additional non-H3K9me3–dependent tethering mechanisms acting on DNA flanking the HBB locus, preventing displacement of HBB >0.5 µm from the periphery.
Figure 8.

H3K9me3 and H3K9me2 double knockdown inhibits peripheral targeting of endogenous human β-globin locus (a) Schematic of an ∼2.5-Mbp region showing human β-globin locus and its flanking LAD regions (Lamin B1 DamID, human fibroblast Tig3 cells, hg19 assembly; University of California, Santa Cruz, Genome Browser). DNA FISH BAC probes shown below the map as black lines are labeled red for HBB and green for LAD sequences. chr, chromosome. (b–e) Representative DNA FISH images showing classes of HBB (red) and adjacent LAD localization (green). Gray, DAPI. (b) HBB and LAD both interior. (c–c″″) HBB and LAD both peripheral. (d and e) Polarized orientation with HBB locus at the periphery and LAD extended into the interior (d) or LAD locus attached at one end to the periphery but extended such that HBB is in the interior (e). (f–i) statistics for these different localization classes (b–e). Suv39H (H3K9me3 [me3] knockdown [KD]) knockdown or G9a (H3K9me2 [me2] knockdown) inhibition did not change the localization of HBB; however, double knockdown of H3K9me3 and H3K9me2 significantly reduces the preferential peripheral localization of HBB. (e and i) The polarized orientation with the flanking LAD attached peripherally at its distal end suggests existence of a third tethering mechanism, independent of Suv39H and G9a (Fig. 10 a). n > 100; data shown are pooled from at least three independent experiments. Bars, 2 µm. Insets are at 2×. Statistical significance: *, P < 0.05.
We identified two GA motif clusters within ±1 Mbp of the HBB gene (Fig. 8 a), using the same motif finder software as used previously (Zullo et al., 2012). The first, in an inter-LAD 800 kb 5′ to HBB (within CTD-2547L21), shows interior localization (unpublished data). The second, 200 kb 3′ to HBB and in an adjacent ∼1 Mbp LAD (within RP11-715G8 BAC; Fig. 8 a), is peripherally located (not depicted). Neither cKrox siRNA single knockdown nor cKrox, Suv39H1, and Suv39H2 triple knockdown disrupted the peripheral localization of HBB or RP11-715G8 BAC probes (Fig. S4). Western blots showed knockdown of ∼95% for cKrox, whereas H3K9me3 knockdown was verified by immunostaining (Fig. S4).
H3K9me2 has been proposed to anchor LADs to the nuclear lamina and is enriched at the nuclear periphery (Kind et al., 2013); however, HBB BAC transgenes (C3 clone) colocalized with H3K9me3 but not H3K9me2 staining (unpublished data). H3K9me3 is also enriched at the nuclear periphery at comparable or even higher levels than H3K9me2 in human Tig3 and WI-38 and mouse NIH 3T3 fibroblasts (Fig. S5). Foci of strong, peripheral H3K9me3 or H3K9me2 immunostaining frequently appear anticorrelated, suggesting independent targeting.
Based on knockout experiments and mass spectroscopy, G9a appears responsible for ∼50% of the total H3K9me2 signal (Peters et al., 2003). This H3K9me2 reduction is comparable to what we observed after G9a drug inhibition (Fig. S4) or shRNA knockdown (not depicted) and similar to that observed by others (Wu et al., 2005; Kind et al., 2013). H3K9me2 knockdown using G9a inhibitors BIX01294 or UNC0638 (not depicted) did not disrupt the peripheral localization of HBB or RP11-715G8 BAC probes (Fig. S4). However, simultaneous G9a inhibition (or shRNA knockdown; not depicted) and H3K9me3 knockdown by Suv39H1/H2 siRNA significantly reduced HBB peripheral localization (Fig. 8 g).
Painting the ∼1-Mbp LAD region with four BAC probes, we simultaneously visualized both LAD and HBB regions (Fig. 8, a–e). In control and single knockdown cells, in a given cell, both the LAD and HBB regions typically were located either at the periphery or the interior (Fig. 8, b, c, f, and g). After double knockdown of H3K9me3 and H3K9me2, the HBB region separated >0.5 µm from the periphery in ∼70% of cells (Fig. 8, f and i). A polarized orientation—peripheral tethering of the distal LAD region, separation of the remainder of the LAD from the periphery, and the HBB locus >0.5 µm from the periphery and more interior than the LAD—was observed in ∼15% of cells (Fig. 8 i).
These results suggest three independent mechanisms for peripheral targeting near the HBB locus—a Suv39H1/H2-dependent mechanism operating over the HBB locus, a G9a-dependent mechanism operating over the proximal region of the adjacent LAD, and likely, a third, uncharacterized mechanism anchoring the distal LAD region (see Fig. 10 a). To more clearly establish anchoring of LAD regions to the periphery through a G9a-dependent, Suv39H1/H2-independent mechanism, we visualized stably integrated, LacO–tagged RP11-715G8 BAC transgenes in a mixed clonal cell population. RP11-715G8 BAC transgenes were peripherally located in 50% of cells. Suv39H1/H2 shRNA had no effect on RP11-715G8 BAC peripheral localization, but BIX01294 G9a inhibition eliminated peripheral targeting to background levels (Fig. 9 c). In contrast, G9a inhibition had no effect on peripheral localization of HBB transgenes (C3 cell clone), but Suv39H1/H2 knockdown eliminated peripheral targeting to background levels (Fig. 9 b).
Figure 10.

Working model for HBB locus nuclear targeting. (a) At least two independent peripheral targeting mechanisms act on adjacent sequences to anchor the HBB locus and surrounding sequences to the periphery: a Suv39H/H3K9me3 (me3)-dependent mechanism mediated by PTR sequences near the HBB locus (red), a G9a/H3K9me2 (me2)-dependent mechanism mediated by sequences in left flanking LAD region (green; possibly also in right flanking LAD, dotted green), and a likely third, uncharacterized mechanism acting to tether distal LAD after combined Suv39H and G9a knockdown/inhibition (dotted blue). KD, knockdown. (b) Peripheral targeting regions (PTRs) induce epigenetic modifications, leading to inhibition of gene expression and, in a fraction of cells, association with the nuclear periphery. Peripheral association may in turn reinforce gene repression. (c) Nucleation of H3K9me3 by PTR and its propagation, presumably with other epigenetic marks, to flanking genomic regions. (d) Epigenetic modifications continuum depicted as a white to black gradient with targeting to the nuclear interior (I; white), chromocenter (C; gray), or periphery (P; black) dependent on position within this continuum. (e) Cis-elements establish epigenetic states characteristic for each BAC transgene, resulting in differential nuclear targeting. (f) Addition of PTR1 shifts this continuum toward the black, altering nuclear targeting. (g) Long-range influence of cis-elements within DHFR BAC transgene shifts the epigenetic state of cointegrated HBB BAC transgenes from black (peripheral) to gray (chromocenter). (h) Reducing H3K9me3 by Suv39H KD shifts the epigenetic state toward white.
Figure 9.
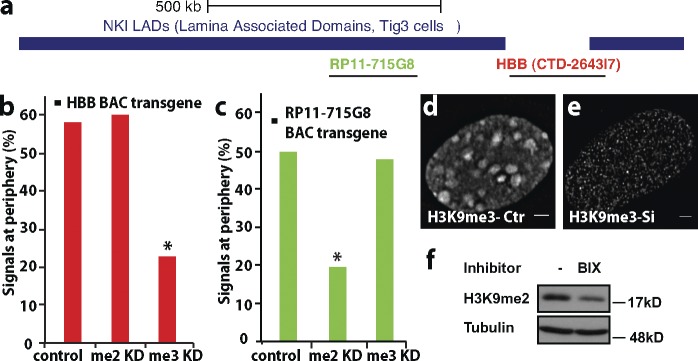
Independent peripheral targeting mechanisms for HBB versus LAD BAC transgenes. (a) Schematic of HBB (CTD-2643I7) and LAD (RP11-715G8) BACs aligned relative to flanking LAD sequence (labeling as in Fig. 8). (b) Peripheral targeting of HBB BAC transgene is inhibited after Suv39H knockdown (H3K9me3 [me3] knockdown [KD]) but not G9a inhibition (H3K9me2 [me2] knockdown). (c) Peripheral targeting of RP11-715G8 BAC transgene is inhibited after G9a inhibition (H3K9me2 knockdown) but not after Suv39H (H3K9me3 knockdown). (d and e) Normal (d) or reduced (e) H3K9me3 immunostaining after scrambled (d) or combined Suv39H1 and Suv39H2 shRNA (e). Ctr, control. (f) H3K9me2 Western Blot before (−) and after (+) BIX01294 G9a inhibition (Tubulin loading control). (b and c) n > 50; data shown are pooled from at least three independent experiments. Bars, 2 µm. Statistical significance: *, P < 0.01.
Discussion
Two independent peripheral targeting mechanisms active in different but adjacent sequences
Large-scale chromatin folding complicates identification of cis- and trans-elements targeting chromosome regions to particular nuclear compartments. Active targeting via a single sequence will cause apparent targeting of 100–1,000 s of kb of adjacent chromosomal sequence, as visualized by conventional light microscopy. Conversely, additional targeting mechanisms distributed across this same 100–1,000 s of kb of adjacent chromosomal sequence will mask the contributions of individual targeting mechanisms.
Here, we used autonomous targeting of randomly integrated BAC transgenes to overcome these problems. Using deletion analysis of a 207-kb HBB BAC, we correlated targeting to the nuclear periphery with elevated levels of H3K9me3 over these BAC transgenes. We next showed that this peripheral targeting was largely eliminated by Suv39H1/H2 knockdown. Because Suv39H1/H2 knockdown had no effect on the peripheral localization of the endogenous HBB locus, we hypothesized a second targeting mechanism acting on flanking sequences.
Double knockdown of G9a and Suv39H1/H2 led to a significant displacement of the endogenous HBB locus away from the periphery. Subsequent FISH analysis suggested a model in which the several hundred kilobase HBB region is tethered to the periphery through a Suv39H1/H2, H3K9me3–dependent mechanism, most of an adjacent ∼1-Mbp LAD is tethered through a G9a, H3K9me2 mechanism, whereas a third, unknown mechanism tethers the distal LAD region to the periphery (Fig. 10 a). These three tethering mechanisms prevent significant displacement of either the LAD or the HBB regions after single knockdown of either H3K9me2 or H3K9me3. Support for this model comes from the G9a-dependent peripheral targeting of a second BAC transgene containing sequence from this adjacent LAD.
Identification of PTRs and an epigenetic basis for peripheral targeting
Focusing on the HBB BAC, an unbiased deletion analysis identified three PTRs, each of which was sufficient to target the remaining ∼100-kb HBB region to the nuclear periphery. Nested deletions narrowed one of these, PTR1, to 6.3 kb. Through this deletion analysis, a tight correlation was demonstrated between peripheral targeting and increased H3K9me3 immunostaining over the BAC transgenes. ChIP against H3K9me3 demonstrated elevated levels over PTR1 itself, plus a PTR-mediated general increase of H3K9me3 over the entire BAC transgene, consistent with our immunofluorescence results. Peripheral targeting activity was subject to position effects, and inhibition of peripheral targeting was again correlated with loss of H3K9me3, either as an elevated peak over PTR1 in plasmid transgenes or as reduced spreading of H3K9me3 over the HBB BAC transgene cointegrated with the DHFR BAC.
In the Introduction, we outlined several models for peripheral targeting. Our results showing PCH targeting of the HBB BAC with all PTRs deleted and interior localization of two BACs containing intergenic regions contradict model 1—default peripheral targeting of transcriptionally inactive chromosome regions. Instead, our results strongly support model 3—binding of particular epigenetic marks to components of the nuclear periphery. In particular, our demonstration that the same PTR1 element, which retargets the HBBD4 BAC from the PCH to the periphery, instead targets the RP11-2I1 BAC from the interior to the PCH strongly supports this epigenetic model, while contradicting model 2, in which targeting is through the binding of specific DNA sequences, and/or the proteins binding to these sequences, to proteins at the nuclear periphery.
A competition between targeting to two different heterochromatin compartments
Our HBB BAC deletion analysis revealed an apparent competition between targeting to the PCH versus the nuclear periphery; this competition is intriguing in light of the known differential targeting of the endogenous locus to either the nuclear periphery or the PCH in different cell types. In particular, the sum of peripheral or PCH targeting for a specific cell clone was near constant, despite consistent differences in the ratio of peripheral versus PCH targeting between the full-length HBB BAC and different HBB BAC derivatives. The PTRs bias this competition toward peripheral versus PCH targeting, rather than targeting to the periphery per se.
Although the molecular mechanism for this competition remains unproven, we propose a working model (Fig. 10, b–g) in which the quantitative levels of specific epigenetic marks averaged over a sufficiently large chromatin domain determine targeting of genomic loci to the nuclear periphery versus the PCH or nuclear interior. H3K9me3-marked chromatin is enriched near the nuclear periphery but also present throughout the nuclear interior. Therefore, we suggest that H3K9me3 is one of these marks and is necessary but not sufficient for targeting; however, we cannot rule out Suv39H1/H2-mediated methylation of a different substrate as the cause of targeting. PTR nucleation and/or the global spreading of these heterochromatin marks is modulated by the epigenetic state of flanking DNA sequences, with the ultimate targeting decision determined by the level of epigenetic modifications over the chromatin domain. At the endogenous locus, it is likely that changes in the Suv39H1/H2-independent targeting activity of flanking regions would also have to occur to redirect the HBB locus away from the periphery to the PCH.
Relationship to other studies
Using a similar autonomous BAC-targeting approach, peripheral targeting of the transcriptionally inactive IgH and Cyp3a multigene loci recently was shown to involve binding of GAGA factor cKrox to GA motif clusters (Zullo et al., 2012). It is unclear how prevalent these GA motif clusters are within the genome. None are present in the HBB BAC, and only two are located within 2 Mbp surrounding the HBB endogenous locus. Neither significantly contributes to peripheral targeting of this chromosome region.
In contrast, recent findings in C. elegans identified two H3K9 histone methyltransferases (HMTs) as required for anchoring multicopy gene arrays. RNAi knockdown of both HMTs resulted in reduced lamin interactions of chromosome arm regions and selected gene loci enriched in H3K9me3 in wild-type embryos (Towbin et al., 2012). The functional redundancy for these two HMTs, only one of which is able to trimethylate H3K9, suggested a single peripheral targeting mechanism in C. elegans embryos.
G9a is a mammalian HMT responsible for roughly 50% of total nuclear H3K9me2 (Peters et al., 2003) and most of the H3K9me2 enriched at the nuclear periphery. G9a knockdown reduces LAD targeting to the periphery approximately twofold based on biochemical assays (Kind et al., 2013). However, G9a knockdown failed to change the peripheral localization of late replicating, G9a-regulated genes based on cytological assays (Wu et al., 2005).
Our results blend aspects of several of these studies: we demonstrate dependence of peripheral targeting through G9a-induced H3K9me2 and Suv39H1/H2-induced H3K9me3, but these pathways act on different genomic regions separated by hundreds of kilobases. Apparent functional redundancy is suggested by cytological assays as a result of the tethering activity of neighboring chromosome regions, but BAC transgenes allow a clear separation of H3Kme2- and H3K9me3-related pathways.
Future directions
Our results reveal two independent pathways for peripheral targeting, each of which can be recapitulated by BAC transgenes that autonomously target to the periphery through a dependence on a single pathway. Focusing on the HBB BAC, we demonstrate the feasibility of using BAC transgenes to identify cis-elements that confer peripheral targeting and ultimately dissect the molecular mechanisms involved.
A current paradox in considering the functional consequences of targeting gene loci to “repressive” nuclear compartments is that only a fraction of alleles, for instance ∼50% for the β-globin locus, show this targeting. Importantly, the fraction of cells with elevated H3K9me3 over the HBB BAC transgenes was significantly higher than the percentage of peripherally located transgenes. These PTRs from the HBB BAC may better be described as elements that confer a particular heterochromatin state that includes spreading of H3K9me3 over a large domain in most cells, resulting in targeting to the PCH and peripheral nuclear compartments in a fraction of these cells. Future dissection of the molecular mechanisms underlying PTR action may therefore reveal how specific sequences establish distinct epigenetic states over large chromatin domains. Such dissection should allow us to distinguish functional consequences of differential nuclear targeting per se from the functional consequences of establishing distinct epigenetic states.
Materials and methods
BAC constructs, transposon insertions, and recombineering
Human BACs CTD-2643I7, CTD-2207K13, RP11-344L6, RP11-2I1, CTD-2547L21, RP11-715G8, RP11-76C5, RP11-678B18, and RP11-982G19 were obtained from Invitrogen. DHFR BAC 057L22 (California Institute of Technology BAC mouse library) was a gift from E. Heard (Curie Institute, Paris, France). Tn5 transposon insertion into BAC DNA was performed using the EZ::TN5 kit (Epicentre Technologies) following the manufacturer’s directions. DNA sequencing from both ends of the transposon mapped transposon insertion sites. A Tn5 transposon carrying the 256 mer LacO direct repeat and the kanamycin/neomycin-selectable marker was excised from p[K/N-PSI-8.32] (Sinclair et al., 2010) and transposed into CTD-26417, inserting 55 kb 5′ from HBB to generate CTD-26417-K/NPSI8.32. Similarly, p[K/N-PSI-8.32] was transposed into RP11-715G8 to generate RP11-715G8-K/NPSI8.32. A Tn5 transposon carrying the 256 mer LacO repeat and a kanamycin-selectable marker was excised from p[Kan-8.32] (Hu et al., 2009) and transposed into BAC 057L22, inserting 75 kb 3′ to the Msh3 transcription initiation site to generate 057L22-K-8.32-C29; a second Tn5 transposon carrying a cytomegalovirus-mRFP reporter gene and a Zeocin-selectable marker was excised from pCRZ (Bian and Belmont, 2010) and transposed into 057L22-K-8.32-C29, inserting at nucleotide 23,426 of the Msh3 gene generating 057L22-K-8.32-C29-C27.
Two Tn5 transposons carrying the 6.3-kb PTR1 sequence were made: p[Kan/Neo-8.32] (Hu et al., 2009) was linearized with SalI and ligated with a 6.3-kb PTR1 region fragment, generated by PCR amplification using the CTD-2643I7 BAC as a substrate with primers 5′-ATCCTCTAGAGTCGACTCTGGAGGCCAAGTGTCTCT-3′ and 5′-CGCCTCTAGAGTCGACCATGTCCTTTGCCCACTTTT-3′, and then cut with SalI, generating p[Kan/Neo-HBB6kb-8.32]. p[Zeo] (Bian and Belmont, 2010) was digested with SalI and ligated with the 6.3-kb fragment excised by SalI digestion of p[Kan/Neo-HBB6kb-8.32] to generate p[Zeo-HBB6kb].
p[Kan/Neo-HBB6kb-8.32] was used to generate a Tn5 transposon carrying the 256 mer LacO direct repeat, 6.3-kb PTR1, and a kanamycin/neomycin-selectable marker, which was transposed into BACs CTD-2207K13 and RP11-2I1 to generate 2I1 HBB6kb LacO-C3 and 2,207 HBB6kb LacO-C2, respectively. p[Kan/Neo-8.32] was used to generate a Tn5 transposon carrying the 256 mer LacO repeat and a kanamycin/neomycin-selectable marker, which was transposed into BACs CTD-2207K13 and RP11-2I1 to generate 2I1-LacO-C1and 2,207 LacO-C2, respectively. p[Zeo-HBB6kb] was used to generate a Tn5 transposon carrying the 6.3-kb PTR1 and Zeocin-selectable marker and transposed into HBBD4 BAC to generate the HBBD4-6kb-C5, which has the transposon inserted 28 kb downstream of HBE1.
BAC deletions using BAC recombineering
λ red–mediated BAC recombineering using a galK-based dual-selection scheme was used to delete specific regions from β-globin BAC CTD-26417-K/NPSI8.32-C4. Sequential rounds of recombination-mediated deletion with galK insertion followed by galK removal using standard recombineering protocols (Warming et al., 2005) allowed generation of BAC derivatives with more than one deleted region. CTD-26417-K/NPSI8.32-C4 was transformed into Escherichia coli strain SW102 in which the λ red recombination machinery is induced by shifting temperature from 32 to 42°C (Warming et al., 2005). Recombination DNA fragments with homology ends were prepared by PCR using primers (Table S1) with 43-bp homology sequences plus 17-bp sequences (forward, 5′-CGACGGCCAGTGAATTG-3′; reverse, 5′-TGCTTCCGGCTCGTATG-3′) for amplifying the galK-selectable marker from plasmid pGalK. After galK insertion, recombinants were selected at 32°C on minimal medium in which galactose was supplied as the only carbon source. Recombinants were screened by PCR using 20-bp primers outside of the target regions. Subsequent removal of galK used DNA fragments generated by PCR using partially overlapping 60-bp primers (GkRm forward and reverse; Table S1). Each of the 60-bp primers consisted of a 52-bp homology region flanking the galK marker and an 8-bp sequence complementary to the last 8 bp of the opposite homology region. Negative selection used minimal medium containing 2-deoxy-galactose, and deletion of galK in recombinants was verified using CHECK forward and reverse primers (Table S1). Integrity of BAC constructs and the LacO repeat length was verified by restriction fingerprinting using an AvaI and HindIII double digest.
Sequence analysis
Searching for GA motifs, as previously described in LAS (Zullo et al., 2012), in PTRs, the HBB locus, and DNA flanking the HBB locus, was performed using the MEME (Multiple EM for Motif Elicitation) software package (Bailey and Elkan, 1994).
Cell culture and establishment of BAC cell lines
NIH 3T3 cells (CRL-1658; ATCC) were grown in DMEM (Invitrogen) plus 10% bovine growth serum (HyClone; Thermo Fisher Scientific) at 37°C with 5% CO2. All transfections were performed using Lipofectamine 2000 (Invitrogen). Stable cell lines were selected and subcloned as described previously (Strukov and Belmont, 2005). To create NIH 3T3 cell clone 3T3_LG_C29 stably expressing the EGFP-dimer LacI-NLS fusion protein, the plasmid p3′SS-EGFP-dLacI (Robinett et al., 1996) was linearized by XmnI and transfected into NIH 3T3 cells. For stable selection, we used 200 µg/ml Hygromycin B (EMD Millipore).
BAC DNA was purified using the large-construct kit (QIAGEN). Purified BAC DNA was linearized, ethanol precipitated, and transfected into 3T3_LG_C29 cells followed by selection of stable transformants using 800 µg/ml G418 and 200 µg/ml Hygromycin B. For β-globin and DHFR BAC cotransfection, both BACs were linearized with PI-SceI and BsiWI, respectively, and transfected into 3T3_LG_C29 as described in the previous paragraph using 800 µg/ml G418, 200 µg/ml Hygromycin B, and 75 µg/ml Zeocin for selection.
BACs CTD-2207K13 and RP11-2I1 were digested with PvuI and AscI, respectively, before transfection into 3T3_LG_C29 cells and selection of stable clones with 600 µg/ml G418 and 200 µg/ml Hygromycin B. BAC HBBD4-6kb-C5 was linearized with PI-SceI before transfection into 3T3_LG_C29 cells, and stable clones were selected with 600 µg/ml G418, 200 µg/ml Hygromycin B, and 75 µg/ml Zeocin. Plasmid p[Kan/Neo-HBB6kb-8.32] was linearized with PvuI before transfection into 3T3_LG_C29 cells, and stable clones were selected with 800 µg/ml G418 and 200 µg/ml Hygromycin B.
The full names for NIH 3T3 cell clones in Fig. 4 b were HBBD4_C40.5, HBBD4_C40.10, and HBBD4_C40.22 (versus HBBD4-5, -10, or -22). For H3K9me3 immunostaining comparisons, we used the following cell clones (Fig. 5): HBB_C3, HBBD5_C43, HBBD10_C32, HBBD5D8_C35, HBBD4_C29.7, D4_C40.10, D4_C40.5, HBBD5D7_C29.17, D5D7_C29.22, HBBD5D7_C40.15, HBBD4-PTR1_B6, and DHFR_HBB_2.12.
BJ-hTERT cells were obtained as a gift from P. Adams (Beatson Institute/Glasgow University, Glasgow, Scotland, UK) and grown in DMEM supplemented with 10% FBS, MEM nonessential amino acids, vitamins, and 0.54 µg/ml puromycin at 37°C with 5% CO2. WI-38 cells (ATCC) and Tig3ET cells (a gift from B. Van Steensel, Netherlands Cancer Institute, Amsterdam, Netherlands) were grown in DMEM plus 10% FBS (Sigma-Aldrich) at 37°C with 5% CO2.
Immunostaining, Western blots, DNA FISH, and immuno-FISH
NIH 3T3 cells were fixed with 3% paraformaldehyde (freshly prepared from powder) in calcium magnesium–free (CMF)-PBS for 10 min at RT and washed 3× with CMF-PBS. Cells were then permeabilized with 0.5% Triton X-100 (Thermo Fisher Scientific) in CMF-PBS for 5 min at RT and blocked with 5% normal goat serum for 30 min. Primary antibody was applied for 4 h at RT or overnight at 4°C in CMF-PBS containing 0.1% Triton X-100 at the following dilutions: mouse monoclonal antibody RL1 against nuclear pore O-linked glycoprotein, 1:500 (Thermo Fisher Scientific); anti–lamin A antibody, 1:500 (obtained from B. Goldman, Northwestern University, Evanston, IL); anti-H3K9me2 (ab1220 [Abcam] or 07–441 [EMD Millipore]), 1:500 ; and rabbit anti-H3K9me3 (ab8898 [Abcam] or 07–442 [EMD Millipore]), 1:500. Cells were washed with CMF-PBS 3× for 5 min and stained with secondary goat anti–rabbit or anti–mouse antibody conjugated with Texas red (Jackson ImmunoResearch Laboratories, Inc.) at 1:500 dilution overnight at 4°C. Cells were washed with CMF-PBS 3× for 5 min and mounted in antifade medium containing 0.3 µg/ml DAPI (Sigma-Aldrich)/10% wt/vol MOWIOL 4–88 (EMD)/1% wt/vol DABCO (Sigma-Aldrich)/25% glycerol/0.1 M Tris, pH 8.5. For Western blotting, we used a 1:100 dilution of mouse anti-cKrox (sc-376250; Santa Cruz Biotechnology, Inc.) or 1:1,000 dilution of anti-H3K9 dimethylation antibodies (ab1220; Abcam).
Anti–lamin A or H3K9me3 immunostaining before DNA FISH used a similar procedure as described in the previous paragraph with a 1:1,000 dilution of anti–lamin A or 1:500 dilution of anti-H3K9me3 primary antibody. After secondary antibody staining, cells were postfixed in 3% paraformaldehyde in CMF-PBS for 10 min at RT and washed in 0.1 M HCl/0.7% Triton X-100 (Thermo Fisher Scientific) in 2× SSC for 10 min on ice.
DNA FISH was performed as described previously (Hu et al., 2009). In brief, paraformaldehyde-fixed cells were permeabilized in 0.5% Triton X-100. Cells were subjected to four freeze–thaw cycles before storing in 50% formamide/2× SSC. FISH probes were prepared using nick translation of BAC DNA (BioNick Labeling System; Invitrogen) using biotin and/or digoxigenin-labeled nucleotides. Probes and cells were codenatured on a 75°C heat block for 2 min followed by overnight hybridization at 37°C followed by washes in 0.4× SSC at 70°C for 2 min and 2× SSC. For detection, we used Streptavidin–Alexa Fluor 594 (Invitrogen) and/or antidigoxigenin-fluorescein antibodies (Roche).
Knockdown experiments
For Suv39H1, Suv39H2, and G9a knockdowns, shRNA lentiviral transduction particles (MISSION; Sigma-Aldrich) were used according to the manufacturer’s protocol. In brief, lentiviruses coding for shRNA for mouse Suv39H1 (GenBank accession no. NM_011514; clone ID TRCN0000097440), mouse Suv39H2 (GenBank accession no. NM_022724; clone ID TRCN0000092815), and mouse G9a (GenBank accession no. NM_145830; clone ID TRCN0000054543) or human Suv39H1 (GenBank accession no. NM_003173.2; clone ID TRCN0000275322) and human Suv39H2 (GenBank accession no. NM_024670.3; clone ID TRCN0000006938) were used at ∼3 × 104 TU each to infect ∼104 NIH 3T3 (mouse) or WI-38 cells (human) overnight in the presence of 6 µg/ml polybrene at 37°C. Scrambled shRNA was used as a control. Media were changed the next day followed by 2 µg/ml puromycin selection 2 d after transduction. Selection media were changed every 3 d; at 7 d of selection, cells fixed with 3% paraformaldehyde were used for immunostaining or immuno-FISH.
Depletion of human cKrox, Suv39H1, and Suv39H2 was performed using siGENOME siRNA SMARTpool (Thermo Fisher Scientific; Table S3). Tig3ET cells were transfected with siRNA using Lipofectamine (Invitrogen) following the manufacturer’s protocol and were cultured for 48 h before being used for analysis.
For the G9a inhibition, Tig3ET or NIH 3T3 cells were treated with BIX01294 (Kubicek et al., 2007) or UNC0638 (Vedadi et al., 2011; both obtained from Sigma-Aldrich) for 2 d, at a final concentration of 1 µM or 500 nM, respectively. Knockdown experiments were performed in at least two biological replicates, and a pooled result is displayed.
Microscopy, image analysis, and statistical analysis
A personal deconvolution microscope system (DeltaVision; Applied Precision) equipped with a charge-coupled device camera (CoolSNAP HQ2; Photometrics) was used with a 60×, 1.4 NA lens for data collection. Deconvolution used an enhanced ratio and an iterative constrained algorithm (Agard et al., 1989), using the softWoRx software (Applied Precision). 2D distances between GFP-LacI spots and the nuclear edge defined by DAPI staining were measured from the optical section in which the GFP-LacI spot was in focus using ImageJ (National Institutes of Health) software. Image segmentation of the DAPI-stained nucleus was then performed using the Otsu thresholding 16 bit and k-mean clustering plugins with the following parameters: number = 2, cluster = 0.00010000, and randomization = 48. For BAC transgenes consisting of multiple GFP-LacI spots, measurements were taken from the spot closest to the nuclear edge. P-values were calculated from two-tailed, two-sample unequal variance t tests. Photoshop and Illustrator programs (Adobe) were used to assemble microscopy images, with bicubic interpolation used to rotate or enlarge images.
RT-qPCR
Total RNA was extracted from NIH 3T3 cells using the RNeasy Mini kit (QIAGEN), with on-column DNase I digestion (New England Biolabs, Inc.) according to the manufacturer’s instructions. cDNA was synthesized from 1 µg total RNA with the cDNA kit (qScript Flex; Quanta BioSciences). Quantitative real-time PCR was performed on a PCR instrument (StepOnePlus; Applied Biosystems) using a 2× SYBR green mix. Real-time PCR reactions were performed in triplicate. β-Actin was used as a reference to obtain the relative fold change for target samples using the comparative cycle threshold 2(−ddct) method.
ChIP
ChIP was performed on NIH 3T3 cell clones containing specific HBB BAC or plasmid transgenes. 10 million cells were cross-linked by adding paraformaldehyde (16% EM grade; Polysciences) directly to the culture medium to a final concentration of 1% at RT for 10 min. Cross-linking was stopped by adding glycine to a final concentration of 0.125 M for 5 min at RT followed by addition of ice-cold PBS. Cells were trypsinized, centrifuged, and washed 3× for 10 min with PBS at 4°C. Cells were lysed on ice for 30 min in L1 buffer (50 mM Tris, pH 8.0, 2 mM EDTA, 0.5% NP-40, and 10% glycerol) supplemented with protease inhibitor cocktail (Roche). Nuclei were pelleted at 700 g and resuspended in SDS lysis buffer (50 mM Tris, pH 8.0, 5 mM EDTA, and 1% SDS) supplemented with protease inhibitors. Chromatin was sheared by sonication (Bioruptor UCD-200TM; Diagenode), centrifuged to pellet debris, and diluted 10× in ChIP dilution buffer (16.7 mM Tris-HCl, pH 8.0, 1.1% Triton X-100, 167 mM NaCl, 0.01% SDS, and 1.2 mM EDTA). Extracts were incubated for 1 h at 4°C with 75 µl Dynabeads protein G (Invitrogen) blocked with salmon sperm DNA and BSA followed by the removal of beads. 5% of the supernatant was saved as an input control. ChIP samples were then incubated with either 10 µg H3K9me3 antibody (ab8898; Abcam) or nonspecific IgG (Jackson ImmunoResearch Laboratories, Inc.) and incubated at 4°C overnight with rotation.
Immunoprecipitations were performed with 100 µl Dynabeads protein G blocked with salmon sperm DNA and BSA for 1 h at 4°C. Immune complexes were then washed with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 150 mM NaCl) followed by high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 500 mM NaCl) followed by LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.0; 5 min each at 4°C). Immune complexes were further washed two times with TE (Tris EDTA) buffer before eluting with the ChIP elution buffer (1% SDS and 0.1 M NaHCO3).
DNA cross-links were reversed by addition of 0.2 M NaCl and incubation at 65°C overnight. Proteins and RNA were removed by 50 µg/ml proteinase K (1 h at 45°C) and 20 µg/ml RNase A treatment (30 min at 37°C), and DNA was purified with spin columns (QIAGEN). Primers for qPCR are listed in Table S2.
Online supplemental material
Fig. S1 shows the experimental approach for identifying peripheral targeting cis-regions. Fig. S2 shows the distance distribution from the nuclear periphery for β-globin endogenous versus BAC transgenes. Fig. S3 lists the genomic coordinates of human β-globin BAC (CTD-2643I7) and various deletions within this BAC. Fig. S4 shows cKrox knockdown experiments that do not support a cKrox-mediated, redundant mechanism for peripheral tethering of HBB locus and flanking sequences in Tig3ET cells. Fig. S5 shows that both H3K9me3 and H3K9me2 are enriched in a peripheral chromatin rim in different human and mouse fibroblast cell lines. Table S1 lists primers used for BAC recombineering. Table S2 lists primers used for RT-qPCR and ChIP. Table S3 lists SMARTpool siRNA target sequences. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201305027/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201305027.dv.
Supplementary Material
Acknowledgments
We thank Dr. Lisa Stubbs (University of Illinois, Urbana) for guidance in ChIP procedures. Light microscopy was performed in the Department of Molecular and Cellular Biology Light Microscopy Facility funded with support from the Carver Foundation.
This work was supported by grant GM58460 from the National Institute of General Medical Sciences awarded to A.S. Belmont. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
Abbreviations used in this paper:
- BAC
- bacterial artificial chromosome
- ChIP
- chromatin immunoprecipitation
- CMF
- calcium magnesium free
- DamID
- DNA adenine methyltransferase identification
- DHFR
- dihydrofolate reductase
- HMT
- histone methyltransferase
- HS
- hypersensitive site
- hTERT
- human telomerase reverse transcriptase
- IAP
- intracisternal A-particle
- LacO
- lac operator
- LAD
- lamina-associated domain
- LAS
- lamin-associated sequences
- LCR
- locus control region
- PCH
- pericentromeric heterochromatin
- PTR
- peripheral targeting region
- qPCR
- quantitative PCR
- UHR
- upstream Hispanic region
References
- Agard D.A., Hiraoka Y., Shaw P., Sedat J.W. 1989. Fluorescence microscopy in three dimensions. Methods Cell Biol. 30:353–377 10.1016/S0091-679X(08)60986-3 [DOI] [PubMed] [Google Scholar]
- Andrulis E.D., Neiman A.M., Zappulla D.C., Sternglanz R. 1998. Perinuclear localization of chromatin facilitates transcriptional silencing. Nature. 394:592–595 10.1038/29100 [DOI] [PubMed] [Google Scholar]
- Bailey T.L., Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- Belmont A.S., Hu Y., Sinclair P.B., Wu W., Bian Q., Kireev I. 2010. Insights into interphase large-scale chromatin structure from analysis of engineered chromosome regions. Cold Spring Harb. Symp. Quant. Biol. 75:453–460 10.1101/sqb.2010.75.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender M.A., Byron R., Ragoczy T., Telling A., Bulger M., Groudine M. 2006. Flanking HS-62.5 and 3′ HS1, and regions upstream of the LCR, are not required for beta-globin transcription. Blood. 108:1395–1401 10.1182/blood-2006-04-014431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Q., Belmont A.S. 2010. BAC TG-EMBED: one-step method for high-level, copy-number-dependent, position-independent transgene expression. Nucleic Acids Res. 38:e127 10.1093/nar/gkq178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.E., Guest S.S., Smale S.T., Hahm K., Merkenschlager M., Fisher A.G. 1997. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. 91:845–854 10.1016/S0092-8674(00)80472-9 [DOI] [PubMed] [Google Scholar]
- Brown K.E., Baxter J., Graf D., Merkenschlager M., Fisher A.G. 1999. Dynamic repositioning of genes in the nucleus of lymphocytes preparing for cell division. Mol. Cell. 3:207–217 10.1016/S1097-2765(00)80311-1 [DOI] [PubMed] [Google Scholar]
- Brown K.E., Amoils S., Horn J.M., Buckle V.J., Higgs D.R., Merkenschlager M., Fisher A.G. 2001. Expression of alpha- and beta-globin genes occurs within different nuclear domains in haemopoietic cells. Nat. Cell Biol. 3:602–606 10.1038/35078577 [DOI] [PubMed] [Google Scholar]
- Cope N.F., Fraser P., Eskiw C.H. 2010. The yin and yang of chromatin spatial organization. Genome Biol. 11:204 10.1186/gb-2010-11-3-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll M.C., Dobkin C.S., Alter B.P. 1989. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5′ beta-globin gene activation-region hypersensitive sites. Proc. Natl. Acad. Sci. USA. 86:7470–7474 10.1073/pnas.86.19.7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Xiang P., Yin W., Stamatoyannopoulos G., Li Q. 2007. Cooperativeness of the higher chromatin structure of the beta-globin locus revealed by the deletion mutations of DNase I hypersensitive site 3 of the LCR. J. Mol. Biol. 365:31–37 10.1016/j.jmb.2006.09.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlan L.E., Sproul D., Thomson I., Boyle S., Kerr E., Perry P., Ylstra B., Chubb J.R., Bickmore W.A. 2008. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4:e1000039 10.1371/journal.pgen.1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francastel C., Magis W., Groudine M. 2001. Nuclear relocation of a transactivator subunit precedes target gene activation. Proc. Natl. Acad. Sci. USA. 98:12120–12125 10.1073/pnas.211444898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer P.K., Vitalini M.W., Wallrath L.L. 2011. Nuclear organization: taking a position on gene expression. Curr. Opin. Cell Biol. 23:354–359 10.1016/j.ceb.2011.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetze S., Mateos-Langerak J., Gierman H.J., de Leeuw W., Giromus O., Indemans M.H., Koster J., Ondrej V., Versteeg R., van Driel R. 2007. The three-dimensional structure of human interphase chromosomes is related to the transcriptome map. Mol. Cell. Biol. 27:4475–4487 10.1128/MCB.00208-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L., Pagie L., Brasset E., Meuleman W., Faza M.B., Talhout W., Eussen B.H., de Klein A., Wessels L., de Laat W., van Steensel B. 2008. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 453:948–951 10.1038/nature06947 [DOI] [PubMed] [Google Scholar]
- Hahm K., Cobb B.S., McCarty A.S., Brown K.E., Klug C.A., Lee R., Akashi K., Weissman I.L., Fisher A.G., Smale S.T. 1998. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev. 12:782–796 10.1101/gad.12.6.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepperger C., Mannes A., Merz J., Peters J., Dietzel S. 2008. Three-dimensional positioning of genes in mouse cell nuclei. Chromosoma. 117:535–551 10.1007/s00412-008-0168-2 [DOI] [PubMed] [Google Scholar]
- Hu Y., Kireev I., Plutz M.J., Ashourian N., Belmont A.S. 2009. Large-scale chromatin structure of inducible genes: transcription on a condensed, linear template. J. Cell Biol. 185:87–100 10.1083/jcb.200809196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J., Pagie L., Ortabozkoyun H., Boyle S., de Vries S.S., Janssen H., Amendola M., Nolen L.D., Bickmore W.A., van Steensel B. 2013. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 153:178–192 10.1016/j.cell.2013.02.028 [DOI] [PubMed] [Google Scholar]
- Kohwi M., Lupton J.R., Lai S.L., Miller M.R., Doe C.Q. 2013. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 152:97–108 10.1016/j.cell.2012.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak S.T., Skok J.A., Medina K.L., Riblet R., Le Beau M.M., Fisher A.G., Singh H. 2002. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science. 296:158–162 10.1126/science.1068768 [DOI] [PubMed] [Google Scholar]
- Kubicek S., O’Sullivan R.J., August E.M., Hickey E.R., Zhang Q., Teodoro M.L., Rea S., Mechtler K., Kowalski J.A., Homon C.A., et al. 2007. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell. 25:473–481 10.1016/j.molcel.2007.01.017 [DOI] [PubMed] [Google Scholar]
- Kumaran R.I., Spector D.L. 2008. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 180:51–65 10.1083/jcb.200706060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldi L., Brickner J.H. 2011. Compartmentalization of the nucleus. Trends Cell Biol. 21:701–708 10.1016/j.tcb.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra R.J., Tolhuis B., Splinter E., Nijmeijer R., Grosveld F., de Laat W. 2003. The beta-globin nuclear compartment in development and erythroid differentiation. Nat. Genet. 35:190–194 10.1038/ng1244 [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D., van Steensel B. 2010. Role of the nuclear lamina in genome organization and gene expression. Cold Spring Harb. Symp. Quant. Biol. 75:517–524 10.1101/sqb.2010.75.014 [DOI] [PubMed] [Google Scholar]
- Peters A.H., Kubicek S., Mechtler K., O’Sullivan R.J., Derijck A.A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., et al. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 12:1577–1589 10.1016/S1097-2765(03)00477-5 [DOI] [PubMed] [Google Scholar]
- Ragoczy T., Bender M.A., Telling A., Byron R., Groudine M. 2006. The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev. 20:1447–1457 10.1101/gad.1419506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K.L., Zullo J.M., Bertolino E., Singh H. 2008. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 452:243–247 10.1038/nature06727 [DOI] [PubMed] [Google Scholar]
- Robinett C.C., Straight A., Li G., Willhelm C., Sudlow G., Murray A., Belmont A.S. 1996. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135:1685–1700 10.1083/jcb.135.6.1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevelyov Y.Y., Nurminsky D.I. 2012. The nuclear lamina as a gene-silencing hub. Curr. Issues Mol. Biol. 14:27–38 [PubMed] [Google Scholar]
- Sinclair P., Bian Q., Plutz M., Heard E., Belmont A.S. 2010. Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J. Cell Biol. 190:761–776 10.1083/jcb.200912167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strukov Y.G., Belmont A.S. 2005. Development of mammalian cell lines with lac operator-tagged chromosomes. Live Cell Imaging: A Laboratory Manual. First edition Spector D.L., Goldman R.D., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: 541–563 [Google Scholar]
- Towbin B.D., González-Aguilera C., Sack R., Gaidatzis D., Kalck V., Meister P., Askjaer P., Gasser S.M. 2012. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 150:934–947 10.1016/j.cell.2012.06.051 [DOI] [PubMed] [Google Scholar]
- Tumbar T., Sudlow G., Belmont A.S. 1999. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J. Cell Biol. 145:1341–1354 10.1083/jcb.145.7.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T.J., Dimaggio P.A., Wasney G.A., Siarheyeva A., et al. 2011. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7:566–574 10.1038/nchembio.599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S., Costantino N., Court D.L., Jenkins N.A., Copeland N.G. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.R., Azuara V., Perry P., Sauer S., Dvorkina M., Jørgensen H., Roix J., McQueen P., Misteli T., Merkenschlager M., Fisher A.G. 2006. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 119:132–140 10.1242/jcs.02727 [DOI] [PubMed] [Google Scholar]
- Wu R., Terry A.V., Singh P.B., Gilbert D.M. 2005. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol. Biol. Cell. 16:2872–2881 10.1091/mbc.E04-11-0997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Fetter R.D., Hu P., Betzig E., Tjian R. 2011. Subnuclear segregation of genes and core promoter factors in myogenesis. Genes Dev. 25:569–580 10.1101/gad.2021411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R., Bodnar M.S., Spector D.L. 2009. Nuclear neighborhoods and gene expression. Curr. Opin. Genet. Dev. 19:172–179 10.1016/j.gde.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zullo J.M., Demarco I.A., Piqué-Regi R., Gaffney D.J., Epstein C.B., Spooner C.J., Luperchio T.R., Bernstein B.E., Pritchard J.K., Reddy K.L., Singh H. 2012. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell. 149:1474–1487 10.1016/j.cell.2012.04.035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.