

Abstract
The Major Immediate Early Promoter (MIEP) of human cytomegalovirus (HCMV) controls viral Immediate Early (IE) gene expression, which must be activated to initiate productive infection and repressed to establish latency. Regulation of the MIEP is critical for both viral spread and persistence. In addition to the Daxx-mediated intrinsic cellular defense that regulates the MIEP, the cell-type specific balance between cellular activators and repressors of the promoter may help dictate whether viral IE genes will be expressed or silenced. For example, in undifferentiated myeloid cells, transcriptional repressors of the MIEP may outnumber transcriptional activators, leading to promoter silencing and latency establishment. We created a recombinant viral genome in which a myeloid-active promoter replaced part of the MIEP. The viable virus generated failed to express the viral IE genes in an undifferentiated myeloid cell line. These observations have mechanistic implications regarding how viral IE gene expression is regulated during latency.
Introduction
The human cytomegalovirus (HCMV) causes disease by productively replicating in epithelial, endothelial, smooth muscle, fibroblast, placental trophoblast, and differentiated myeloid lineage cells, such as macrophage and dendritic cells (Sinzger, Digel, and Jahn, 2008). It persists for the life of the infected host by establishing latent infections in undifferentiated cells of the myeloid lineage (Goodrum, Caviness, and Zagallo, 2012). Thus, HCMV must be equipped to express its lytic phase genes in multiple cell types, silence them to establish and maintain latent reservoirs, and activate (animate) them to reactivate from latency to productive replication (Penkert and Kalejta, 2011). Much of the lytic phase transcriptional program is initiated by the viral Immediate Early (IE) 1 and IE2 proteins, encoded within the viral genome's unique long (UL) region by the UL123 and UL122 genes (respectively). They are among the first genes to be expressed upon initiation of a lytic infection, but are not expressed during latency. Expression (or repression) of IE1 and IE2 is controlled by the Major Immediate Early Promoter (MIEP) (Meier and Stinski, 2006).
The MIEP consists of a core promoter (P) (nucleotide positions +1 (the transcription start site) to -39), an enhancer (E) divided into proximal (-39 to -300) and distal (-300 to -550) components, a unique region (U) (-500 to -750) and a modulator (M) (-750 to -1140) (Fig. 1). Truncated versions of the MIEP containing the promoter, proximal enhancer, and sometimes the distal enhancer are ubiquitously present in expression vectors for use in mammalian cells, where they are often referred to as simply “the CMV promoter”. A substantial number of transcription factor binding sites have been identified by both bioinformatic and experimental approaches, and presumably these sites contribute to both the wide array of cell types in which the promoter is active, as well as the comparative strength of the promoter within those cells. Despite the awesome power of this promoter, it has also evolved to sustain transcriptional repression during latency.
Fig 1. Promoter region of the major immediate early locus of wild type HCMV and SFFV-LTR recombinants.
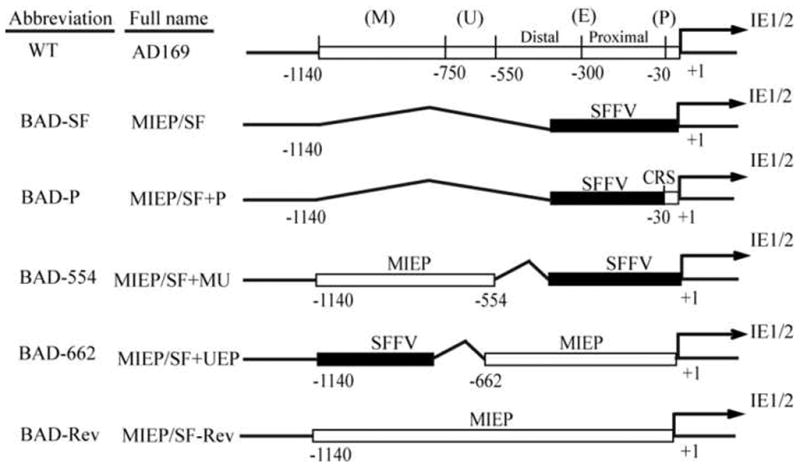
Functional regions of the MIEP are numbered relative to the transcription start site (+1). These include the core promoter (P), the enhancer (E), which is divided into proximal and distal components, the unique region (U), and the modulator (M). Recombinants are given full names based on the location of the insertion (MIEP) and the inserted sequences (P, E, U, and M). Abbreviated names are used for simplicity. BAD-SF represents a complete deletion of the MIEP with insertion of the SFFV LTR. BAD-P retains a portion of the core promoter with insertion of the SFFV LTR. BAD-554 retains the modulator and unique regions with an insertion of the SFFV LTR. BAD-662 retains the core promoter, proximal and distal enhancers, and a portion of the unique region with an insertion of the SFFV LTR. BAD-Rev represents a revertant of BAD-SF to the wild type sequence. See materials and methods for details.
At least three mechanisms conspire to silence the MIEP during latency. These are a cellular intrinsic immune defense found in all cells tested to date (Saffert and Kalejta, 2006; Saffert and Kalejta, 2007; Saffert, Penkert, and Kalejta, 2010), an unidentified viral function (or functions) encoded only by certain strains of the virus (Saffert, Penkert, and Kalejta, 2010), and the potential activity of the promoter, likely controlled by the balance between positive and negative cellular regulators of the MIEP expressed in a cell type-specific manner (Reeves, 2011; Saffert, Penkert, and Kalejta, 2010; Sinclair, 2010). The cellular Daxx intrinsic immune protein silences viral gene expression by instituting a repressive chromatin structure at the MIEP (Tavalai and Stamminger, 2011; Woodhall et al., 2006). It is inactivated in cells destined to initiate a lytic infection when the tegument-delivered pp71 protein traffics to the nucleus and degrades Daxx, but remains active during the establishment of latency because tegument-delivered pp71 localizes to the cytoplasm in these cells (Penkert and Kalejta, 2012). Artificial inactivation of the Daxx defense with histone deacetylase (HDAC) inhibitors is sufficient to activate the MIEP and drive IE gene expression in cells where the virus would otherwise establish latency, but only when the laboratory adapted AD169 virus strain is used for infections (Albright and Kalejta, 2013; Penkert and Kalejta, 2013; Qin, Penkert, and Kalejta, 2013; Saffert and Kalejta, 2007). IE gene expression from clinical virus strains that encode additional genes in the ULb' genomic locus is not rescued by HDAC inhibition, indicating that these viral strains impose an additional impediment to viral IE gene expression during latency above and beyond that instituted by Daxx (Saffert, Penkert, and Kalejta, 2010). Finally, in the context of a recombinant adenoviral genome, a truncated MIEP is 10-1000-fold less active in undifferentiated cell types where the virus establishes latency as compared to differentiated cell types where it initiates a lytic infection (Saffert, Penkert, and Kalejta, 2010). This finding seems to support the long-held contention (Reeves, 2011; Sinclair, 2010) that the balance between cellular MIEP activating and repressing factors is tipped in favor of repression in cell types where the virus establishes latency.
In attempts to alter this balance, we generated recombinant HCMVs in which all or part of the MIEP was replaced with the Long Terminal Repeat (LTR) of the Spleen Focus Forming Virus (SFFV), a promoter known to be active in undifferentiated cells of the myeloid lineage (Baum et al., 1995; Carter et al., 2010; Gutsch et al., 2011). Furthermore, we employed the AD169 strain to avoid the complication of the additional clinical strain-specific restriction. Recombinants in which the SFFV promoter replaced the entire MIEP, or the proximal and distal enhancers, were not viable. A recombinant in which the SFFV promoter replaced part of the unique region and the modulator grew with wild type kinetics in fully differentiated fibroblasts, but failed to express viral IE genes in THP-1 monocytes, incompletely differentiated myeloid cells that are used to study viral transcriptional silencing during experimental latency. As our recombinant viruses apparently failed to shift the equilibrium between positive and negative MIEP regulators enough to overcome the intrinsic defense and perhaps other cellular responses to foreign DNAs, the promoter elements that control HCMV IE gene repression during latency remain enigmatic, while appreciation for the controlling contribution of intrinsic cellular silencing mechanisms is reinforced.
Materials and Methods
Cells and viruses
Human foreskin fibroblasts (HFs) were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% (vol/vol) fetal bovine serum (Gemini), 100 U/ml penicillin, and 100 µg/ml streptomycin plus 0.292 mg/ml glutamine (Gibco), in a 5% CO2 atmosphere at 37°C. THP-1 cells were cultured in RPMI 1640 medium (Invitrogen) supplemented as above. Wild type and recombinant HCMVs were rescued from the AD169 BAC (Terhune et al., 2007) and tittered by plaque assay. Recombinant HCMV lacking the unique region and modulator (-582/-1108) was previously described (Meier and Pruessner, 2000).
Viral Reverse Genetics
HCMV BACs were mutagenized as previously described (Tischer, Smith, and Osterrieder, 2010). The MIEP (-1140 to -1, relative to transcription start site +1) was replaced with the SFFV LTR (from pSR-CMV-MLV-SIN) (Wu and Lu, 2010) to make BAD-MIEP/SF. A revertant (BAD-MIEP/SF-Rev) was made from BAD-MIEP/SF by replacing SFFV sequences with the MIEP. The MIEP (-1140 to -30) was replaced with SFFV to make BAD-MIEP/SF+P. The MIEP (-1140 to -554) was added back to BAD-MIEP/SF to make BAD-MIEP/SF+MU. The MIEP (-662 to -1) was added back to BAD-MIEP/SF to make BAD-MIEP/SF+UEP. See Figure 1 for schematic representation and nomenclature abbreviations. All primers used for making BACs are listed in Table 1. BACs were confirmed by restriction digest and sequencing. To reconstitute virus, HFs were transfected with 20 ug of BACs and 5 ug of plasmid pCGN-pp71 in 350 ul Ingenio Electroporation solution (Mirus) in 4 mm Cuvette at 260 Volts, 950 uF (Bio-Rad, GenePulser X cell™) and cultured up to 30 days.
Table 1.
Oligonucleotides used for the construction of plasmids, recombinant BACs, and PCR.
| Name | Sequence | Purpose |
|---|---|---|
| KanR For | CAACCCTCAGCAGTTTCTTAAGACCCATCAGATGTTTCCAGGCT CCCCCAAGGACCTGAAATGACCTAGAGAACCCACTGCTTACTGG |
KanR |
| KanR Rev | AACTGCTGAGGGCCAGTGTTACAACCAATTAACC | |
| BAD-SF For | GAGAGCGTTAAAAAACACAAACGGCTGGATGTGTGCCGCGCTA AAATGGGCTATATGCTGGTAACGCCATTTTGCAAGGCATGGA |
BAD-SF |
| BAD-SF Rev | CGGTCCCGGTGTCTTCTATGGAGGTCAAAACAGCGTGGATGG CGTCTCCAGGCGATCTGACGACTCAGTCTGTCGGAGGACTGG |
|
| BAD-P For | GAGAGCGTTAAAAAACACAAACGGCTGGATGTGTGCCGCGC TATCTCCAGGCGATCTGACGACTCAGTCTGTCGGAGGACTGG |
BAD-P |
| BAD-P Rev | ACAGCGTGGATGGCGTCTCCAGGCGATCTGACGGTTCACTAA ACGAGCTCTGCTTATATA GAGCTCGGGAAGCAGAAGCG |
|
| MIEP For | GGATCAACCTGGAATACGACAAG | MIEP |
| MIEP Rev | CTCTATAGGCGGTACTTACGTC | |
| MIEP-K For | CAACTGTACATTTATATTGGCTCATGTCCAACATTACCGCCATG TTGACATTGATTATTGACTAGTTAGAGAACCCACTGCTTACTGG |
KanR |
| MIEP-K Rev | AACTTGTACAGCCAGTGTTACAACCAATTAACC | |
| BAD-Rev For | GAGAGCGTTAAAAAACACAAACGGCTGGATGTGTGCCGCGCTA AAATGGGCTATATGCTGCAGTGAATAATAAAATGTGTGTTTGT |
BAD-Rev |
| BAD-Rev Rev | CGGTCCCGGTGTCTTCTATGGAGGTCAAAACAGCGTGGATGGC GTCTCCAGGCGATCTGACGGTTCACTAAACGAGCTCTGCTT |
|
| BAD-554 For | GAGAGCGTTAAAAAACACAAACGGCTGGATGTGTGCCGCGCTA AAATGGGCTATATGCTGCAGTGAATAATAAAATGTGTGTTTGT |
BAD-554 |
| BAD-554 Rev | GATCTGAACTTCTCTATTCTTGGTTTGGTATTTTTCCATGCCTTG CAAAATGGCGTTACCCCCGTAATTGATTACTATTAATA |
|
| BAD-662 For | TTTGAATTAACCAATCAGCCTGCTTCTCGCTTCTGTTCGCGCG CTTCTGCTTCCCGAGCTCACGTTGTATCCATATCATAATATG |
BAD-662 |
| BAD-662 Rev | CGGTCCCGGTGTCTTCTATGGAGGTCAAAACAGCGTGGATGG CGTCTCCAGGCGATCTGACGGTTCACTAAACGAGCTCTGCTT |
|
| IE1 For | CGTCCTTGACACGATGGA | IE1 |
| IE1 Rev | TCTCCTCGAAAGGCTCATGA | |
| GAPDH For | GAGCCAAAAGGGTCATC | GAPDH |
| GAPDH Rev | GTGGTCATGAGTCCTTC |
Indirect immunofluorescence
Transfected HFs were grown on coverslips. Infected, suspension THP-1 cells were collected by low-speed centrifugation, treated with trypsin (0.5 mg/ml) for 5 min at 37°C, collected again by low-speed centrifugation, resuspended in cold phosphate-buffered saline (PBS), and allowed to attach to water-washed coverslips for 60 min at room temperature. Cells on coverslips were fixed with 2% paraformaldehyde in PBS. After washing with PBST (PBS plus 0.1% Triton X-100 and 0.05% Tween 20) coverslips were blocked with PBST plus 5% goat serum and 0.5% bovine serum albumin for 30 min. Cells were then incubated for 1 hour with primary antibodies specific for IE1 (1B12), IE2 (3H9), UL44 (CA006-100; Virusys), or pp28 (CMV157), washed three times with PBST, and then incubated for an additional hour with secondary antibodies. Immunostained cells were washed a final three times with PBS, and mounted on slides with ProLong® reagent with DAPI (4′, 6-diamidino-2-phenylindole dihydrochloride) (Invitrogen). Images were captured with a Zeiss microscope and camera (Axiovert 200 M).
Western Blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer as described previously (Saffert and Kalejta, 2006). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immobilized on Optitran nitrocellulose membranes. The membrane was blocked for 15 min with 5% non-fat skim milk in Tris-buffered saline (20 mM Tris, 137 mM NaCl [pH 7.6]) containing 0.1% Tween-20 (TBS-T) and then incubated for 4 hours with primary antibodies (see above) in TBS-T containing 1% milk. Blots were washed three times for 15 min each with TBS-T, followed by a 4 hour incubation with HRP-conjugated secondary antibodies in TBS-T containing 1% milk. Blots were washed three times and developed with the ECL enhanced chemiluminescence system (Thermo Scientific).
PCR and RT-PCR
To monitor viral DNA replication, total DNA was extracted by mini DNA extraction kit (Qiagen) one or thirteen days after BAC transfection. Aliquots were digested with Dpn I, the MIEP regions was amplified by PCR with the primers indicated in Table 1, and products were separated by ethidium bromide agarose gel electrophorsesis. To analyze the transcription of IE gene in undifferentiated cells, THP-1 cells (1 × 106) were pretreated with or without VPA (100ug/ml) and then infected with wild type AD169 or BAD-662 at an MOI of 2. Total RNA was extracted with the RNeasy minikit (Qiagen) at 20 hpi. Isolated RNA was quantified, and equivalent amount (2 ug) of each sample was treated with RNase-free DNase (Promega) following the manufacturer's protocol. Equivalent amount (0.1ug) of RNA was subsequently used in a reverse transcription (RT) as described in the manual of SuperScript III First Strand Synthesis kit (Invitrogen). cDNA was used for PCR with Taq flexi polymerase (Promega). All primers used were listed in table 1.
Luciferase assays
A Dual luciferase reporter assay system (Promega) was employed. Wild type and SFFV-hybrid MIEPs were amplified by PCR from BAC genomes and inserted into pGL-3. THP-1 cells (1 × 106) were transfected with Lipofectamine 2000 (Invitrogen) in Opti-MEM-1 (Invitrogen) with 200ng of pGL-3 based Firefly reporters and 400ng pRL-TK-Renilla as a normalization control. HFs (3.5 × 105) were transfected as above. Media was changed at 24h and lysates were harvested at 48h in 40 ul of Passive Lysis Buffer (Promega). Luciferase activities were quantitated with a Veritas microplate luminometer (Turner Biosystems) and are expressed as Firefly relative to Renilla. Values were compared with Student's T test.
Results
The SFFV promoter cannot replace the MIEP
HCMV recombinants in which mutant MIEPs with deleted segments drive substantially lower expression levels of IE transcripts display moderate to severe growth defects, but nonetheless are viable (Isomura, Tsurumi, and Stinski, 2004; Meier and Pruessner, 2000; Meier and Stinski, 1997; Stinski and Isomura, 2008). Therefore we reasoned that replacing all or part of the MIEP with the SFFV promoter may result in a virus crippled yet competent for productive replication in differentiated fibroblasts, and because the hybrid promoter now driving IE1 and IE2 expression contains elements geared for activation in myeloid lineage cells (Baum et al., 1995; Carter et al., 2010; Gutsch et al., 2011), this recombinant may fail to silence IE genes and thus fail to establish latency upon infection of incompletely differentiated myeloid cells. Infecting THP-1 cells with a virus harboring a hybrid SFFV-MIEP and comparing the propensity for IE gene expression to wild type virus tests the hypothesis that the balance between binding sites for positive and negative regulators within the promoter has a dominant controlling effect on viral IE gene expression during the establishment of latency. However, recombinant viruses rendered competent for IE gene expression in such a manner are still not expected to productively replicate, as it has been clearly shown that forcing IE gene expression in undifferentiated cells does not result in productive replication (Saffert and Kalejta, 2007; Yee, Lin, and Stinski, 2007). Such an abortive infection would likely prove immunogenic, providing an additional reason (other than the seeding of the latent reservoir) for HCMV IE gene expression to be silenced upon infection of undifferentiated myeloid cells.
Replacing the entire MIEP with the SFFV promoter (BAD-SF; Fig. 1) resulted in a recombinant genome (Fig. 2A-B) that failed to generate infectious progeny virions upon transfection with a pp71-expressing plasmid into fully permissive human fibroblasts, judged by the inability to observe a spreading plaque by GFP fluorescence (Fig. 2C). This was somewhat surprising as the SFFV promoter and the MIEP showed statistically indistinguishable activity in a reporter assay in the same population of fibroblasts (Fig. 2D), reinforcing the notion that promoters are regulated differently within infecting viral genomes and transfected plasmids. Replacing the SFFV promoter in BAD-SF with the MIEP (BAD-Rev; Fig. 1) generated an infectious, revertant genome (Fig. 2A-C) indicating that the lesion at the MIEP was responsible for the growth defect of BAD-SF.
Fig 2. Recombinant genomes differ in abilities to reconstitute infectious virus.
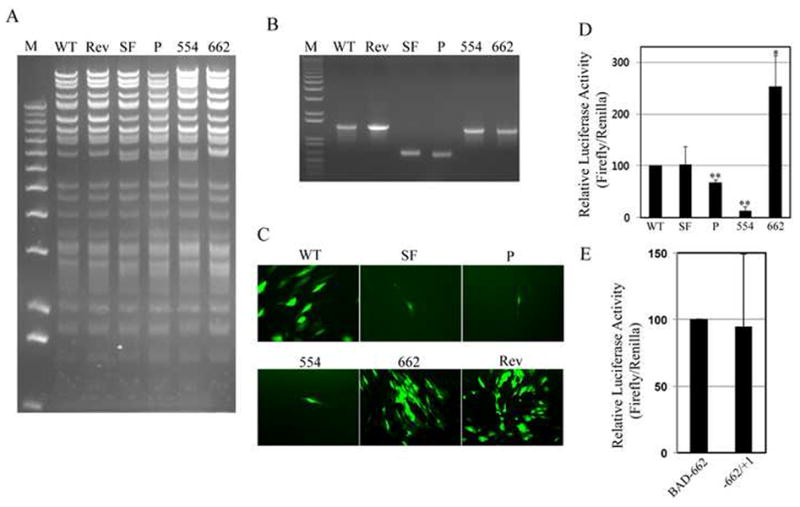
A. Recombinant BACs were digested with Bbv CI, bands were separated by agarose gel electrophoresis and stained with ethidium bromide. M, size markers. B. The MIEP region of each recombinant was amplified by PCR, products were separated by agarose gel electrophoresis and stained with ethidium bromide. M, size markers. C. Recombinant genomes were transfected into fully permissive human fibroblasts. Thirty days after transfection, representative images were captured by fluorescence microscopy. D. HFs were transfected with firefly luciferase reporter plasmids driven by the indicated promoter. Firefly luciferase activity relative to a co-transfected Renilla luciferase reporter is displayed with standard deviation. WT and SF were not statistically different (p = 0.91). P (p < 0.01), 554 (p < 0.01) and 662 (p < 0.05) were statistically different than WT. E. Luciferase assay as in panel D with the indicated promoters (p = 0.84).
Supplementing the SFFV sequences in BAD-SF by replacing most of the core promoter without (BAD-P) or with (BAD-554) the unique region and the modulator (Fig. 1) also generated viral genomes (Fig. 2A-B) that were not converted to expanding plaques or infectious virus upon transfection with a pp71-expressing plasmid into fibroblasts (Fig. 2C). Inserting the unique region and the modulator, elements known to contain binding sites for repressive transcription factors including Yin-Yang-1 (YY1), ETS2-repressor factor (ERF) and silencing binding protein (SBP) (Bain, Mendelson, and Sinclair, 2003; Huang et al., 1996; Liu et al., 1994) actually impaired the activity of the hybrid promoter in a reporter assay (Fig. 2D). Inserting the MIEP core promoter, proximal and distal enhancers, and part of the unique region in between the SFFV promoter and the transcription start site (BAD-662; Fig. 1) generated an infectious, recombinant genome (Fig. 2A-C) and a hybrid promoter with increased activity in reporter assays compared to the full length MIEP (Fig. 2D), and similar activity to a promoter containing only the extant MIEP sequences (Fig. 2E).
Interestingly, transfected recombinant genomes (BAD-SF, BAD-P, and BAD-552) for non-infectious viruses resulted in the expression of IE1 and IE2, and the early protein UL44 (Fig. 3). Qualitative examination suggested a similar percentage of cells in each transfection went on to express IE and UL44 proteins. However, no viral DNA replication was observed. All viral genomes were detectable as transfected BAC clones by PCR in total DNA prepared from cells one day after transfection (Fig. 4). Prior digestion with DpnI (which digests methylated, bacterially-produced DNAs, but not hemi- or unmethylated DNA produced in mammalian cells) (Lacks and Greenberg, 1977) prevented their detection. By thirteen days after transfection, wild type and BAD-662 viral genomes had replicated, as they were detectable in both DpnI-treated and untreated DNA preparations. Other recombinant viral genomes were undetectable even in the absence of DpnI digestion, likely indicating that the transfected genomes, in the absence of replicative amplification, were degraded. Not surprisingly, recombinants unable to replicate their DNA (BAD-SF, BAD-P, and BAD-554) also failed to express the late UL99 gene that encodes the pp28 protein (Fig. 3), and never showed any signs of generating spreading plaques, although single IE1-positive cells could still be detected as long as 58 days after transfection in one experiment (data not shown). Thus, non-viable recombinants in which the SFFV promoter replaced all or part of the MIEP were competent for the expression of the major IE genes and at least one early gene, but failed to replicate their DNA or express late genes.
Fig 3. Non-viable recombinants express representative immediate early and early, but not late genes.
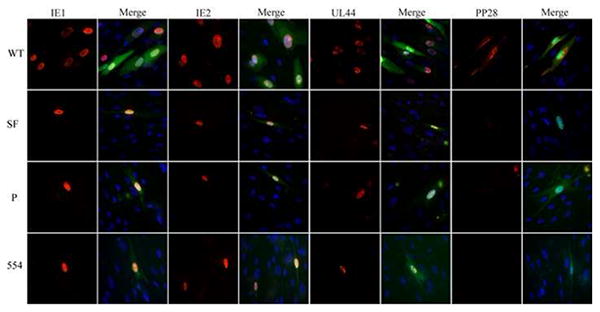
Fibroblasts were transfected with the indicated recombinant genomes and a pp71-expression plasmid and thirteen days later, cells grown on coverslips were stained for the indicated viral antigen (red) and visualized by indirect immunofluorescence microscopy. Nuclei were stained with DAPI (blue) and are displayed in merged images to the left of each viral antigen. Representative images are shown.
Fig 4. Non-viable recombinants fail to replicate viral DNA.
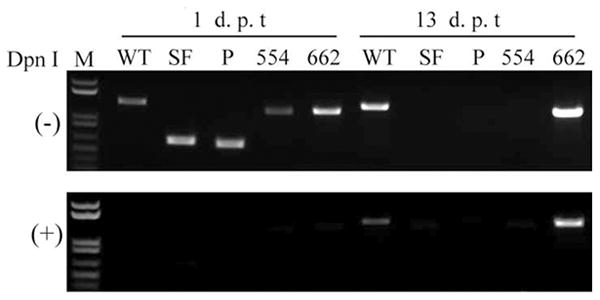
Total DNA was extracted either one or thirteen days post transfection (dpt) of fibroblasts with the indicated recombinant BAC genomes. Aliquots were digested (+) or not (-) with DpnI. The MIEP region was amplified by PCR, products were separated by agarose gel electrophoresis and stained with ethidium bromide. M, size markers.
Virus with a chimeric MIEP/SSFV promoter (BAD-662) replicates with wild type kinetics in fibroblasts
The only viral genome we created with a chimeric MIEP/SFFV promoter driving expression of the MIE locus that generated a spreading plaque contained the entire SFFV promoter and the promoter, enhancer and unique region of the MIEP (BAD-662, Fig. 1). This virus was recoverable, and stocks were prepared. The kinetics of viral IE, E, and L protein expression between BAD-662 and wild type virus were indistinguishable (Fig. 5A). Likewise, progeny virion production between the two viruses was essentially identical at both high (Fig. 5B) and low (Fig. 5C) multiplicities of infection. Therefore we conclude that BAD-662 productively infects and replicates in fibroblasts as well as wild type virus.
Fig 5. BAD-662 displays gene expression and viral replication kinetics indistinguishable from wild type virus.
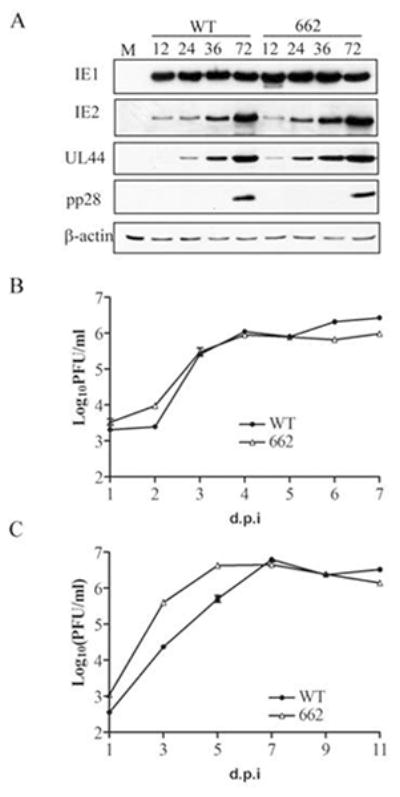
A. Lysates prepared at the indicated hour post infection from fibroblasts infected at a multiplicity of infection (MOI) of 1 with the indicated virus were analyzed by Western blotting with the indicated antibodies. Actin serves as a loading control. M, mock infection. B. Samples were prepared at the indicated day post infection (dpi) from supernatants and attached cells infected at an MOI of 1 with the indicated virus and tittered by plaque assay. Error bars represent standard deviations. C. Growth curves as described in panel B were conducted after infections at an MOI of 0.1.
Viral IE gene expression from BAD-662 is silenced upon infection of THP-1 cells
Our ability to produce BAD-662 virus allowed us to test whether inserting myeloid active promoter elements into the MIEP could disrupt the balance between factors that promote or repress transcription, and thus drive viral IE gene expression in undifferentiated myeloid cells where they are normally silenced. Viral IE RNA accumulation was inhibited upon infection of THP-1 monocytes with both wild type and BAD-662 (Fig. 6). The HDAC inhibitor VPA promoted viral gene expression from both viruses, indicating not only that viral genomes successfully entered the nucleus, but also that they were efficiently silenced by the cellular intrinsic defense. Similar results were obtained when viral IE1 protein accumulation was monitored (Fig. 7). Interestingly, this hybrid promoter showed similar activity in a reporter assays in THP-1 cells when compared to either the full length (Fig. 8A) or a truncated (Fig. 8B) MIEP. This further confirms the inability of reporter assays to accurately reflect promoter activity within HCMV genomes. The combined removal of binding sites for repressive factors and the insertion of myeloid active promoter elements were insufficient to drive viral IE gene expression in incompletely differentiated myeloid lineage cells. This could indicate that intrinsic, perhaps catholic repressive mechanisms play a dominant role to specific activating promoter elements when HCMV gene expression is silenced during the establishment of experimental latency, and that sequences in the unique region or the modulator are not required for the suppression of MIEP activity observed in these cells. The latter speculation is confirmed by the observation that an HCMV mutant lacking the unique region and the modulator is silenced upon infection of THP-1 cells (Fig. 8C).
Figure 6. Transcription of the major immediate early locus of BAD-662 in THP-1 cells requires HDAC inhibition.
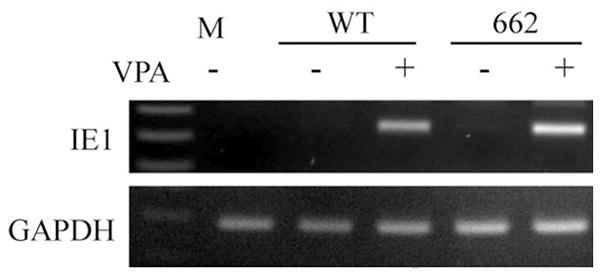
THP-1 cells pretreated (+) or not (-) with VPA for three hours were infected with the indicated virus at an MOI of 2. Total RNA was extracted twenty hours later and analyzed for the expression of the indicated viral (IE1) or cellular (GAPDH) gene by RT-PCR. Products were separated by agarose gel electrophoresis and stained with ethidium bromide. M, mock infection.
Figure 7. Viral IE protein expression from BAD-662 in THP-1 cells requires HDAC inhibition.
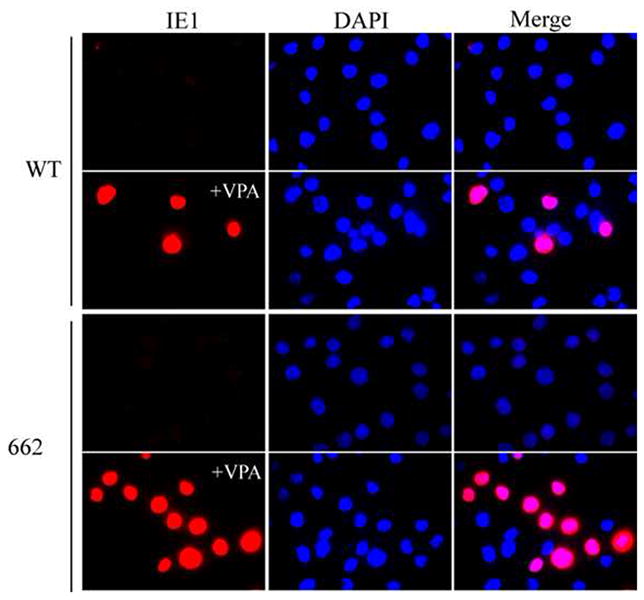
THP-1 cells pretreated with VPA where indicated (+VPA) for three hours were infected with the indicated virus at an MOI of 2 and, twenty hours later were attached to coverslips, stained for the viral IE1 protein (red) and visualized by indirect immunofluorescence microscopy. Nuclei stained with DAPI (blue) are displayed alone and in merged images.
Figure 8. Wild type and SFFV-hybrid MIEPs are active on transfected plasmids in THP-1 cells.
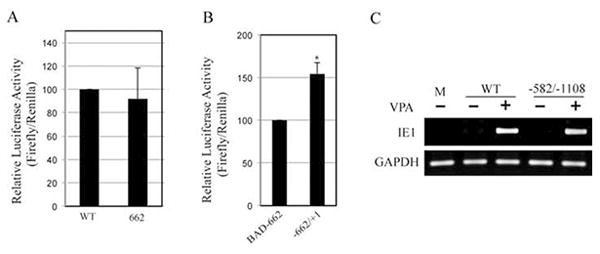
A. THP-1 cells were transfected with firefly luciferase reporter plasmids driven by the indicated promoter. Firefly luciferase activity relative to a co-transfected Renilla luciferase reporter is displayed with standard deviation. WT and BAD662 were not statistically different (p = 0.63). B. Luciferase assay as in panel A with the indicated promoters (p = 0.003). C. THP-1 cells pretreated (+) or not (-) with VPA for three hours were infected with the indicated virus at an MOI of 2. Total RNA was extracted twenty hours later and analyzed for the expression of the indicated viral (IE1) or cellular (GAPDH) gene by RT-PCR. Products were separated by agarose gel electrophoresis and stained with ethidium bromide. M, mock infection.
Discussion
Substantial efforts to understand how the overall structure of, or individual elements within the HCMV MIEP, modulates its activity have failed to produce a clear picture of how this sequence either activates or represses transcription. Early approaches with reporter assays mapped regions and transcription factor binding sites that, when deleted or mutated, altered promoter function. However, these lesions, in general, fail to produce the same effects when incorporated into recombinant viral genomes and tested under more physiologic assay conditions (Stinski and Isomura, 2008). We obtained similar results here, where the SFFV-modified promoters showed similar (Fig. 2E) or enhanced (Fig. 2D) activity compared to the MIEP in reporter assays but failed to support virus propagation (Fig. 2C), and while active in reporter constructs (Fig. 8) were silenced in the viral genome (Fig. 6) in THP-1 cells. Thus, the only appropriate test for physiologically relevant promoter function is in the context of a recombinant virus.
Therefore, we addressed the question of how the balance between sites for positively- and negatively-acting transcription factors modulates MIEP repression during the establishment of experimental HCMV latency by testing chimeric promoters in the context of the viral genome during infection of THP-1 cells. The recombinant virus we created (BAD-662) harbors a hybrid MIEP retaining native features critical for promoter activity (Isomura et al., 2005; Isomura et al., 2008; Isomura, Tsurumi, and Stinski, 2004) but missing numerous binding sites for transcriptional repressors (Bain, Mendelson, and Sinclair, 2003; Huang et al., 1996; Liu et al., 1994). Furthermore, binding sites for positively acting transcriptional activators in hematopoietic cells (SP1, ETS, GATA, CBF, E-box, etc.) have been inserted in the form of the SFFV promoter (Baum et al., 1997; Wahlers et al., 2002; Zychlinski et al., 2008). Despite this substantial change in the balance between sequences able to bind transcriptional activators or repressors, this hybrid promoter in the context of the HCMV genome was still repressed upon infection of THP-1 cells and the establishment of experimental latency.
Neither inserting complete, heterologous viral promoters at different locations within the HCMV genome (Qin, Penkert, and Kalejta, 2013), nor altering the balance between the binding sites for activating and repressing transcription factors at the MIEP, at least in the proportion we have here, is sufficient to avoid transcriptional silencing during the establishment of latency. Such silencing occurs in part through a cellular intrinsic defense mediated by Daxx and HDACs (Saffert and Kalejta, 2007; Saffert, Penkert, and Kalejta, 2010), and here we have confirmed HDACs contribute to repression of the SFFV-hybrid MIEP during the establishment of experimental latency in THP-1 cells. We therefore hypothesize that initial silencing during the establishment of latency occurs through a broad, likely promoter element-independent mechanism instituted by cellular intrinsic immune factors and sensors of foreign DNA. While specific promoter elements may help perpetuate repression of the MIEP during latency, they do not appear to play dominant roles during latency establishment. Identifying components of intrinsic, restrictive defenses that transcriptionally silence infecting viral genomes (in addition to Daxx and HDACs) will be required before more detailed mechanisms can be delineated.
Interestingly, all of our non-viable recombinant genomes were capable of expressing IE1 and IE2 when transfected into fully permissive fibroblasts (Fig. 3). Though quantitative comparisons of such experiments are difficult, we speculate that the level of IE gene expression was reduced compared to wild type. However, sufficient levels of IE proteins were generated to activate the expression of at least one early gene, UL44 (Fig. 3). Thus these recombinant genomes, though non-viable, were able to progress past the IE stage and into the early stage of HCMV infection. Why the gene expression achieved with these non-viable recombinants was sufficient to progress into, but not complete the early stage of infection (Fig. 4) is unclear. Recently, it was demonstrated that the rate of IE protein accumulation, independent of absolute levels, provides a replication advantage for HCMV (Teng et al., 2012). Accelerated accumulation of IE1 and IE2 likely establishes replication competence prior to the activation of innate immunity, while maintaining reasonably low levels of these proteins likely mitigates their toxicity. Thus, perhaps the SFFV sequences in our mutant viruses can produce the required amounts of IE proteins to activate early gene expression, but can not produce them fast enough to allow the early phase of the viral life cycle sufficient time to establish itself prior to inhibition by cellular innate immune proteins. Thus, while specific promoter elements appear to be unimportant for silencing during the establishment of latency, they seem to be critical for orchestrating the precise pattern of IE gene expression required for de novo lytic infection, and perhaps the animation of viral gene expression that occurs during reactivation from latency.
Highlights.
Myeloid active promoter elements in context of HCMV genome repressed by THP-1 cells
Cellular intrinsic defense dominates virus silencing when HCMV establishes latency
Balance of MIEP activators and repressors may control maintenance or reactivation
Acknowledgments
We thank Phil Balandyk for expert technical assistance, Rhiannon Penkert for crucial experimental advice and helpful comments on the manuscript, Yuanan Lu (University of Hawaii) for providing plasmid pSR-CMV-MLV-SIN, and Jeff Meier (University of Iowa) for the -582/-1108 virus. This work was supported by a grant from the NIH (AI074984) to RFK who is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albright ER, Kalejta RF. Myeloblastc Cell Lines Mimic Some but Not All Aspects of Human Cytomegalovirus Experimental Latency Defined in Primary CD34+ Cell Populations. J Virol. 2013;87(17):9802–12. doi: 10.1128/JVI.01436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain M, Mendelson M, Sinclair J. Ets-2 Repressor Factor (ERF) mediates repression of the human cytomegalovirus major immediate-early promoter in undifferentiated non-permissive cells. J Gen Virol. 2003;84(Pt 1):41–9. doi: 10.1099/vir.0.18633-0. [DOI] [PubMed] [Google Scholar]
- Baum C, Hegewisch-Becker S, Eckert HG, Stocking C, Ostertag W. Novel retroviral vectors for efficient expression of the multidrug resistance (mdr-1) gene in early hematopoietic cells. J Virol. 1995;69(12):7541–7. doi: 10.1128/jvi.69.12.7541-7547.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum C, Itoh K, Meyer J, Laker C, Ito Y, Ostertag W. The potent enhancer activity of the polycythemic strain of spleen focus-forming virus in hematopoietic cells is governed by a binding site for Sp1 in the upstream control region and by a unique enhancer core motif, creating an exclusive target for PEBP/CBF. J Virol. 1997;71(9):6323–31. doi: 10.1128/jvi.71.9.6323-6331.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell Jt, Bixby D, Savona MR, Collins KL. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16(4):446–51. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum F, Caviness K, Zagallo P. Human cytomegalovirus persistence. Cell Microbiol. 2012;14(5):644–55. doi: 10.1111/j.1462-5822.2012.01774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutsch R, Kandemir JD, Pietsch D, Cappello C, Meyer J, Simanowski K, Huber R, Brand K. CCAAT/enhancer-binding protein beta inhibits proliferation in monocytic cells by affecting the retinoblastoma protein/E2F/cyclin E pathway but is not directly required for macrophage morphology. J Biol Chem. 2011;286(26):22716–29. doi: 10.1074/jbc.M110.152538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TH, Oka T, Asai T, Okada T, Merrills BW, Gertson PN, Whitson RH, Itakura K. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 1996;24(9):1695–701. doi: 10.1093/nar/24.9.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H, Stinski MF, Kudoh A, Daikoku T, Shirata N, Tsurumi T. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J Virol. 2005;79(15):9597–607. doi: 10.1128/JVI.79.15.9597-9607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H, Stinski MF, Kudoh A, Nakayama S, Murata T, Sato Y, Iwahori S, Tsurumi T. A cis element between the TATA Box and the transcription start site of the major immediate-early promoter of human cytomegalovirus determines efficiency of viral replication. J Virol. 2008;82(2):849–58. doi: 10.1128/JVI.01593-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H, Tsurumi T, Stinski MF. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J Virol. 2004;78(23):12788–99. doi: 10.1128/JVI.78.23.12788-12799.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacks S, Greenberg B. Complementary specificity of restriction endonucleases of Diplococcus pneumoniae with respect to DNA methylation. J Mol Biol. 1977;114(1):153–68. doi: 10.1016/0022-2836(77)90289-3. [DOI] [PubMed] [Google Scholar]
- Liu R, Baillie J, Sissons JG, Sinclair JH. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 1994;22(13):2453–9. doi: 10.1093/nar/22.13.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JL, Pruessner JA. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J Virol. 2000;74(4):1602–13. doi: 10.1128/jvi.74.4.1602-1613.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JL, Stinski MF. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J Virol. 1997;71(2):1246–55. doi: 10.1128/jvi.71.2.1246-1255.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JL, Stinski MF. Major Immediate-early Enhancer and its Gene Products. In: Reddehase MJ, editor. “Cytomegalovirus”. Caister Academic Press; Norfolk, UK: 2006. pp. 151–166. [Google Scholar]
- Penkert RR, Kalejta RF. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae. 2011;2(1):3. doi: 10.1186/2042-4280-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert RR, Kalejta RF. Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 2012;7(9):855–869. doi: 10.2217/fvl.12.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert RR, Kalejta RF. Human embryonic stem cell lines model experimental human cytomegalovirus latency. MBio. 2013;4(3) doi: 10.1128/mBio.00298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Penkert RR, Kalejta RF. Heterologous Viral Promoters Incorporated into the Human Cytomegalovirus Genome Are Silenced during Experimental Latency. J Virol. 2013;87(17):9886–94. doi: 10.1128/JVI.01726-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB. Chromatin-mediated regulation of cytomegalovirus gene expression. Virus Res. 2011;157(2):134–43. doi: 10.1016/j.virusres.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert RT, Kalejta RF. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol. 2006;80(8):3863–71. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert RT, Kalejta RF. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol. 2007;81(17):9109–20. doi: 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert RT, Penkert RR, Kalejta RF. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol. 2010;84(11):5594–604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta. 2010;1799(3-4):286–95. doi: 10.1016/j.bbagrm.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Sinzger C, Digel M, Jahn G. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol. 2008;325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- Stinski MF, Isomura H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med Microbiol Immunol. 2008;197(2):223–31. doi: 10.1007/s00430-007-0069-7. [DOI] [PubMed] [Google Scholar]
- Tavalai N, Stamminger T. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 2011;157(2):128–33. doi: 10.1016/j.virusres.2010.10.002. [DOI] [PubMed] [Google Scholar]
- Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell. 2012;151(7):1569–80. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol. 2007;81(7):3109–23. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer BK, Smith GA, Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol. 2010;634:421–30. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- Wahlers A, Zipfel PF, Schwieger M, Ostertag W, Baum C. In vivo analysis of retroviral enhancer mutations in hematopoietic cells: SP1/EGR1 and ETS/GATA motifs contribute to long terminal repeat specificity. J Virol. 2002;76(1):303–12. doi: 10.1128/JVI.76.1.303-312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem. 2006;281(49):37652–60. doi: 10.1074/jbc.M604273200. [DOI] [PubMed] [Google Scholar]
- Wu C, Lu Y. High-titre retroviral vector system for efficient gene delivery into human and mouse cells of haematopoietic and lymphocytic lineages. J Gen Virol. 2010;91(Pt 8):1909–18. doi: 10.1099/vir.0.020255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee LF, Lin PL, Stinski MF. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytc THP-1 cells. Virology. 2007;363(1):174–88. doi: 10.1016/j.virol.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E, Mishra A, Baum C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16(4):718–25. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]