

Abstract
he Asian citrus psyllid Diaphorina citri is a notorious agricultural pest that transmits the phloem-inhabiting alphaproteobacterial ‘Candidatus Liberibacter asiaticus’ and allied plant pathogens, which cause the devastating citrus disease called Huanglongbing or greening disease. D. citri harbors two distinct bacterial mutualists in the symbiotic organ called bacteriome: the betaproteobacterium ‘Candidatus Profftella armatura’ in the syncytial cytoplasm at the center of the bacteriome, and the gammaproteobacterium ‘Candidatus Carsonella ruddii’ in uninucleate bacteriocytes. Here we report that a putative amino acid transporter LysE of Profftella forms a highly supported clade with proteins of L. asiaticus, L. americanus, and L. solanacearum. L. crescens, the most basal Liberibacter lineage currently known, lacked the corresponding gene. The Profftella-Liberibacter subclade of LysE formed a clade with proteins from betaproteobacteria of the order Burkholderiales, to which Profftella belongs. This phylogenetic pattern favors the hypothesis that the Liberibacter lineage acquired the gene from the Profftella lineage via horizontal gene transfer (HGT) after L. crescens diverged from other Liberibacter lineages. K A/K S analyses further supported the hypothesis that the genes encoded in the Liberibacter genomes are functional. These findings highlight the possible evolutionary importance of HGT between plant pathogens and their insect vector’s symbionts that are confined in the symbiotic organ and seemingly sequestered from external microbial populations.
Introduction
The Asian citrus psyllid Diaphorina citri (Hemiptera: Psyllidae) is an important agricultural pest that transmits a serious citrus disease, Huanglongbing (HLB) or greening disease. This insect is widely distributed in Asia, and is spreading into other citrus growing regions worldwide [1]. The causative agents of HLB are considered to be three species of a fastidious phloem-inhabiting alphaproteobacterial lineage of the genus Candidatus Liberibacter: L. asiaticus, L. americanus, and L. africanus [2], [3]. D. citri vectors L. asiaticus and L. americanus in Asia and the Americas, and the African citrus psyllid Trioza erytreae (Hemiptera: Triozidae) vectors L. africanus in Africa [1], [2], [3], [4]. Similar diseases have been found in potatoes, tomatoes and other solanaceous crops infected with L. solanacearum (also known as L. psyllaurous) [5]. These Liberibacter species are very fastidious, but L. crescens, the species recovered from mountain papaya, has recently been reported to be readily culturable [6]. Complete genome sequences have been determined for L. asiaticus [7], L. solanacearum [5], and L. crescens [6], whereas draft genome sequence is available for L. americanus [8].
In its abdomen, D. citri possesses a large yellow symbiotic organ called the bacteriome, where two distinct symbionts are harbored [9]. The betaproteobacterium ‘Candidatus Profftella armatura’ is located in the syncytial cytoplasm at the center of the bacteriome, whilst the gammaproteobacterium ‘Candidatus Carsonella ruddii’ is found in uninucleate bacteriocytes on the surface of the bacteriome. Our previous study revealed that Profftella is a toxin-producing defensive symbiont that potentially protects D. citri from natural enemies, while Carsonella_DC is a nutritional symbiont that provides the host with essential amino acids, which are scarce in the psyllid’s diet of phloem sap [10].
Here we report that the Liberibacter lineage horizontally acquired a putative transporter gene from a bacterium closely related to the extant Profftella.
Materials and Methods
HGT candidates in the Profftella genome were extracted by BLASTP searches [11] against NCBI nr database, using deduced amino acid sequences of all protein coding genes on the Profftella genome as queries. Amino acid sequences were aligned using MAFFT 6.847 [12], followed by manual refinement. Amino acid sites corresponding to alignment gap(s) were omitted from the data set. The best fitting amino acid substitution model for the alignment was estimated using ProtTest3 [13]. For the present analysis, ProtTest selected LG with a gamma distribution (+G), a proportion of invariable sites (+I) and empirical base frequencies (+F) as the best fitting substitution model, followed by WAG with the options +I +G +F. Phylogenetic trees were inferred by the Maximum Likelihood (ML) [14] and the Bayesian Inference (BI) [15] methods. ML trees were constructed using RAxML7.2.1 [16] with LG + G + I + F model. The support values for the internal nodes were inferred by 1,000 bootstrap replicates. In the BI, we used the program MrBayes 3.1.2 [15]. Since the LG model is not implemented in MrBayes, WAG as the next best available model was used with the options +I +G +F. In total, 18,000 trees were obtained (Nruns = 2, Ngen = 900000, Samplefreq = 100), and the first 2,000 of each run were considered as the “burn in” and discarded. The posterior probability of each node was used as the support value of the node. We checked that the potential scale reduction factor was approximately 1.00 for all parameters and that the average standard deviation of split frequencies converged towards zero.
K S and K A values were calculated as described previously [17]. Statistical significance of the obtained K A/K S value was tested against a bootstrap distribution of K A/K S values, which was generated by 10,000 bootstrap resamplings of codons from the original alignment. When K S values calculated from resampled alignments were close to saturation values (larger than 2.0 per site), the K S values was set as 2.0 for the estimation of K A/K S value.
To analyze the structural organization, the genomic sequences of L. asiaticus str. psy62 [accession no. NC_012985], L. asiaticus str. gxpsy [NC_020549], L. solanacearum CLso-ZC1 [NC_014774], L. americanus PW_SP [AOFG01000001-22], and L. crescens BT-1 [NC_019907] were obtained from GenBank.
Results
BLASTP searches against the NCBI nr database demonstrated that the putative LysE protein (accession no: YP_008343788) of Profftella is significantly similar to its counterparts of Liberibacter spp. The top BLAST hit was the “putative homoserine/homoserine lactone efflux protein” of L. asiaticus str. psy62 and str. gxpsy (accession nos: YP_003065395 and YP_007599438, respectively. E = 2e-47 for the both strains). Subordinate hits were “putative homoserine/homoserine lactone efflux protein” of L. americanus (WP_007557425, E = 2e-43) and L. solanacearum (YP_004063007, E = 3e-42), followed by LysE superfamily proteins, such as “lysine transporter LysE”, “threonine transporter RhtB”, and “homoserine/homoserine lactone efflux protein”, of various betaproteobacterial species belonging to the order Burkholderiales. Putative orthologs of the Profftella lysE were observed in L. asiaticus, L. americanus, and L. solanacearum, but not in L. crescens, the most basal Liberibacter lineage currently known [6]. The LysE of Profftella was 40–43% identical to its orthologs of Liberibacter spp (Fig. 1). No other HGT candidates were found between Profftella and Liberibacter spp.
Figure 1. Alignment of amino acid sequences of LysEs.
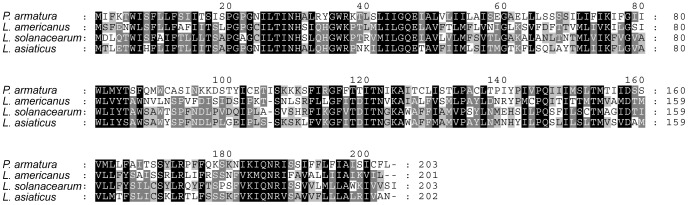
Residues conserved in all lineages, three lineages, and two lineages are shaded black, dark gray, and light gray, respectively.
Molecular phylogenetic analysis demonstrated that the LysE of Profftella forms a highly supported clade with the proteins of Liberibacter spp. (Fig. 2). The Profftella-Liberibacter subclade was placed within a clade that largely consisted of the LysE sequences of betaproteobacteria and gammaproteobacteria that are paraphyletic to Betaproteobacteria [18]. Moreover, this subclade formed a clade with proteins from betaproteobacteria of the order Burkholderiales, to which Profftella belongs [19]. This phylogenetic pattern, together with the presence/absence of the orthologous genes in Liberibacter spp., is most simply explained by the hypothesis that the Liberibacter lineage acquired the transporter gene from the Profftella lineage via horizontal gene transfer (HGT) after L. crescens diverged from other Liberibacter lineages. The structural organizations of the lysE flanking regions were partially conserved among genomes of L. asiaticus, L. americanus and L. solanacearum (Fig. 3), which were all assembled de novo without reference to one another [5], [6], [7], [8], further supporting a single acquisition of this gene in the Liberibacter lineage.
Figure 2. Phylogenetic position of Profftella LysE in related transporter proteins.

A total of 185 unambiguously aligned amino acid sites were subjected to the analysis. A maximum likelihood phylogeny is shown, whereas a Bayesian analysis inferred essentially the same result. On each node, support values of maximum-likelihood analysis/Bayesian posterior probabilities are shown. Scale bar indicates substitutions per site. Source organisms are shown with higher bacterial taxa in brackets. α, β, and γ indicate classes of the Proteobacteria, respectively. Accession numbers of proteins are shown in parentheses. The Profftella-Liberibacter cluster is highlighted in red.
Figure 3. Gene order of lysE flanking regions in the Liberibacter genomes.
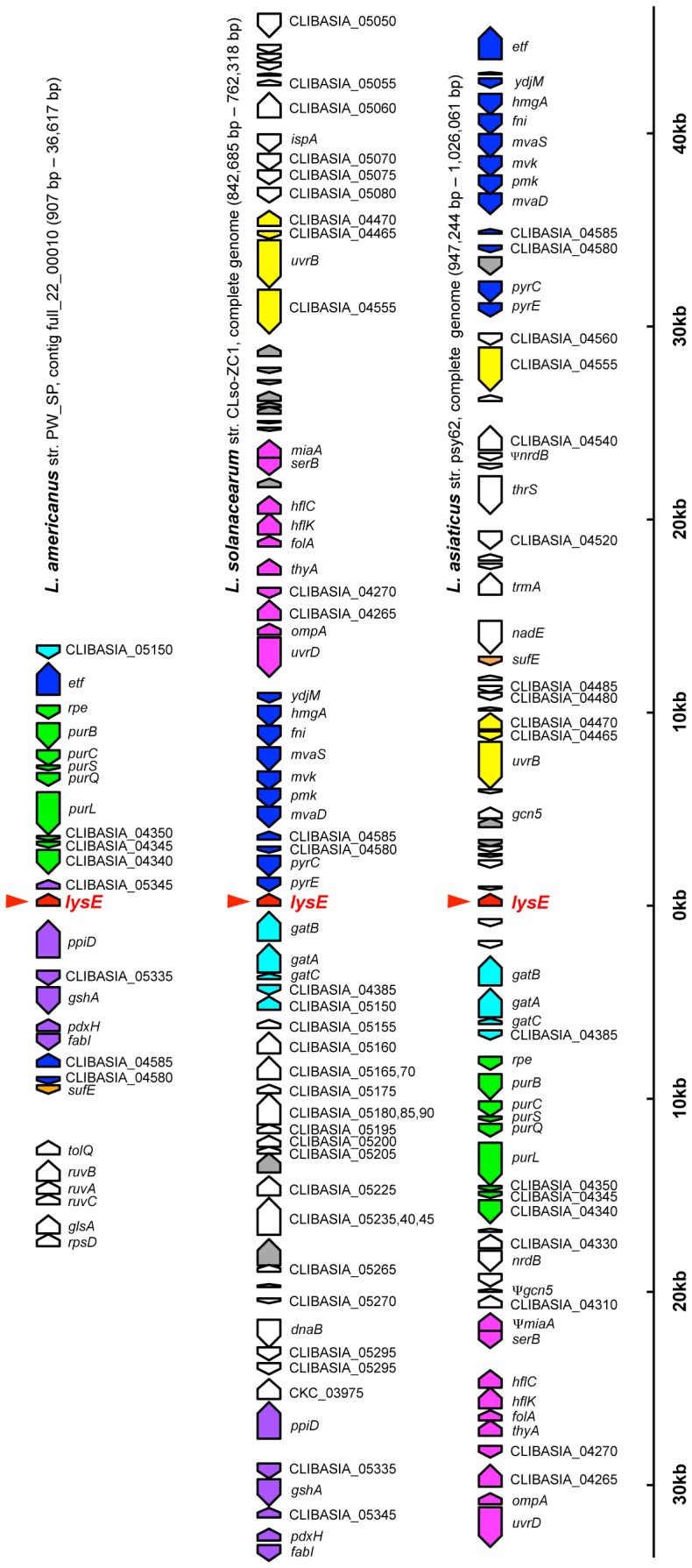
Pentagons indicate genes with coding directions. Colored pentagons are conserved among the three Liberibacter genomes. Genes with the same color indicate gene clusters. lysE-type protein genes are indicated by arrowheads. Gray pentagons are phage related genes. For L. asiaticus, structural organization of the str. psy 62 genome is shown, whereas that of str. gxpsy was essentially the same.
The K A/K S ratio between lysE genes of L. asiaticus and L. solanacearum was significantly lower than 1 (K A = 0.24, K S = 1.61, K A/K S = 0.15, p < 0.0001). Whereas the K S values both between L. asiaticus and L. americanus and between L. solanacearum and L. americanus were saturated (> 3.00), the K A values were still as low as 0.42 and 0.39, respectively. These results support the hypothesis that the lysE genes of Liberibacter spp. are under purifying selection and thus are functional.
Discussion
The present study demonstrated that the Liberibacter lineage horizontally acquired a lysE-type transporter gene from the Profftella lineage, an endosymbiont of their vector insect. K A/K S analyses further supported the hypothesis that the genes encoded in the Liberibacter genomes are functional. Although their true functions are yet to be identified, LysE superfamily proteins of various bacteria are generally involved in exporting substrates, playing important roles in resistance to toxic substances, in maintenance of optimum intracellular concentration of metabolites, and in excretion of regulatory molecules [20], [21]. Thus, it is probable that Liberibacter have acquired novel functions through this HGT. Whereas HGTs are rampant among bacteria [22], [23], such transfers of genes are rare in intracellular bacteria that are harbored in insects’ symbiotic organ and are seemingly sequestered from external microbial populations [24], [25], [26]. Apparently, Profftella, the putative donor lineage of the lysE gene, is this type of endosymbiont. In this context, infection style of Liberibacter, the putative accepter of the gene, would be noticeable. As Liberibacter spp. are transmitted by psyllids in a persistent manner, exhibiting near systemic infection of various organs and tissues [27], they may also intrude into the bacteriome of the vector psyllids, having opportunity of HGT with endosymbionts therein. The present findings highlight the previously unrecognized possible evolutionary importance of HGT between plant pathogens and their vector’s mutualists that are confined in symbiotic organs.
Funding Statement
This work was supported by JSPS KAKENHI grant numbers 21687020 and 24117510 to AN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Grafton-Cardwell EE, Stelinski LL, Stansly PA (2013) Biology and management of Asian citrus psyllid, vector of the huanglongbing pathogens. Annu Rev Entomol 58: 413–432. [DOI] [PubMed] [Google Scholar]
- 2. Jagoueix S, Bove JM, Garnier M (1994) The phloem-limited bacterium of greening disease of citrus is a member of the alpha subdivision of the Proteobacteria . International journal of systematic bacteriology 44: 379–386. [DOI] [PubMed] [Google Scholar]
- 3. Teixeira Ddo C, Saillard C, Eveillard S, Danet JL, da Costa PI, et al. (2005) 'Candidatus Liberibacter americanus', associated with citrus huanglongbing (greening disease) in Sao Paulo State, Brazil. Int J Syst Evol Microbiol 55: 1857–1862. [DOI] [PubMed] [Google Scholar]
- 4. Gottwald TR (2010) Current epidemiological understanding of citrus Huanglongbing. Annual review of phytopathology 48: 119–139. [DOI] [PubMed] [Google Scholar]
- 5. Lin H, Lou B, Glynn JM, Doddapaneni H, Civerolo EL, et al. (2011) The complete genome sequence of ‘Candidatus Liberibacter solanacearum', the bacterium associated with potato zebra chip disease. PLoS One 6: e19135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leonard MT, Fagen JR, Davis-Richardson AG, Davis MJ, Triplett EW (2012) Complete genome sequence of Liberibacter crescens BT-1. Stand Genomic Sci 7: 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duan Y, Zhou L, Hall DG, Li W, Doddapaneni H, et al. (2009) Complete genome sequence of citrus huanglongbing bacterium, 'Candidatus Liberibacter asiaticus' obtained through metagenomics. Mol Plant Microbe Interact 22: 1011–1020. [DOI] [PubMed] [Google Scholar]
- 8.Lin H, Coletta-Filho HD, Han CS, Lou B, Civerolo EL, et al. (2013) Draft genome sequence of "Candidatus Liberibacter americanus" bacterium associated with citrus Huanglongbing in Brazil. Genome Announc 1. [DOI] [PMC free article] [PubMed]
- 9. Subandiyah S, Nikoh N, Tsuyumu S, Somowiyarjo S, Fukatsu T (2000) Complex endosymbiotic microbiota of the citrus psyllid Diaphorina citri (Homoptera : Psylloidea). Zool Sci 17: 983–989. [Google Scholar]
- 10. Nakabachi A, Ueoka R, Oshima K, Teta R, Mangoni A, et al. (2013) Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol 23: 1478–1484. [DOI] [PubMed] [Google Scholar]
- 11. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9: 286–298. [DOI] [PubMed] [Google Scholar]
- 13. Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27: 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17: 368–376. [DOI] [PubMed] [Google Scholar]
- 15. Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- 16. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- 17. Miyata T, Yasunaga T (1980) Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J Mol Evol 16: 23–36. [DOI] [PubMed] [Google Scholar]
- 18. Williams KP, Gillespie JJ, Sobral BW, Nordberg EK, Snyder EE, et al. (2010) Phylogeny of Gammaproteobacteria. J Bacteriol 192: 2305–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCutcheon JP, Moran NA (2010) Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol 2: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aleshin VV, Zakataeva NP, Livshits VA (1999) A new family of amino-acid-efflux proteins. Trends Biochem Sci 24: 133–135. [DOI] [PubMed] [Google Scholar]
- 21. Eggeling L, Sahm H (2003) New ubiquitous translocators: amino acid export by Corynebacterium glutamicum and Escherichia coli . Arch Microbiol 180: 155–160. [DOI] [PubMed] [Google Scholar]
- 22. Ochman H, Lawrence JG, Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation. Nature 405: 299–304. [DOI] [PubMed] [Google Scholar]
- 23. Pallen MJ, Wren BW (2007) Bacterial pathogenomics. Nature 449: 835–842. [DOI] [PubMed] [Google Scholar]
- 24. Moran NA, McCutcheon JP, Nakabachi A (2008) Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190. [DOI] [PubMed] [Google Scholar]
- 25. McCutcheon JP (2010) The bacterial essence of tiny symbiont genomes. Curr Opin Microbiol 13: 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCutcheon JP, Moran NA (2012) Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26. [DOI] [PubMed] [Google Scholar]
- 27. Ammar ED, Shatters RG, Hall DG (2011) Localization of Candidatus Liberibacter asiaticus, associated with citrus Huanglongbing disease, in its psyllid vector using fluorescence in situ hybridization. J Phytopathol 159: 726–734. [Google Scholar]