

Abstract
Dyslipidaemias play a key role in determining cardiovascular risk; the discovery of statins has contributed a very effective approach. However, many patients do not achieve, at the maximal tolerated dose, the recommended goals for low-density lipoprotein-cholesterol (LDL-C), non-high-density lipoprotein-cholesterol, and apolipoprotein B (apoB). Available agents combined with statins can provide additional LDL-C reduction, and agents in development will increase therapeutic options impacting also other atherogenic lipoprotein classes. In fact, genetic insights into mechanisms underlying regulation of LDL-C levels has expanded potential targets of drug therapy and led to the development of novel agents. Among them are modulators of apoB containing lipoproteins production and proprotein convertase subtilisin/kexin type-9 inhibitors. Alternative targets such as lipoprotein(a) also require attention; however, until we have a better understanding of these issues, further LDL-C lowering in high and very high-risk patients will represent the most sound clinical approach.
Keywords: Dyslipidaemia, Pharmacology, PCSK9, Apolipoprotein B
Introduction
Dyslipidaemias play a key role in determining cardiovascular disease (CVD); the lowering of low-density lipoproteins (LDL) by statins effectively reduces cardiovascular risk as documented by the results obtained in clinical trials and in clinical practice. Experimental evidence, however, clearly suggests that other lipoprotein classes beyond LDL play important roles in determining cardiovascular risk; furthermore, the current efficacy of statin comes short of providing the benefit that could derive from an optimal reduction of LDL cholesterol (LDL-C) in high-risk and very high-risk patients. For these reasons, a number of new potential drugs are under development in this area.
Here, we review the new drugs in clinical evaluation in the field of dyslipidaemias with attention to LDL-C-lowering drugs but also to those that could potentially modulate other lipoprotein classes intimately linked to LDL such as Lp(a).
Emerging therapeutic agents for low-density lipoprotein-cholesterol lowering
New agents for LDL-C-lowering should demonstrate their efficacy principally as add-on therapy to statins, and therefore should integrate their mechanisms of action with the promotion of LDL-receptor activity induced by statins following the inhibition of HMG-CoA reductase activity. The emerging therapeutic agents could be classified in two categories: those interfering with lipoprotein synthesis such as apolipoprotein B (apoB) production or microsomal triglyceride transfer protein (MTP) inhibitors and those promoting lipoprotein catabolism such as proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors.
Interfering with lipoprotein synthesis
Hepatic production of very low-density lipoprotein (VLDL) heavily depends on two dominant proteins, namely apoB and MTP. Apolipoprotein B is an obligatory structural component of VLDL and requires progressive lipidation, mediated by the resident endoplasmic reticulum chaperone MTP, to maintain conformational integrity and folding during the process of lipoprotein assembly. Interfering with this process is therefore an attractive approach for reducing lipoprotein production and decreasing plasma LDL-C concentration.
Apolipoprotein B production inhibitors
The possibility of targeting apoB during the process of gene translation is under extensive investigation. One approach is to block mRNA translation of a gene through the use of a single-strand antisense oligonucleotide (ASO) that is complementary to the mRNA. Following hybridization to the mRNA, the ASO inhibits translation and splicing, leading to mRNA degradation by RNase.1 As ASO kinetics are characterized by a large and rapid distribution to the liver, this approach is quite attractive to reduce synthesis of proteins, such as apoB, in the liver2 (Figure 1).
Figure 1.
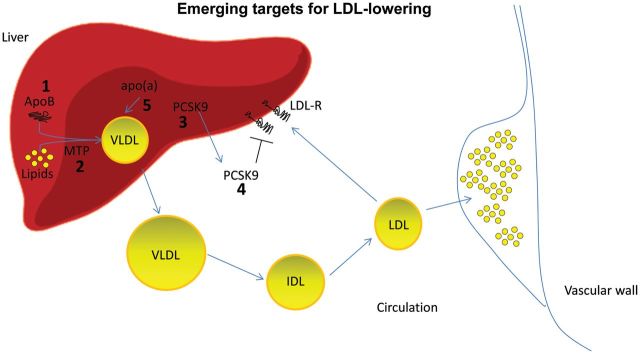
Emerging targets for dyslipidaemia. The novel drugs that are under development for the treatment of dyslipidaemia present several mechanisms of action. Emerging therapeutic agents for low-density lipoprotein-cholesterol lowering will: (a) interfere with lipoprotein synthesis in the liver by silencing apolipoprotein B expression (1) or inhibiting microsomal triglyceride transfer protein (MTP) activity (2); (b) promote low-density lipoprotein-receptor activity by silencing (3) or blocking (4) proprotein convertase subtilisin/kexin type-9. Specific silencing of apolipoprotein(a) is also under investigation (5).
ASOs targeting apoB are quite effective in mice in reducing apoB mRNA liver levels in a dose–response manner3 followed by a reduction of LDL-C, LDL particle number, circulating TG, and lipoprotein(a) [Lp(a)], while chylomicrons, which contain apoB-48, were spared, probably a consequence of the ASOs preferential uptake by the liver.
Mipomersen is an ASO targeting apoB which leads to a dose-dependent reduction in apoB and total cholesterol4; the results from clinical trials are summarized in Table 1.
Table 1.
Effect of apolipoprotein B silencing by mipomersen therapy (200 mg/weekly) on low-density lipoprotein-cholesterol levels (for details see Visser et al.4)
| Patient population | Trial phase | Conventional lipid-lowering therapy | Duration of treatment (weeks) | Baseline LDL-C levels (mg/dL) | Change in LDL-C, % |
|---|---|---|---|---|---|
| Heterozygous FH | II | Y | 6 | 207 | −21 |
| Heterozygous FH | II | Y | 13 | 155 | −22 |
| High-CV risk statin-intolerant patients | II | N | 26 | 244 | −47 |
| LDL-C ≥130 mg/dL | II | N | 13 | 171 | −45 |
| Homozygous FH | III | Y | 26 | 439 | −25 |
| Heterozygous FH (with CAD) | III | Y | 26 | 153 | −28 |
| Severe hypercholesterolaemia | III | Y | 26 | 276 | −36 |
| High CV risk | III | Y | 26 | 123 | −37 |
FH, familial hypercholesterolaemia; CAD, coronary artery disease; CV, cardiovascular.
As monotherapy, mipomersen was tested in individuals with mild-to-moderate hyperlipidaemia,5 familial hypercholesterolaemia (FH),6 and in statin-intolerant patients.7 Mipomersen was studied (13 weeks) at various dosages in patients (n = 18) with mildly to moderately elevated LDL-C (130 mg/dL) who had not taken any lipid-lowering therapy in the past month. LDL-C reductions were greater with increasing dosages (from –7 to –71%, from 50 to 400 mg/week), but adverse effects also increased.5 The most common was injection-site reaction, which occurred in all patients treated with mipomersen. ALT elevation >3 times ULN occurred in nine patients.5 In a phase II study, 39 patients with heterozygous FH were randomized to mipomersen 50 mg/week, 100 mg/week, or 200 mg/week or placebo for 6 weeks, or to mipomersen 300 mg/week or placebo for 13 weeks. In the 6-week study, apoB and LDL-C were reduced by 23 and 21%, respectively, with mipomersen 200 mg/week and by 33 and 34%, respectively, with mipomersen 300 mg/week.6 Lipoprotein(a) and triglyceride changes were not significantly different between treatment groups. The most common adverse effect with mipomersen was a transient injection-site erythema (97%), which in 19–33% of cases was accompanied by pain, pruritus, and discolouration at the injection site. In four mipomersen-treated patients, (three receiving 300 mg/week), transaminase elevations were >3 times ULN and hepatic steatosis was demonstrated on computed tomography.6 Mipomersen 200 mg/week was also compared with placebo as monotherapy in 33 high-risk statin-intolerant patients with baseline LDL-C at least 130 mg/dL (mean 244 mg/dL).7 After 26 weeks of treatment, LDL-C was reduced by 47%. Common adverse effect in patients receiving mipomersen were injection-site reactions (95%), ALT elevations >3 times ULN (33%).7
In a key study, 51 patients with homozygous FH, taking maximal lipid-lowering therapy, were randomized to mipomersen 200 mg/week or placebo for 26 weeks. LDL-C decreased 25% from 440 mg/dL.8 Significant reductions in apoB (24%), non-HDL-C (21%), and Lp(a) (23%) were also observed. Injection-site reactions occurred in 76% of mipomersen and 24% of placebo patients and was accompanied with haematoma, pain, or pruritus in 30% of the patients on mipomersen. ALT elevations ≥3 times ULN occurred in 12% of mipomersen patients, including one who had significantly increased hepatic fat on magnetic resonance imaging.8
The efficacy of mipomersen also in combination with statin therapy in individuals with LDL-C 100–220 mg/dL on a maximal tolerated statin dose with or without ezetimibe, bile-acid sequestrant, and/or niacin and in FH patients was also confirmed in phase II and phase III studies.4 Overall, mipomersen provided significant further reduction in LDL-C (∼30%; range −21 to −37%) and other lipids when added to conventional lipid therapy. Injection-site reactions and flu-like symptoms were the most common adverse effects. Liver fat accumulation was also observed, as expected from the mechanism of the action of the drug. Additional studies are needed to provide more information on the incidence of adverse effects, especially on fat accumulation and potential inflammatory response(s) in the liver, as well as compliance. The ongoing Evaluating the saFety and atherOgeniC lipoprotein redUction of mipomerSen in FH (FOCUS FH) study will randomize approximately 480 patients with severe heterozygous FH to treatment with mipomersen once or thrice weekly or to placebo for 6 weeks, with change in LDL-C as the primary endpoint (www.clinicaltrials.gov/ct2/show/NCT01475825). To date, no patients treated with mipomersen had concomitant bilirubin and AST increase consistent with drug-induced liver injury. Whether the liver steatosis will further develop into fibrotic changes is unknown, but so far no such change has been reported.
In January 2013, mipomersen has been approved by the FDA for use in homozygous FH with a warning for the possible clinical consequences of the liver fat accumulation (http://ir.isispharm.com/phoenix.zhtml?c=222170&p=irol-news&nyo=0); however, the EMA Committee for Medicinal Products for Human Use (CHMP) issued a negative opinion on mipomersen based on safety issues and further assessment is awaited.
Microsomal triglyceride transfer protein inhibitors
Microsomal triglyceride transfer protein, found in the endoplasmic reticulum of hepatocytes and enterocytes, mediates the formation of apoB-containing lipoproteins in the liver and in the intestine.9 Mutations in the MTP gene can cause abetalipoproteinaemia, a rare genetic disease characterized by the absence of apoB-containing lipoproteins, severe malabsorption of fat and fat-soluble vitamins associated during the first few months of life with failure to thrive, diarrhoea, acanthocytosis, steatorrhoea, and in most cases steatohetapatosis.9 The genetic defect underlying abetalipoproteinaemia suggests that inhibiting MTP may reduce circulating concentrations of cholesterol and apoB-containing lipoproteins (Figure 1).
The MTP inhibitor lomitapide has completed phase III testing. The drug, in monotherapy or in combination with conventional lipid-lowering therapy in homozygous FH10 or in patients with hypercholesterolaemia (LDL-C 130–250 mg/dL),11 reduced LDL-C, apoB, non-HDL-C, and Lp(a) levels. In an earlier open-label dose escalation study in six patients with homozygous FH, patients were placed on a low-fat diet (10% of calories from fat) to avoid potential steatorrhoea caused by fat malabsorption with MTP inhibition and received lomitapide at dosages of 0.03, 0.1, 0.3, and 1.0 mg/kg/day for 4 weeks each.10 LDL-C was reduced from a baseline of 614 mg/dL by 25% with lomitapide 0.3 mg/kg/day to 51% with lomitapide 1.0 mg/kg/day. At the highest dosage, apoB levels were reduced by 56% and triglyceride level by 65%, respectively. The most serious adverse events were levated aminotransferase levels, which were dosage-dependent and occurred in four of the six patients, and accumulation of hepatic fat, which occurred in all patients and ranged from <10 to <30%. Potentially drug-related adverse events were primarily gastrointestinal, especially increased stool frequency (five of six patients), which was often attributable to a high-fat meal. In a 12-week study, 84 patients with hypercholesterolaemia (LDL-C 130–250 mg/dL) were placed on a low-fat diet (<20% of calories from total fat) and randomized to three treatment groups: ezetimibe 10 mg/day and placebo for 12 weeks; lomitapide 5.0, 7.5, and 10 mg for 4 weeks at each dosage; or ezetimibe 10 mg/day for 12 weeks and lomitapide 5.0, 7.5, and 10 mg for 4 weeks at each dosage.11 LDL-C was reduced by 20% with ezetimibe monotherapy, 30% with lomitapide monotherapy, and 46% with combination therapy. Both groups receiving lomitapide also had significant reductions in total cholesterol (23–34%), non-HDL-C (27–41%), apoB (24–37%), and Lp(a) (16–17%). The most common side effects were gastrointestinal. Elevated liver transaminases led to discontinuation of study treatment in nine of the 56 patients receiving lomitapide either alone or in combination; none of the patients receiving ezetimibe alone had elevated liver enzymes.
In a recent study, lomitapide added to conventional lipid-lowering therapy in 29 patients with homozygous FH with the dose escalated from 5 to 60 mg a day, decreased LDL-C (baseline 336 mg/dL) by 40% at 26 weeks and by 44% at 56 weeks with a median dose of 40 mg/day.12
The most common adverse events were gastrointestinal. Elevations in liver transaminases (5–11 times ULN) occurred in four patients and resolved with dose reduction or temporary suspension of the drug. No discontinuation due to liver function abnormalities occurred, and no changes in bilirubin or alkaline phosphatase were observed. Liver fat content assessed by nuclear magnetic resonance spectroscopy increased from 0.9% at baseline to 9.0% at 26 weeks and 7.3% at 56 weeks.
Adverse effects such as elevated liver enzymes and hepatic fat accumulation (expected from the mechanism of action) occur and may restrict the patient population for this drug. However, for patients with homozygous FH that cannot be controlled with conventional lipid-lowering therapy, MTP inhibition may be a beneficial approach.
In December 2012, lomitapide was approved by the FDA for the treatment of patients with homozygous FH, approval from EMA is pending. To date, no cases of suspected drug-induced liver injury have been observed in lomitapide-treated subjects. As the liver lipid accumulation may vary greatly from patient to patient, this matter requires careful consideration and longer-term follow-up to definitely exclude specific changes leading to fibrosis and cirrhosis, especially in the light of the very low-fat diet that the patients were adhering to and the low fat liver content at baseline.
Promoting low-density lipoprotein-receptor activity: proprotein convertase subtilisin/kexin type-9 inhibitors
Proprotein convertase subtilisin/kexin type-9 inhibitors
Cholesterol homeostasis is regulated by the LDL receptor (LDL-R) through its binding and uptake of circulating apoB-containing lipoproteins (mainly LDL) that are then internalized into the hepatocyte. The key mechanism associated with statin-mediated LDL reduction involves the increase of LDL-R expression on the hepatocyte surface, followed by increased LDL turnover and reduction of plasma cholesterol levels. This mechanism is partially dampened by a negative feedback response associated with the induction of the expression and secretion of PCSK9 by statins,13 a serine protease which promotes the degradation of LDL-R14 thus attenuating, at least in part, the lipid-lowering efficacy of statins and ezetimibe.15 Mutations in PCSK9 that reduce its activity are associated with low LDL-C levels, and protection against coronary heart disease,16,17 while gain-of-function mutations result in an increase of LDL-C cholesterol.18 Also several single-nucleotide polymorphisms in PCSK9 are associated with increased LDL-C levels.15,19
Given that PCSK9 acts both intracellularly, as a chaperone directing the LDL-R to the lysosomes, and in the circulation, by promoting LDL-R internalization,15 the possibility of inhibiting PCSK9 represents an attractive approach to enhance the lipid-lowering effect of statins15 (Figure 1). To this end, at least five different human monoclonal antibodies and three gene-silencing approaches are under development. Among a series of antibodies against PCSK9, clinical trial results are available for two of them, SAR236553/REGN72720–23 and AMG145.24–28 A number of additional anti-PCSK9 monoclonal antibodies, in earlier clinical development, are currently being investigated for potential use in humans, including 1B20, PF-04950615/RN-316, and LGT 209.
Phase I studies conducted in individuals with hypercholesterolaemia (including heterozygous FH) showed that LDL-C was significantly reduced at all dosages and no patients discontinued study treatment because of adverse events for both antibodies.20,24 Adverse events occurred in similar proportions of patients receiving REGN727/SAR236553 intravenously and placebo, but in a higher proportion of patients receiving REGN727/SAR236553 subcutaneously compared with placebo.20 The overall incidence of treatment-emergent AEs was similar in AMG 145 subcutaneous vs. placebo.24
A number of reports have recently appeared on phase 2 trials for both antibodies on the top of maximal-tolerated doses of statins in hypercholesterolaemic patients including heterozygous familial hypercholesterolaemia,21,23,25,26 and, in some cases, of statins plus ezetimibe.22,27 PCSK9 antibodies were effective also as monotherapy in hypercholesterolaemic26 and in statin-intolerant patients.28 The dosing of the antibody by subcutaneous injection with a frequency of twice a month appears to be the optimal approach. The results indicate that a further LDL-C reduction of 50–60% range is achieved (Table 2). Assuming a benefit that is linear with the LDL reduction, a reduction of 40–50% of the relative cardiovascular risk can be expected.
Table 2.
Effect of proprotein convertase subtilisin/kexin type-9 inhibition (REGN727/SAR236553 or AMG145) on low-density lipoprotein cholesterol levels during phase II trials
| Patient population | Trial | In study lipid-lowering therapy | Doses and duration of treatment | Baseline LDL-C average level (mg/dL) | Change in LDL-C, % |
|---|---|---|---|---|---|
| Hypercholesterolaemia (LDL-C 100–190 mg/dL) | MENDELa | None | 70–480 mg s.c. every 2 or 4 weeks for 12 weeks | 143 | −41 to −51 |
| High-CV risk statin intolerant patients | GAUSSa | ±Ezetimibe | 280–480 mg s.c. every 4 weeks for 12 weeks | 193 | −41 to −63 |
| Hypercholesterolaemia (LDL-C ≥100 mg/dL) | NCT01288443b | Statins | 50–300 mg s.c. every 2 or 4 weeks for 12 weeks | ∼125 | −40 to −72 |
| Heterozygous FH | NCT01266876b | Statins ± ezetimibe | 150–300 mg s.c. every 2 or 4 weeks for 12 weeks | ∼152 | −29 to −68 |
| Heterozygous FH | RUTHERFORDa | Statins ± ezetimibe | 350–480 mg s.c. every 4 weeks for 12 weeks | 156 | −43 to −55 |
| Hypercholesterolaemia (LDL-C ≥85 mg/dL) | LAPLACE-TIMI 57a | Statins | 70–480 mg s.c. every 2 or 4 weeks for 12 weeks | 146 | −42 to −66 |
FH, familial hypercholesterolaemia; CAD, coronary artery disease; CV, cardiovascular; s.c. subcutaneously administered.
aStudy with AMG145.
bStudy with REGN727/SAR236553.
The safety results for PCSK9 monoclonal antibodies are encouraging and, generally, are well tolerated, with no drug-related adverse effects on liver function tests or other laboratory parameters, and no serious treatment-emergent adverse effects.20–22 The number of injection-site reactions (including erythema, pruritis, swelling, haematoma, and rash) were low and mild in severity. However, to date, the trials have been relatively short in duration and on relatively small patient populations. Further trials are therefore required to test the long-term safety and efficacy of PCSK9 monoclonal antibodies in larger and more varied patient populations. In this context, given that statin treatment increases PCSK9 levels, it should be considered that the frequency of injection or the dose administered may need to be increased in statin-treated patients for optimal PCSK9 inhibition. In addition, the possibility that a low-degree engagement of the immune system may occur needs to be actively investigated ; furthermore, as PCSK9 was suggested to modulate hepatitis C virus infectivity, attention must be paid in patients with viral hepatitis.29 In clinical practice, the initial use of these drugs may be limited to patients at great distance from their targets such as the patients with heterozygous familial hypercholesterolaemia or with acute coronary syndrome (ACS) and high LDL-C. Whether homozygote patients will benefit from this class of drug may depend on whether the mutations present in these subjects allow a minimal LDL-R expression. Finally, statin-intolerant patients may benefit from this therapeutic approach.
PCSK9 protein levels can also be reduced through the inhibition of gene translation taking advantage of nucleic-acid-based therapies that promote gene silencing.30 Among these, the development of SPC5001, a locked nucleic-acid-based inhibitor, and of BMS-844421, an antisense RNA therapy, were terminated during phase I clinical trials. ALN-PCS02, an RNA interference molecule, is being tested in an ongoing phase I study in healthy volunteers to evaluate safety and tolerability of various doses. In preliminary data on 20 subjects, robust target protein knockdown was observed at the highest dose tested, with a mean 60% reduction in plasma PCSK9 levels 3–5 days after administration, with a mean 39% reduction in LDL-C, with no drug-related discontinuations or liver enzyme elevations (www.clinicaltrials.gov/ct2/show/NCT01437059).
Other agents
Ezetimibe, by decreasing cholesterol absorption in the intestine, triggers a two-fold response in the liver, increases cholesterol synthesis, and increases the expression of LDL receptors.31,32 The further LDL-C reduction after statin therapy is in the range of 23–25%.32 Clinical endpoint data on the effect of ezetimibe itself with head-to-head comparison with a statin arm have long be awaited and the IMPROVE IT trial data will answer this question, although the trial is investigating an area of LDL-C where only subgroup analyses have been performed with average LDL values in the range of 60–50 mg/dL.33 Data from SEAS subgroups34 and from the SHARP trial35 are consistent with an effect related to the LDL-C lowering of the drug. The second-line therapy approach indicated by the ESC/ EAS guidelines on dyslipidaemias is consistent with this available evidence.36
Among the emerging agents in controlling hypercholesterolaemia, a dual modulator of AMP-kinase and ATP–citrate lyase, ETC-1002 therapeutically modulates the pathways of cholesterol, carbohydrate, and fatty acid metabolism and might effectively treat the cluster of interrelated risk factors associated with CVD and diabetes. This agent was tested in patients with elevated LDL-C (130–220 mg/dL) stratified by baseline TG (<150 or 150–399 mg/dL). ETC-1002 treatment was associated with an LDL-C reduction up to 27% across a broad range of baseline TG levels and was generally safe and well tolerated.37
Another class of molecules, initially described to improve HDL plasma levels, are the cholesteryl ester transfer protein (CETP) inhibitors, which were shown also to reduce VLDL and LDL plasma levels, the mechanisms by which this effect occurs is not completely understood and may not be related only to the decreased flux of cholesterol into apo B containing lipoproteins from HDL. While the development of the first two compounds was halted either for an off-target effect (torcetrapib)38,39 or for the absence of a significant reduction in cardiovascular adverse events in patients with ACS (dalcetrapib),40 two inhibitors with the highest potency towards CETP inhibition (anacetrapib and evacetrapib) are being tested in endpoint clinical trials.
Anacetrapib at 100 mg/day in patients with coronary heart disease or risk equivalent conditions (peripheral artery disease, cerebrovascular disease, diabetes, or a 10-year Framingham risk score >20%) reduced LDL-C levels by 40% and increased HDL-C levels by 138% with no changes in blood pressure or serum aldosterone levels.41,42 This study set the stage for the REVEAL phase III study which will examine major coronary events in 30 000 patients with coronary heart disease, cerebrovascular atherosclerotic diseases, or peripheral artery disease. The completion of the study is estimated for 2017, and the results will be critical to support the clinical relevance of CETP inhibition.
Evacetrapib is a benzoazepine compound selectively inhibiting CETP activity. In a phase II study, evacetrapib (30, 100, or 500 mg daily) increased HDL-C levels (+53.6 to +128.8%) and reduced LDL-C (−13.6 to −35.9%) compared with placebo. Evacetrapib 100 mg daily in combination with statins increased HDL-C levels by 80% and decreased LDL-C levels by 12% above statin monotherapy.43 The effects of evacetrapib on cardiovascular outcomes is being studied in a large phase III trial, ACCELERATE. Evacetrapib, like anacetrapib and dalcetrapib, is well tolerated with no adverse effects on blood pressure and mineralocorticoid levels.43 Ultimately, the benefits of each of these novel CETP inhibitors must be determined through prospective, randomized, clinical outcome trials. While CETP inhibitors were developed to increase HDL-C more than any therapy currently available, the benefit may still be due to the incremental lowering of LDL-C owing the mounting evidence against a causal role for HDL in causing atherosclerosis,44 and this requires careful consideration for the transfer of these drugs in the clinical practice.
Lipoprotein(a)-lowering drugs
Major advances in understanding the causal role of elevated Lp(a) in premature CVD have been achieved.45 Although the benefits of lowering Lp(a) per se are still not demonstrated, a number of clinical and experimental studies, including mendelian randomization studies, indicate that this lipoprotein is causal in CVD.46,47 Whether this occurs by proatherogenic mechanisms, by enhancing coagulation or both remains to be addressed. Compared with LDL, Lp(a) is relatively refractory to both lifestyle and drug intervention. The data on the effects of statins and fibrates on Lp(a) are limited and highly variable. Overall, statins have, however, been shown to modestly decrease elevated Lp(a) in patients with heterozygous familial hypercholesterolaemia. In the Coronary Drug Project trial, niacin was shown to reduce Lp(a) levels by up to 40% in a dose-dependent manner in addition to the other potential beneficial effects of reducing LDL-C, total cholesterol, TG, and remnant cholesterol and raising HDL-C.45 Data from the HPS2-THRIVE trial were recently released (http://www.thrivestudy.org/). The trial was aimed at investigating in more than 25 000 patients with pre-existing atherosclerotic vascular disease, who were all receiving simvastatin 40 mg daily (plus, if indicated, ezetimibe 10 mg daily), the effects of raising blood HDL cholesterol on the risk of major vascular events among patients receiving effective LDL-lowering therapy. The preliminary results indicated that the nicotinic acid/laropripant combination is not associated with beneficial effects on combined cardiovascular events on the top of maximal LDL-C lowering therapy. These results prompted the producer to withdraw the drug worldwide. Given the effects of nicotinic acid on HDL-C, this trial further supports the negative data on the role of HDL-C as causal in CVD as indicated by recent Mendelian randomization study.44 In addition, a significant increase of non-fatal serious adverse events (including intracranial and GI bleeding) was reported in the group receiving nicotinc acid/laropripant. Whether laropiprant might be responsible for these findings is unknown. Ultimately, it will also be of interest to know if benefits have occurred in specific subgroups with elevated Lp(a) levels. Future analysis will provide this much needed information. The overall results of the study are in agreement with those of the AIM-HIGH study that reported no effect for nicotinic acid on the top of statins in patients with established CVD and low HDL.48,49 A recent meta-analysis including also the AIM-HIGH trial , however, still indicates a CV benefit of nicotinic acid.50 Whether patients with low HDL and high triglycerides would benefit from this treatment, as it has been suggested for fibrates in the Accord trial,51 remains to be addressed.
Controlled intervention trials with selective reduction in plasma Lp(a) levels aimed to reduce CVD are still needed; selective Lp(a) apheresis may represent such an approach.45 Other agents reported to decrease Lp(a) to a minor degree (10%) include aspirin, l-carnitine, ascorbic acid combined with l-lysine, calcium antagonists, angiotensin-converting enzyme inhibitors, androgens, oestrogen and its replacements (e.g. tibolone), and anti-estrogens (e.g. tamoxifen), while the development of a thyroxine derivative such as eprotirome, although effective in reducing Lp(a), was halted because of long-term cartilage damage in pre-clinical studies.52
Given that a proper lipidation of apoB is a pre-requisite for Lp(a) assembly and secretion in plasma, it was not unexpected that both mipomersen and lomitapide resulted in Lp(a) reduction in the range of 40–50% the first4 and up to 17% the second.11
Finally, PCSK9 inhibition results in a reduction of Lp(a) in the range of 25–30%.20 The mechanism of this effect is unclear but may be related to the increased expression of LDL receptors that will make fewer LDL available for the interaction with apo(a) to form the lipoprotein.
More recently, early pre-clinical studies suggest that targeting liver expression of apo(a) with ASOs may provide a highly effective approach to lower elevated Lp(a) levels in humans. The development of ASOs directed to KIV-2 repeats to lower Lp(a) levels might offer the unique opportunity to address the relevance of lowering Lp(a) levels in the therapy and prevention of CVD.
It is clear that more detailed studies of the metabolism of Lp(a) are required to aid in the design and development of selective and potent therapies for lowering Lp(a).45 Given the critical role of Lp(a) synthesis in determining the plasma concentration of Lp(a), targeting either the synthesis of apo(a) and/or the formation of Lp(a) would appear worthwhile.
Conclusions
Although statins provide effective and substantial reductions in LDL-C, non-HDL-C, and apoB, many patients do not achieve the recommended goals despite maximal therapy, and some patients cannot tolerate high-dose statin therapy thus remaining at unacceptably elevated risk. Available agents combined with statins can provide additional benefit on LDL-C reduction, and agents in development may increase therapeutic options. Genetic insights into mechanisms underlying regulation of LDL-C levels have expanded potential targets of drug therapy such as PCSK9 and led to the development of novel agents.
Lp(a) may also represent an attractive target; however, with currently available intervention, it will be difficult to address, whether decreasing Lp(a) provides a reduction in cardiovascular risk. Until we have a better understanding of these issues, further LDL-C lowering in high-risk and very high-risk individuals remains the pillar for lipid-lowering intervention in decreasing cardiovascular risk.
Funding
A.L.C. received grants from Astra Zeneca, Genentech, Merck, Novartis, Roche, Sanofi-Synthelabo and C.M.B. received grants from Abbott, Amarin, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Genentech, Merck, Novartis, Roche, Sanofi-Synthelabo, Takeda, ADA, AHA, and NIH.
Conflict of interest: A.L.C. is a consultant for AstraZeneca, Bristol-Myers Squibb, Genentech, Kowa, Novartis, Pfizer, Roche, Sanofi-Synthelabo, Takeda and received honoraria from Abbott, AstraZeneca, Genentech, GlaxoSmithKline, Kowa, Merck, Novartis, Roche, Sanofi-Synthelabo, Takeda. C.M.B. is a consultant for Abbott, Amarin, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Genentech, Adnexus, Cerenis, Esperion, Idera Pharma, Kowa, Merck, Novartis, Omthera, Pfizer, Resverlogix, RocheSanofi-Synthelabo, Takeda, received lecture fee from Abbott and GlaxoSmithKline, and received honoraria from Abbott, Adnexus, Amarin, AstraZeneca, Cerenis, Esperion, Genentech, GlaxoSmithKline, Idera Pharma, Kowa, Merck, Novartis, Omthera, Resverlogix, Roche, Sanofi-Synthelabo, and Takeda.
References
- 1.Crooke ST. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 2.Yu RZ, Kim TW, Hong A, Watanabe TA, Gaus HJ, Geary RS. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35:460–468. doi: 10.1124/dmd.106.012401. [DOI] [PubMed] [Google Scholar]
- 3.Crooke RM, Graham MJ, Lemonidis KM, Whipple CP, Koo S, Perera RJ. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Visser ME, Witztum JL, Stroes ES, Kastelein JJ. Antisense oligonucleotides for the treatment of dyslipidaemia. Eur Heart J. 2012;33:1451–1458. doi: 10.1093/eurheartj/ehs084. [DOI] [PubMed] [Google Scholar]
- 5.Akdim F, Tribble DL, Flaim JD, Yu R, Su J, Geary RS, Baker BF, Fuhr R, Wedel MK, Kastelein JJ. Efficacy of apolipoprotein B synthesis inhibition in subjects with mild-to-moderate hyperlipidaemia. Eur Heart J. 2011;32:2650–2659. doi: 10.1093/eurheartj/ehr148. [DOI] [PubMed] [Google Scholar]
- 6.Akdim F, Visser ME, Tribble DL, Baker BF, Stroes ES, Yu R, Flaim JD, Su J, Stein EA, Kastelein JJ. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1413–1419. doi: 10.1016/j.amjcard.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Visser ME, Wagener G, Baker BF, Geary RS, Donovan JM, Beuers UH, Nederveen AJ, Verheij J, Trip MD, Basart DC, Kastelein JJ, Stroes ES. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2012;33:1142–1149. doi: 10.1093/eurheartj/ehs023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL, Flaim JD, Crooke ST. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 9.Calandra S, Tarugi P, Speedy HE, Dean AF, Bertolini S, Shoulders CC. Mechanisms and genetic determinants regulating sterol absorption, circulating LDL levels, and sterol elimination: implications for classification and disease risk. J Lipid Res. 2011;52:1885–1926. doi: 10.1194/jlr.R017855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuchel M, Bloedon LT, Szapary PO, Kolansky DM, Wolfe ML, Sarkis A, Millar JS, Ikewaki K, Siegelman ES, Gregg RE, Rader DJ. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–156. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 11.Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5:497–505. doi: 10.1038/ncpcardio1250. [DOI] [PubMed] [Google Scholar]
- 12.Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK, Gaudet D, Stefanutti C, Vigna GB, Du Plessis AM, Propert KJ, Sasiela WJ, Bloedon LT, Rader DJ. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6. doi: 10.1016/S0140-6736(12)61731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, Liau G, Yowe D, Harris JL. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J. 2007;406:203–207. doi: 10.1042/BJ20070664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tibolla G, Norata GD, Artali R, Meneghetti F, Catapano AL. Proprotein convertase subtilisin/kexin type 9 (PCSK9): from structure-function relation to therapeutic inhibition. Nutr Metab Cardiovasc Dis. 2011;21:835–843. doi: 10.1016/j.numecd.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 17.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 18.Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D, Bocchi L, Valenti V, Bonardi R, Guardamagna O, Averna M, Tarugi P. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007;27:677–681. doi: 10.1161/01.ATV.0000255311.26383.2f. [DOI] [PubMed] [Google Scholar]
- 19.Norata GD, Garlaschelli K, Grigore L, Raselli S, Tramontana S, Meneghetti F, Artali R, Noto D, Cefalu AB, Buccianti G, Averna M, Catapano AL. Effects of PCSK9 variants on common carotid artery intima media thickness and relation to ApoE alleles. Atherosclerosis. 2010;208:177–182. doi: 10.1016/j.atherosclerosis.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. doi: 10.1056/NEJMoa1105803. [DOI] [PubMed] [Google Scholar]
- 21.McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. doi: 10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 22.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, Wu R, Pordy R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 23.Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–1900. doi: 10.1056/NEJMoa1201832. [DOI] [PubMed] [Google Scholar]
- 24.Dias CS, Shaywitz AJ, Wasserman SM, Smith BP, Gao B, Stolman DS, Crispino CP, Smirnakis KV, Emery MG, Colbert A, Gibbs JP, Retter MW, Cooke BP, Uy ST, Matson M, Stein EA. Effects of AMG 145 on low-density lipoprotein cholesterol levels: results from 2 randomized, double-blind, placebo-controlled, ascending-dose phase 1 studies in healthy volunteers and hypercholesterolemic subjects on statins. J Am Coll Cardiol. 2012;60:1888–1898. doi: 10.1016/j.jacc.2012.08.986. [DOI] [PubMed] [Google Scholar]
- 25.Giugliano RP, Desai NR, Kohli P, Rogers WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald ST, Abrahamsen TE, Wasserman SM, Scott R, Sabatine MS. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380:2007–2017. doi: 10.1016/S0140-6736(12)61770-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koren MJ, Scott R, Kim JB, Knusel B, Liu T, Lei L, Bolognese M, Wasserman SM. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006. doi: 10.1016/S0140-6736(12)61771-1. [DOI] [PubMed] [Google Scholar]
- 27.Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the reduction of LDL-C with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–2417. doi: 10.1161/CIRCULATIONAHA.112.144055. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan D, Olsson AG, Scott R, Kim JB, Xue A, Gebski V, Wasserman SM, Stein EA. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012:1–10. doi: 10.1001/jama.2012.25790. [DOI] [PubMed] [Google Scholar]
- 29.Labonte P, Begley S, Guevin C, Asselin MC, Nassoury N, Mayer G, Prat A, Seidah NG. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology. 2009;50:17–24. doi: 10.1002/hep.22911. [DOI] [PubMed] [Google Scholar]
- 30.Norata GD, Tibolla G, Catapano AL. Gene silencing approaches for the management of dyslipidaemia. Trends Pharmacol Sc. 2013 doi: 10.1016/j.tips.2013.01.010. in press; http://dx.doi.org/10.1016/j.tips.2013.01.010 . [DOI] [PubMed] [Google Scholar]
- 31.Norata GD, Catapano AL. Lipid lowering activity of drugs affecting cholesterol absorption. Nutr Metab Cardiovasc Dis. 2004;14:42–51. doi: 10.1016/s0939-4753(04)80046-2. [DOI] [PubMed] [Google Scholar]
- 32.Grigore L, Norata GD, Catapano AL. Combination therapy in cholesterol reduction: focus on ezetimibe and statins. Vasc Health Risk Manag. 2008;4:267–278. doi: 10.2147/vhrm.s1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cannon CP, Giugliano RP, Blazing MA, Harrington RA, Peterson JL, Sisk CM, Strony J, Musliner TA, McCabe CH, Veltri E, Braunwald E, Califf RM. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimbe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J. 2008;156:826–832. doi: 10.1016/j.ahj.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 34.Holme I, Boman K, Brudi P, Egstrup K, Gohlke-Baerwolf C, Kesaniemi YA, Malbecq W, Rossebo AB, Wachtell K, Willenheimer R, Pedersen TR. Observed and predicted reduction of ischemic cardiovascular events in the Simvastatin and Ezetimibe in Aortic Stenosis trial. Am J Cardiol. 2010;105:1802–1808. doi: 10.1016/j.amjcard.2010.01.363. [DOI] [PubMed] [Google Scholar]
- 35.Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, Neal B, Jiang L, Hooi LS, Levin A, Agodoa L, Gaziano M, Kasiske B, Walker R, Massy ZA, Feldt-Rasmussen B, Krairittichai U, Ophascharoensuk V, Fellstrom B, Holdaas H, Tesar V, Wiecek A, Grobbee D, de Zeeuw D, Gronhagen-Riska C, Dasgupta T, Lewis D, Herrington W, Mafham M, Majoni W, Wallendszus K, Grimm R, Pedersen T, Tobert J, Armitage J, Baxter A, Bray C, Chen Y, Chen Z, Hill M, Knott C, Parish S, Simpson D, Sleight P, Young A, Collins R. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–2192. doi: 10.1016/S0140-6736(11)60739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Catapano AL, Reiner Z, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Perrone Filardi P, Riccardi G, Storey RF, Wood D. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Atherosclerosis. 2011;217(Suppl. 1):S1–S44. doi: 10.1016/j.atherosclerosis.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 37.Ballantyne CM, Davidson M, MacDougall D, Margulies J, DiCarlo L. ETC-1002 lowers LDL-C and beneficially modulates other cardiometabolic risk factors in hypercholesterolemic subjects with either normal or elevated triglycerides. J Am Coll Cardiol. 2012;59(13s1):E1625. [Google Scholar]
- 38.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 39.Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey AM, Ehrhart J, Hershey JC, Keller WJ, Ma X, McPherson HE, Messina E, Peterson LB, Sharif-Rodriguez W, Siegl PK, Sinclair PJ, Sparrow CP, Stevenson AS, Sun SY, Tsai C, Vargas H, Walker M, III, West SH, White V, Woltmann RF. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 41.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 42.Davidson M, Liu SX, Barter P, Brinton EA, Cannon CP, Gotto AM, Jr, Leary ET, Shah S, Stepanavage M, Mitchel Y, Dansky HM. Measurement of LDL-C after treatment with the CETP inhibitor anacetrapib. J Lipid Res. 2013;54:467–472. doi: 10.1194/jlr.M032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicholls SJ, Brewer HB, Kastelein JJ, Krueger KA, Wang MD, Shao M, Hu B, McErlean E, Nissen SE. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA. 2011;306:2099–2109. doi: 10.1001/jama.2011.1649. [DOI] [PubMed] [Google Scholar]
- 44.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, Konig IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O'Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 47.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 48.Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 49.Michos ED, Sibley CT, Baer JT, Blaha MJ, Blumenthal RS. Niacin and statin combination therapy for atherosclerosis regression and prevention of cardiovascular disease events: reconciling the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes) trial with previous surrogate endpoint trials. J Am Coll Cardiol. 2012;59:2058–2064. doi: 10.1016/j.jacc.2012.01.045. [DOI] [PubMed] [Google Scholar]
- 50.Lavigne PM, Karas RH. The current state of niacin in cardiovascular disease prevention: a systematic review and meta-regression. J Am Coll Cardiol. 2012;61:440–446. doi: 10.1016/j.jacc.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 51.Ginsberg HN, Elam MB, Lovato LC, Crouse JR, III, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr, Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–1574. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolski B, Tsimikas S. Emerging therapeutic agents to lower lipoprotein (a) levels. Curr Opin Lipidol. 2012;23:560–568. doi: 10.1097/MOL.0b013e3283598d81. [DOI] [PubMed] [Google Scholar]