

Abstract
Iron–sulfur (Fe-S) clusters are ancient enzyme cofactors found in virtually all life forms. We evaluated the physiological effects of chronic Fe-S cluster deficiency in human skeletal muscle, a tissue that relies heavily on Fe-S cluster-mediated aerobic energy metabolism. Despite greatly decreased oxidative capacity, muscle tissue from patients deficient in the Fe-S cluster scaffold protein ISCU showed a predominance of type I oxidative muscle fibers and higher capillary density, enhanced expression of transcriptional co-activator PGC-1α and increased mitochondrial fatty acid oxidation genes. These Fe-S cluster-deficient muscles showed a dramatic up-regulation of the ketogenic enzyme HMGCS2 and the secreted protein FGF21 (fibroblast growth factor 21). Enhanced muscle FGF21 expression was reflected by elevated circulating FGF21 levels in the patients, and robust FGF21 secretion could be recapitulated by respiratory chain inhibition in cultured myotubes. Our findings reveal that mitochondrial energy starvation elicits a coordinated response in Fe-S-deficient skeletal muscle that is reflected systemically by increased plasma FGF21 levels.
INTRODUCTION
Enzyme cofactor deficiencies underlie many of the world's most prevalent diseases. In recent years, Fe-S cluster deficiency has been found to underlie the pathogenesis of a rare and unique set of human disorders that includes Friedreich's ataxia (1), sideroblastic anemia (2), several severe infantile metabolic syndromes (3) and a rare mitochondrial myopathy (4,5). ‘Myopathy with deficiency of succinate dehydrogenase and aconitase’, also known as ISCU (iron–sulfur cluster scaffold homolog) myopathy, is a rare autosomal recessive Fe-S cluster deficiency syndrome found mainly in individuals of northern Swedish descent that is characterized by life-long exercise intolerance (6,7). ISCU myopathy is caused by deficiency of the Fe-S cluster scaffold protein ISCU, which works in concert with the cysteine desulfurase NFS1 as well as the proteins ISD11 and frataxin to catalyze the formation of nascent Fe-S clusters for transfer to recipient apo-proteins (8). The symptoms of ISCU myopathy appear to be restricted to skeletal muscle due in part to tissue-specific mis-splicing of ISCU mRNA (9–11), which renders Fe-S cluster-dependent enzymes in muscle unable to recover from oxidative stress because of ISCU protein deficiency (11).
Trivial bouts of exercise in ISCU myopathy patients cause dramatically increased heart rate, dyspnea, muscle fatigue and lactic acidosis (6,12). Calf hypertrophy is frequently observed in ISCU myopathy patients (6,13,14), and many individuals have experienced activity-related episodes of rhabdomyolysis and myoglobinuria (6,15). Skeletal muscle biopsies from ISCU myopathy patients have revealed biochemical hallmarks that are also found in other Fe-S cluster deficiency diseases, including deficiency of critical Fe-S cluster-containing enzymes in the mitochondrial respiratory chain, as well as insoluble mitochondrial iron deposits indicative of chronic mitochondrial iron overload (13,16). The most striking Fe-S enzyme deficiencies in ISCU myopathy are found in aconitase and mitochondrial complex II/succinate dehydrogenase (SDH), where normalized enzyme activity values were reported to be just 10–20% of healthy controls (13,14,17,18), though milder deficiencies in mitochondrial complex I and the Rieske protein of complex III were also reported (13,17).
In this study, we evaluated global changes in gene expression in ISCU myopathy patient muscle biopsies in order to explore the structural and metabolic responses to mitochondrial energy deficit caused by chronic Fe-S cluster deficiency (Table 1). Our results demonstrate that Fe-S cluster-deficient human skeletal muscle shows unique features that include some similarities to the physiological remodeling seen during endurance exercise training. Additionally, we found striking induction of starvation response pathways that were originally described in liver, including increased expression of ketogenic enzymes and the secreted hormone fibroblast growth factor 21 (FGF21).
Table 1.
Summary of patient and control subjects included in the study
| Subject | Designation | Age (at biopsy) | Sex | Nature of mutation | Biochemical features |
|---|---|---|---|---|---|
1A 
|
Control | 42 | F | – | – |
2A 
|
Control | 35 | M | – | – |
3A 
|
Control | 42 | M | – | – |
4A 
|
Control | 47 | M | – | – |
5A 
|
Control | 22 | F | – | – |
1B 
|
ISCU myopathy | 66 | F | ISCU g.7044 G>C | Fe-S enzyme deficiency, especially of aconitase and complex II |
2B 
|
ISCU myopathy | 42 | M | ISCU g.7044 G>C | Fe-S enzyme deficiency, especially of aconitase and complex II |
3B 
|
ISCU myopathy | 42 | M | ISCU g.7044 G>C | Fe-S enzyme deficiency, especially of aconitase and complex II |
4B 
|
ISCU heterozygote | 43 | M | −/ISCU g.7044 G>C | – |
1C 
|
Mitochondrial myopathy | 63 | M | mtDNA: A5543C tRNAtrp | Multiple respiratory chain deficiencies, especially complex IV |
2C 
|
Mitochondrial myopathy | 37 | F | mtDNA: cytochrome b mutation | Selective complex III deficiency |
3C 
|
Mitochondrial myopathy | 31 | M | mtDNA: T10010C tRNAgly | Multiple respiratory chain deficiencies, especially complex IV |
4C 
|
Mitochondrial myopathy | 26 | F | mtDNA: large-scale mtDNA deletion | Multiple respiratory chain deficiencies |
5C 
|
Mitochondrial myopathy | 29 | F | mtDNA: cytochrome b mutation | Selective complex III deficiency |
RESULTS
The muscle respiratory deficiencies in ISCU myopathy patients are attributed to deficiency of Fe-S cluster cofactors due to depletion of the Fe-S cluster scaffold protein ISCU (4,5), and the disease is restricted to skeletal muscle due in part to muscle-specific ISCU mRNA mis-splicing and ISCU protein instability (10,11). Northern blot hybridization of 32P-labeled antisense DNA probes with total RNA isolated from vastus lateralis biopsies from healthy controls, ISCU myopathy patients and several un-related mitochondrial myopathy patients revealed decreased expression of the normally spliced ISCU mRNA (Fig. 1A) in the ISCU myopathy patients, as well as expression of two patient-specific mRNA splice isoforms (Fig. 1A and B) that have also been described elsewhere (11). Abnormal splicing of ISCU in skeletal muscle led to deficiencies in the activity of the [4Fe-4S] cluster-containing mitochondrial and cytosolic aconitases (Fig. 1C), as well as depletion of the Fe-S cluster-containing mitochondrial SDH/complex II subunit B (SDH-B; Fig. 1D), thus illustrating some of the most prominent biochemical defects in these ISCU myopathy patients.
Figure 1.

Phenotype and global gene expression analysis of ISCU myopathy patient skeletal muscle. Northern blot analysis: ISCU-specific 32P-labeled DNA probes specific for exon 2–4 sequence (A) or ISCU intron 4 sequence (B) were hybridized with control and patient vastus lateralis biopsy RNA, demonstrating normal (I) and additional patient-specific ISCU mRNA bands containing 100 bp (II) or the entire span (III) of ISCU intron 4. (C) In-gel activity assay demonstrated that mitochondrial (m-) and cytosolic (c-) aconitase activities were decreased in ISCU myopathy patient muscle biopsies. (D) Protein levels of the Fe-S cluster-containing SDH-B subunit of mitochondrial complex II were decreased in ISCU myopathy patients. Informatics tools were used to identify cellular structures and processes that were affected in patient biopsies based on microarray gene expression analysis. (E) The DAVID bioinformatics tool identified sub-cellular structures in which organelle-specific gene clusters were significantly altered in patient biopsies. P < 0.001 for all groups shown. (F) Gene clusters representing significantly altered cellular processes were identified using Ingenuity Pathway Analysis. At least 20% of the genes in each functional group shown were significantly up- or down-regulated.
Global gene expression analysis in ISCU myopathy patient muscle biopsies
We performed microarray analysis on ISCU myopathy patient muscle biopsies to identify transcriptional modulation of pathways involved in the cellular response to Fe-S cluster deficiency. We included RNA samples from five healthy control individuals as well as three unrelated ISCU myopathy patients (Table 1). For subsequent quantitative real-time polymerase chain reaction (qRT-PCR) analyses, we also included muscle biopsy RNA from a healthy heterozygous offspring of one ISCU myopathy patient, and RNA from five additional patients affected by un-related mitochondrial myopathies (Table 1). These patients had heteroplasmic mtDNA mutations causing either selective deficiency of complex III (attributable to cytochrome b mutations in two patients, denoted as light green symbols in the figures) or deficiency of multiple respiratory chain complexes, especially complex IV (in two patients with mitochondrial tRNA mutations and one patient with a single large-scale mitochondrial DNA deletion, denoted as darker green symbols).
Analysis of the microarray data using iProXpress, a tool for exploration of large-scale expression data (19), suggested that there were alterations in oxidative phosphorylation, glycolysis, fatty acid metabolism and peroxisome proliferator-activated receptor (PPAR) signaling in ISCU myopathy patient muscles (Supplementary Material, Table S1). DAVID ontology analysis demonstrated that a large number of mitochondrial genes were up-regulated, whereas groups of cytoskeletal genes responsible for muscle contraction were down-regulated (Fig. 1E), likely reflecting the changes in the proportions of muscle fiber types (discussed below). Finally, Ingenuity Pathway Analysis also indicated remodeling of glycolysis, oxidative phosphorylation and fatty acid metabolism, the latter two of which were largely up-regulated (Fig. 1F). Subsets of the genes representing these functional pathways were selected for analysis by qRT-PCR and other methods reported below.
Up-regulation of key iron metabolism genes in ISCU-depleted biopsies and cells
ISCU depletion results in deficiency of Fe-S protein activities, altered cellular iron homeostasis and mitochondrial iron overload in cultured cells (20) and in affected ISCU myopathy patient skeletal muscle (4). Our microarray analysis suggested that expression of several iron homeostasis-related genes was altered in ISCU myopathy patient muscle biopsies (Supplementary Material, Table S2). qRT-PCR confirmed that expression of the mitochondrial iron importer mitoferrin 2 (MFRN2) mRNA was significantly increased in the ISCU myopathy patients (Supplementary Material, Fig. S1A), as was expression of FBXL5, the ubiquitin ligase responsible for targeted degradation of IRP1 and IRP2 (Supplementary Material, Fig. S1B). Moreover, two enzymes involved in sulfur metabolism, namely TST (rhodanese) and cysteine desulfurase NFS1, were also significantly increased (Supplementary Material, Fig. S1C and D), as was ALAS1, the initial and rate-limiting enzyme in heme biosynthesis (Supplementary Material, Fig. S1E).
ISCU myopathy patient muscles experience a chronic state of Fe-S cluster deficiency (15). In order to assess differences in gene expression between chronic and acute ISCU deficiency, siRNA treatment was performed on cultured human myoblasts. Short-term depletion of ISCU resulted in decreased Fe-S protein levels in three different cellular compartments, namely mitochondrial matrix (FECH, ferrochelatase), cytosol (PPAT, phosphoribosyl-pyrophosphate amidotransferase) and nucleus (NTHL1, nth endonuclease III-like 1), and also decreased cytosolic aconitase enzyme activity (Supplementary Material, Fig. S1F). Moreover, cellular iron homeostasis was disrupted by ISCU depletion, as assessed by induction of iron regulatory element (IRE)-binding activities of iron regulatory proteins IRP1 and IRP2, and increased IRP2 and transferrin receptor 1 (TFR1) protein levels (Supplementary Material, Fig. S1G). qRT-PCR analysis of gene expression in myoblasts revealed significant increases in mRNA levels of MFRN2 and ALAS1 (Supplementary Material, Fig. S1H), although of lesser magnitude than the increases seen in ISCU patient muscle biopsies. We were unable to observe reproducible changes in FBXL5, TST or NFS1 levels during ISCU depletion in the myoblasts, perhaps due to the relatively short duration of Fe-S cluster deficiency in the siRNA knockdown experiment when compared with the chronic Fe-S cluster deficiency in ISCU myopathy patient muscles.
Type I muscle fiber predominance and enhanced expression of mitochondrial oxidative metabolism genes in ISCU myopathy
Skeletal muscles consist of a mixture of different fiber types, including slow-twitch type I fibers, which are rich in myoglobin and mitochondria and are resistant to fatigue, and fast-twitch type II fibers, which rely more heavily on glycolysis to generate contractile force of moderate (type IIa) or very brief (type IIx) duration (21). The bioinformatics analyses presented above indicated that changes in expression of myofibril structural components in ISCU myopathy patient muscles occurred coordinately, which suggested that alterations in skeletal muscle fiber proportions might contribute to the ISCU myopathy disease phenotype. We found a large group of down-regulated genes that were annotated as structural components of type II fast-twitch glycolytic muscle fibers (Supplementary Material, Table S3). We also found a smaller group of up-regulated muscle-specific genes that included the cardiac myosin MYH6 (Supplementary Material, Table S3), which is reported to co-express with slow-twitch MHCI in human facial muscles (22), and in rabbits during glycolytic to oxidative skeletal muscle fiber-type transition (23). MYH7B, which was previously shown to be expressed in slow-tonic fibers of rat extraocular muscle and in muscle spindle bag fibers (24), was also significantly up-regulated (Supplementary Material, Table S3). Moreover, over 30 genes encoding components of the mitochondrial respiratory chain were increased significantly in ISCU myopathy patient muscle biopsies, but to a level below our cut-off of 1.5-fold (data not shown).
qRT-PCR analysis confirmed profound decreases in type II muscle fiber contractile motor protein myosin heavy chain 1 (MYH1; Fig. 2A) and the F-actin crosslinking protein α-3 actinin (ACTN3; Fig. 2B) in ISCU patient biopsies. The creatine biosynthetic enzyme glycine amidinotransferase (GATM) was also strongly down-regulated (Fig. 2C), whereas expression of MYH6 was increased in ISCU patient biopsies (Fig. 2D). Monocarboxylate transporters MCT1 and MCT2, which transport lactate and pyruvate as well as ketone bodies and are highly expressed in human type I oxidative fibers (25,26), were also significantly increased in ISCU patients (Fig. 2E and F). Interestingly, mitochondrial uncoupling protein UCP2, which might function to dissipate the proton gradient formed by the mitochondrial electron transport chain, was also significantly increased in ISCU myopathy and mitochondrial myopathy patient biopsies (Fig. 2G). Moreover, we observed a profound 16.8-fold average increase in quinolinate phosphoribosyltransferase (QPRT), a key enzyme in the pathway for de novo synthesis of nicotinamide adenine dinucleotide (NAD) (27) in ISCU patient muscle biopsies (Fig. 2H), possibly indicating a compensatory attempt to increase NAD+ production in patient muscles.
Figure 2.

Structural and metabolic remodeling during chronic Fe-S cluster deficiency results in a predominance of type I muscle fibers and elevated fatty acid oxidation genes. qRT-PCR demonstrated decreased expression of type II muscle fiber contractile protein MYH1 (A: average 0.25-fold), the structural protein ACTN3 (B: average 0.21-fold) and the creatine biosynthetic enzyme GATM (C: average 0.13-fold). (D) Cardiac myosin MYH6 expression was increased by an average of 3.8-fold in ISCU myopathy patient biopsies. (E and F) Expression of lactate/pyruvate transporters MCT1 and MCT2 was increased in ISCU myopathy patient biopsies. (G) Mitochondrial uncoupling protein 2 (UCP2) was increased by an average of 7.2-fold in ISCU myopathy patients and 2.7-fold in the mitochondrial myopathy patients. (H) Quinolinate phosphoribosyltransferase (QPRT) was elevated by an average of 16.8-fold in ISCU myopathy patient biopsies. (I) Liver-specific carnitine palmitoyltransferase I (CPT1A) mRNA was increased in two of the three ISCU myopathy patients evaluated. (J) The acyl-carnitine translocase SLC25A20 was significantly increased by an average of 4.5-fold in ISCU myopathy patients. (K) Carnitine palmitoyltransferase II (CPT2) was significantly increased by an average of 4.6-fold. (L) Cytosolic fatty acid-binding protein 4 (FABP4) was increased by an average of 5.5-fold in ISCU myopathy patients. The non-transformed 2−ΔΔCt values were analyzed by one-way ANOVA with Holm–Sidak post hoc test to determine statistical significance. (M) Muscle fiber composition in control and ISCU myopathy patient muscle biopsy sections was quantified histochemically by myosin ATPase staining. Type I fibers showed a predominance in patients (n = 3) compared with controls (n = 13), whereas the number of type IIa fibers was significantly lower, and the number of type IIx (IIb) fibers trended lower. Two-tailed, unpaired t-tests were used here to determine statistical significance. *P < 0.05; **P < 0.01.
Functional profiling of the microarray output data using IPA and iProXpress suggested that expression of PPAR-regulated genes in fatty acid utilization pathways increased significantly (Supplementary Material, Table S1). Several genes in this group were directly involved in cytosolic fatty acid transport (FABP3, 4 and 6), mitochondrial fatty acid uptake (CPT1A, SLC25A20 and CPT2) and mitochondrial and peroxisomal fatty acid β-oxidation (ETFB, EHHADH, ACAA2, ECH1, ACOX2, MCEE and PCCB) (Supplementary Material, Table S4). qRT-PCR verified up-regulation of the functional units of the mitochondrial carnitine palmitoyltransferase system, CPT1A, SLC25A20 and CPT2 (28) (Fig. 2I–K), as well as the cytosolic fatty acid-binding protein 4 (FABP4; Fig. 2L), which has also been found to be increased in the muscles of endurance-trained athletes (29). Our microarray analysis revealed decreased expression of several key glycolytic enzymes, including muscle-specific phosphofructokinase, phosphoglycerate kinase 1, aldolase and muscle-specific enolase (Supplementary Material, Table S4). Notably, expression of the muscle-specific lactate dehydrogenase isoform LDHA was significantly decreased by −2.1-fold, whereas the heart LDH isoform LDHB was increased by 3.4-fold (Supplementary Material, Table S4).
To assess whether the altered gene expression reported above reflected actual changes in muscle fiber composition in ISCU myopathy patients, vastus lateralis biopsy sections were examined by differential myosin ATPase histochemistry (30). This revealed an average 22% relative increase in fibers staining at low pH in ISCU myopathy patients, indicative of type I fibers, accompanied by a significantly decreased total fraction of type II fast-twitch fibers (Fig. 2M), compared with control biopsies. More specifically, type IIa fibers, which show fast shortening speed and the capacity for both aerobic and anaerobic contractile activity, were significantly decreased by 13%, whereas type IIx fast-twitch fibers (formally classified as type IIb by the ATPase method) decreased non-significantly by 9% (Fig. 2M). Together, these methods suggested that the overall fraction of fast-twitch muscle fibers in ISCU myopathy patient biopsies was decreased, whereas the proportion of type I slow-twitch fibers was increased.
Increased peroxisome proliferator-activated receptor gamma, coactivator 1 alpha protein expression and capillary abundance in ISCU myopathy patient muscles
The coordinately increased expression of fatty acid transport and oxidation genes in the ISCU myopathy patient muscles led us to analyze cellular factors that modulate transcription of these genes. Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α) is known to positively co-activate the expression of many of the fatty acid oxidation and fiber type-specific genes that we found to be up-regulated in our microarray and qPCR analysis. Although we saw no significant increase in PGC-1α mRNA expression in the patients (Supplementary Material, Fig. S2A), western blot analysis of control and patient muscle biopsy protein extracts showed that PGC-1α protein expression was increased in both vastus lateralis and gastrocnemius muscle biopsies in two ISCU myopathy patients (Fig. 3A). Quantification and normalization of PGC-1α protein levels in a separate experiment confirmed that endogenous PGC-1α expression was increased in three different ISCU patients relative to healthy controls (Supplementary Material, Fig. S2B and C).
Figure 3.

Nuclear expression of PGC-1α is increased in Fe-S cluster-deficient patient muscle fibers. (A) PGC-1α protein expression was increased in patient muscle biopsy protein extracts. Actin served as protein loading control. (B) PGC-1α (green), COX1 (red: mitochondrial complex IV, subunit I) and nuclei (DAPI) were visualized by immunofluorescence microscopy of vastus lateralis biopsy sections from two ISCU myopathy patients and two healthy controls. (C) Numbers of PGC-1α-positive nuclei were counted in non-overlapping frames. An average of 250 nuclei per sample were counted. (D) Abundant centralized PGC-1α-positive myonuclei were observed in one of the ISCU myopathy patient biopsies by immunohistochemistry. (E) Immunofluorescence microscopy demonstrated strong nuclear PGC-1α expression in a centralized myofiber nucleus. The inset frame is a magnified centralized PGC-1α-positive myonucleus in a patient.
To further characterize the potential role of PGC-1α in the transcriptional changes we observed in ISCU myopathy patient muscles, we performed immunofluorescence microscopy to ascertain the sub-cellular localization of PGC-1α in patient and control vastus lateralis biopsies (Fig. 3B). The mitochondrial DNA-encoded gene COX1 provided a qualitative indicator of mitochondrial position and morphology, and nuclei were counterstained stained with DAPI. Immunofluorescence demonstrated both cytosolic and nuclear pools of PGC-1α, a localization that we and others have also observed by western blot [data not shown; (31)]. Quantitative analysis revealed that patient biopsy sections tended to have a greater proportion of PGC-1α-positive nuclei compared with the two control biopsies (Fig. 3C). Additionally, mitochondrial COX1 staining was decreased in some patient fibers (Fig. 3C), which is consistent with previous studies demonstrating a relatively mild deficiency in cytochrome oxidase activity in ISCU myopathy patient muscles (13,14,17). Further immunohistochemical analysis of PGC-1α revealed that one ISCU myopathy patient biopsy showed appreciable numbers of centralized PGC-1α-positive nuclei (Fig. 3D, arrows), indicative of previous fiber regeneration. Immunofluorescence analysis confirmed strong nuclear PGC-1α staining in these centralized nuclei (Fig. 3E). Together, these data show that PGC-1α protein expression was increased in ISCU myopathy patient muscles, and that a greater proportion of myonuclei were PGC-1α-positive in these patients.
A recent study reported increased capillaries localized around respiration-incompetent skeletal muscle fibers in biopsies from patients with heteroplasmic mitochondrial DNA mutations (32), possibly mediated by activation of PGC-1α in these fibers due to energy deficiency (32,33). Microarray and qRT-PCR analysis of our ISCU patient muscle biopsies demonstrated significant increases in vascular endothelial growth factor B (VEGFB; Fig. 4A), the VEGF receptor KDR (Fig. 4B), hypoxia-inducible factor 2α (EPAS1/HIF-2A; Fig. 4C) and basic fibroblast growth factor (FGF2; Fig. 4D) (34). We also found increased VEGF protein levels in both vastus lateralis and gastrocnemius biopsy protein extracts from ISCU myopathy patients (Fig. 4E). This increased angiogenesis-related gene expression suggested that enhanced capillary formation might be taking place in our ISCU myopathy patients. Accordingly, we stained gastrocnemius sections from an ISCU patient and the healthy heterozygous offspring of this patient for the capillary endothelial marker protein, CD31. A clear increase in capillary abundance was observed in the patient sections (Fig. 4F), compared with the unaffected heterozygote (Fig. 4G), even though the unaffected subject's oxidative capacity in cycle exercise was 3-fold higher than that of the patient. Normalization of capillary numbers to fiber numbers revealed an average of 3.4 capillaries per fiber in the ISCU myopathy patient sections versus 2.3 capillaries per fiber in the healthy heterozygote sections. The ISCU myopathy patient muscle section also showed greater heterogeneity in muscle fiber sizes, as well as a smaller average fiber cross-section (data not shown).
Figure 4.
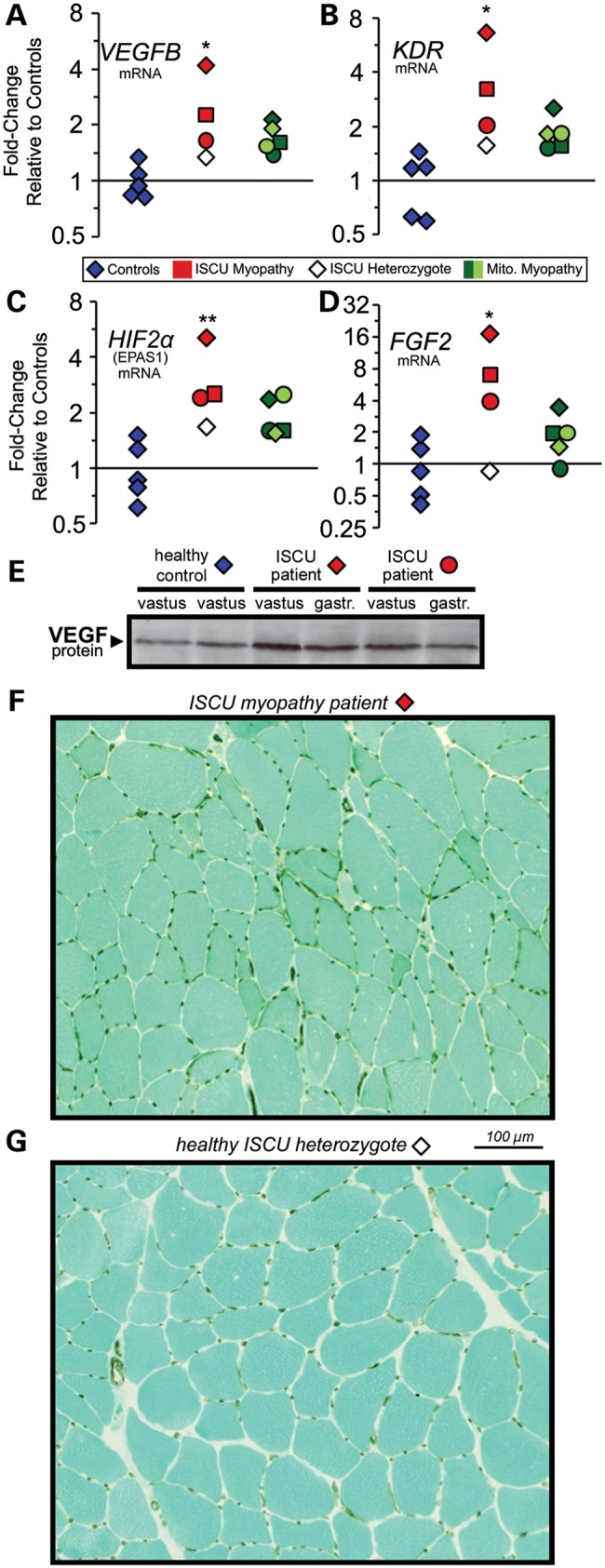
Increased angiogenesis-related gene expression and enhanced capillary abundance in ISCU myopathy patient muscles. qRT-PCR revealed significant increases in (A) VEGFB (average 2.7-fold), (B) the VEGF receptor KDR (average 4-fold), (C) hypoxia-inducible factor 2α (EPAS1/HIF-2α; average 3.3-fold) and (D) fibroblast growth factor 2 (FGF2; average 9-fold) in ISCU myopathy patient biopsies. The non-transformed 2−ΔΔCt values were analyzed by one-way ANOVA with Holm–Sidak post hoc test. *P < 0.05; **P < 0.01. (E) Immunoblot analysis of muscle biopsy protein extracts demonstrated increased VEGF protein expression in ISCU myopathy patients compared with control. Immunohistochemistry showed increased abundance of brown-colored CD31-positive capillaries perfusing myofibers in muscle sections taken from an ISCU myopathy patient (F), compared with the healthy heterozygous offspring of this individual (G).
Ketogenic enzyme expression in ISCU myopathy patient muscles
Our microarray data suggested that the rate-limiting mitochondrial enzyme involved in ketone body formation (Fig. 5A), HMGCS2, was up-regulated in ISCU myopathy patient muscles (Supplementary Material, Table S4). In fact, HMGCS2 was the most highly up-regulated gene in our microarray data set. Accordingly, qRT-PCR quantified an average 27-fold induction of HMGCS2 in the ISCU myopathy patients (Fig. 5B), as well as in one of the non-ISCU mitochondrial myopathy patients (Fig. 5B). Expression of 3-hydroxybutyrate dehydrogenase (BDH1), which interconverts the two major ketone body species 3-hydroxybutyrate and acetoacetate, was also increased by an average of 3.2-fold in ISCU myopathy patient biopsies (Fig. 5C). Further evidence of this striking increase in HMGCS2 expression was obtained by performing northern blots using patient and control mRNA samples and a 32P-labeled RNA probe specific for the HMGCS2 gene (Fig. 5D). We found that high HMGCS2 mRNA expression was restricted to the ISCU myopathy patient muscle biopsies, and was absent in patient-derived myoblasts, fibroblasts and lymphoblasts (Fig. 5E), despite varying degrees of ISCU depletion in all of these cell types (11).
Figure 5.

Induction of ketogenic enzyme expression in ISCU myopathy patient muscle biopsies. (A) Schematic diagram depicting the metabolic pathway of ketone body formation. mRNA levels of the ketogenic enzymes hydroxymethyglutaryl coenzyme-A synthase 2 (B: HMGCS2) and BDH1 (C ) were increased by an average of 27- and 3.2-fold, respectively, in ISCU myopathy patient biopsies. (D) Increased HMGCS2 expression was confirmed by northern blot using a 32P-labeled RNA probe synthesized from a fragment of the HMGCS2 gene. (E) HMGCS2 mRNA expression levels were evaluated by northern blot in skeletal muscle biopsies, primary myoblasts, fibroblasts and lymphoblasts derived from an ISCU patient and a healthy control. (F) Western blot showed that HMGCS2 protein levels were significantly elevated in patient skeletal muscle biopsies. (G) Western blot of control and patient skeletal muscle biopsies alongside mouse tissue protein extracts shows that HMGCS2 protein levels in ISCU myopathy patient muscles approach the level of HMGCS2 expression observed in the mouse liver. (H) Plasma ketones, measured enzymatically as β-hydroxybutyrate, were assessed in healthy controls, ISCU myopathy patients (samples taken on two different days) and in patients with un-related mitochondrial myopathies.
Western blot analysis confirmed that HMGCS2 protein expression was greatly increased in the three different ISCU myopathy patients (Fig. 5F). To obtain a qualitative assessment of the relative level of HMGCS2 protein expression in the ISCU patients, we ran control and patient biopsy protein extracts alongside tissue protein extracts from a mouse that was starved for 18 h before sacrifice (Fig. 5G). The data show that the HMGCS2 protein level in ISCU myopathy patient muscle approached that of mouse liver, suggesting that HMGCS2 protein accumulated to biologically significant levels within the patient muscles. However, despite the dramatically increased HMGCS2 levels in patient muscles, circulating plasma ketones measured in two ISCU myopathy patients were not elevated to clinically significant levels (Fig. 5H).
Elevated muscle FGF21 expression in patient skeletal muscles leads to high plasma FGF21 concentrations
Long-term energy starvation has been shown to up-regulate hepatic expression of the secreted endocrine hormone FGF21 (35). Our microarray data showed that FGF21 expression was robustly increased in patient muscle samples (Supplementary Material, Table S4), and qRT-PCR analysis confirmed an average 25-fold induction of FGF21 mRNA in ISCU myopathy patient muscle biopsies, whereas the un-related mitochondrial myopathy patient results ranged from 2- to 67-fold induction of FGF21 mRNA (Fig. 6A). Western blots of control and patient vastus lateralis biopsy protein extracts also showed a robust increase in FGF21 protein expression in two ISCU myopathy patients (Fig. 6B). Finally, measurement of circulating FGF21 concentrations in plasma taken from two ISCU myopathy patients as well as five non-ISCU mitochondrial myopathy patients and controls by ELISA demonstrated that the increased skeletal muscle expression of FGF21 in these patients is associated with elevated systemic levels of circulating FGF21 (Fig. 6C).
Figure 6.

Enhanced FGF21 secretion in ISCU myopathy patient muscles can be recapitulated in vitro with mitochondrial respiratory chain inhibitors. (A) Starvation response hormone FGF21 mRNA was significantly increased by an average of 25-fold in ISCU myopathy patient biopsies, and was also elevated in the mitochondrial myopathy patients. (B) Western blot of muscle biopsy protein extracts demonstrated elevated FGF21 protein expression in two ISCU myopathy patients compared with controls. (C) Plasma FGF21 protein concentrations, measured by ELISA, were elevated in ISCU myopathy patients (samples taken on two different days) and in the un-related mitochondrial myopathy patients. (D) Western blot of mouse tissue extracts showed greatest expression of FGF21 protein in heart tissue, whereas skeletal muscle showed FGF21 protein levels comparable with those in liver. (E) FGF21 protein expression was induced during C2C12 myoblast differentiation. (F and G) FGF21 mRNA expression in human and mouse myotubes was significantly increased following treatment with mitochondrial respiratory chain inhibitors. *P < 0.05; n = 4–8 experimental replicates. (H) C2C12 myoblasts were differentiated with low serum medium and then treated with metabolic inhibitors for 18 h. FGF21 secretion into serum-free medium was measured by western blot as described in Materials and Methods. (I) FGF21 secretion was increased following treatment with the complex IV inhibitor sodium azide, whereas intracellular FGF21 protein levels remained nearly constant. Total secreted protein levels were assessed by ponceau-S staining. (J) Sodium azide-stimulated FGF21 secretion was blocked by inclusion of the secretory pathway inhibitor brefeldin A in the secretion medium.
The above data suggest that sustained skeletal muscle expression of FGF21 in mitochondrial myopathy patients might lead to measurable increases in circulating FGF21 concentrations. To assess the validity of this hypothesis, we first compared FGF21 protein expression in various tissues derived from mice. We found that FGF21 protein expression in quadriceps and soleus muscle protein extracts was comparable with expression levels in the liver and brain, while the kidney and heart showed even higher FGF21 expression (Fig. 6D). We also monitored FGF21 protein expression during differentiation of mouse C2C12 myoblasts into multi-nucleated myotubes, and we found that FGF21 protein levels increased substantially during the latter stage of differentiation following myoblast fusion, reaching expression levels comparable with those of mature skeletal muscle and liver (Fig. 6E).
FGF21 secretion in mitochondrial myopathy patient muscles can be recapitulated in vitro with mitochondrial respiratory chain inhibitors.
To explore the hypothesis that plasma FGF21 in ISCU myopathy and other mitochondrial myopathy patients is secreted directly from respiratory chain-deficient skeletal muscle, we treated cultured myotubes with compounds known to target distinct components of the mitochondrial respiratory chain. Treatment of differentiated primary human myotubes with an inhibitor of complex II (TTFA, theonoyltrifluoroacetone), a specific inhibitor of complex III (antimycin A) or a respiratory chain uncoupler [FCCP, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone] resulted in significant and reproducible increases in FGF21 mRNA levels (Fig. 6F). Similarly, FGF21 mRNA expression was increased in differentiated mouse C2C12 myotubes treated with TTFA, FCCP or the complex IV inhibitor sodium azide (Fig. 6G). To measure FGF21 protein expression and secretion, we again treated differentiated C2C12 myotubes with respiratory chain inhibitors for 18 h, followed by a 1 h incubation in serum-free protein secretion medium (Fig. 6H). Our results show that although total intracellular FGF21 protein levels were relatively unaffected by treatment with increasing concentrations of sodium azide, FGF21 protein secretion was dramatically increased in a dose-dependent manner (Fig. 6I). Similar results were observed when cells were treated with TTFA (Supplementary Material, Fig. S3A), or with FCCP (Supplementary Material, Fig. S3B). Finally, we assessed the cellular pathway of FGF21 secretion by including brefeldin A, an inhibitor of the classical secretory pathway (36), during the 1 h secretion period. Brefeldin A strongly inhibited FGF21 protein secretion in both control and sodium azide-treated myotubes (Fig. 6J), and in TTFA-treated myotubes (Supplementary Material, Fig. S3C). Together, these results demonstrate that inhibition of the mitochondrial respiratory chain in skeletal muscle leads directly to increased FGF21 expression and secretion via the classical constitutive secretory pathway.
DISCUSSION
Skeletal muscle plays a critical role in organism-level energy metabolism, and is unique in its capacity to adapt to the energy demands of exercise (37). Fe-S cluster deficiency in the skeletal muscle of ISCU myopathy patients causes profoundly decreased activities of key mitochondrial Fe-S enzymes (4,13), leading to life-long exercise intolerance and sporadic periods of severe weakness and rhabdomyolysis (6). We hypothesized that chronic Fe-S cluster deficiency would elicit remodeling of gene expression in ISCU myopathy patient muscles. Indeed, we detected up-regulation of several key iron metabolism genes, as well as striking morphological and metabolic remodeling in skeletal muscle as a consequence of chronic Fe-S cluster deficiency. Despite the well-known TCA cycle enzyme deficiencies (13,17), we found increased type I muscle fibers and oxidative metabolism gene expression in ISCU myopathy patients, as well as increased expression and nuclear localization of the transcriptional co-activator PGC-1α (Fig. 7). Chronic Fe-S cluster deficiency strongly induced the rate-limiting ketogenic enzyme HMGCS2 and the secreted hormone FGF21, leading to elevated plasma FGF21 levels (Fig. 7). Our data demonstrate that elevated plasma FGF21 will be a useful indicator of mitochondrial function in ISCU myopathy and confirm its utility in diagnosing other muscle-manifesting mitochondrial disorders (38).
Figure 7.

Morphological and biochemical alterations caused by Fe-S cluster depletion in ISCU myopathy patient skeletal muscles. A schematic diagram summarizing the morphological differences between (A) control and (B) ISCU myopathy patient muscle biopsies, as inferred from gene expression analysis, biochemistry, histochemical data and previous studies. ISCU myopathy patient muscle biopsies showed a predominance of type I fibers as well as increased capillary abundance. Previous studies have demonstrated marked mitochondrial proliferation as well as insoluble mitochondrial iron deposits. (C) Biochemical alterations in ISCU myopathy patient muscles due to prolonged Fe-S cluster deficiency and TCA cycle deficits include enhanced mitochondrial fatty acid uptake capacity due to increased expression of fatty acid-binding proteins (FABP), the components of the carnitine palmitoyltransferase system (CPT1A, SLC28A25 and CPTII) and of mitochondrial and peroxisomal fatty acid β-oxidation genes. Up-regulation of the rate-limiting ketogenic enzyme HMGCS2 may reflect an effort to dispose of excess acetyl-CoA as ketone bodies. These alterations in gene expression are likely driven in part by transcriptional activation of PPARα/γ and PGC-1α, which can be activated by low ATP levels, high NAD+ levels and/or by autocrine stimulation via increased expression and secretion of the starvation-induced hormone FGF21, which is dramatically increased in the muscles and bloodstream of the patients.
Altered muscle fiber-type composition and capillary content in ISCU myopathy patients are unique in that they occur in the absence of increased neural stimulation of muscle contraction (39,40) or regular exercise (41–43), and thus likely reflect a cellular response to muscle energy deficiency due to decreased mitochondrial energy production. In contrast to the limited muscle oxidative capacity that accompanies habitual physical inactivity, impaired oxidative metabolism in ISCU myopathy patients leads to increased PGC-1α and HIF-2α expression, which likely drive the increase in type I oxidative fibers and capillary formation that we have observed here (44). Activation of PGC-1α in patient muscle tissue would lead to stimulated expression of PPARα target genes (45) such as the components of the carnitine palmitoyltransferase system (46), the peroxisomal and mitochondrial β-oxidation systems (47), HMGCS2 (48) and FGF21 (49), all of which we have observed here in the skeletal muscle tissues of ISCU myopathy patients.
The differences in gene expression patterns between the ISCU myopathy patients and the non-ISCU mitochondrial myopathy patients could be attributed to several factors. Several of the biochemical defects in ISCU myopathy such as profound deficiency in aconitase and SDH activities are unique to these patients. Furthermore, histochemical studies have demonstrated a strikingly homogeneous biochemical defect in ISCU patient muscle biopsies, likely due to robust depletion of ISCU in nearly all patient muscle fibers (4,13–15,18). In contrast, the non-ISCU mitochondrial myopathy patients in our study possessed heteroplasmic mtDNA deletions, and the individual myofibers in their muscle biopsies likely exhibit highly differing levels of biochemical impairment (50), which could impede our ability to observe robust changes in gene expression. Nevertheless, the biopsy from patient 1C in our study showed patterns of elevated expression of mitochondrial fatty acid uptake genes and robust HMGCS2 expression, suggesting that at least some of the effects reported here are not unique to ISCU myopathy. Notably, a previous muscle biopsy from this patient showed a very high percentage (>90% T5543C) of mutant mtDNA (51).
The dramatically increased expression of the rate-limiting ketogenic enzyme HMGCS2 that we found in patient muscles might reflect an effort to dispose of excess mitochondrial acetyl-CoA and/or NADH produced from fatty acid oxidation when functions of the TCA cycle and/or respiratory chain function are impaired. In ISCU myopathy patient muscles, where chronic Fe-S cluster deficiency reduces the critical enzyme activities of aconitase and SDH, it is likely that rising acetyl-CoA concentrations diminish the pool of free coenzyme A and limit the activities of pyruvate dehydrogenase and pyruvate carboxylase (52). Thus, ketone body formation could provide a pathway for the continued oxidation of fatty acids in patient muscles despite severe impairment of the mitochondrial respiratory chain by disposing of acetyl-CoA and by regenerating NAD+. The rapid kinetics of ketone clearance by the heart, brain, kidneys and other organs which retain normal metabolic capacity in ISCU myopathy patients may preclude accumulation of muscle-derived ketones in blood (53); however, functional studies are needed first to validate ketone formation in muscle tissue with high HMGCS2 expression levels. Interestingly, HMGCS2 over-expression has been shown to increase FGF21 mRNA expression in HepG2 cells (54), whereas FGF21 over-expression increased HMGCS2 protein levels in the mouse liver (35), suggesting that a feed-forward mechanism might drive expression of these genes in ISCU myopathy patient muscles.
Increased circulating FGF21 levels cause beneficial physiological responses such as decreased plasma triglycerides, elevated insulin sensitivity and enhanced liver fatty acid oxidation (49,55,56). FGF21 can also induce PGC-1α expression (57), and thus might serve as an autocrine regulator of PGC-1α levels in ISCU myopathy patient muscles. Recently, FGF21 expression was implicated as a key feature of a starvation-like response in skeletal muscle of a mouse model exhibiting progressive mtDNA deletions and adult-onset respiratory chain deficiency (58,59). Moreover, increased FGF21 mRNA expression was associated with autophagy deficiency and mitochondrial dysfunction due to Atg7 deletion in mouse skeletal muscle (60). Thus, FGF21 is emerging as a factor that is responsive to muscle mitochondrial dysfunction.
To our knowledge, our experiments with cultured C2C12 myotubes represent the first direct measurement of increased FGF21 secretion in response to respiratory chain inhibition and energy insufficiency. Our data provide a direct link between the observations of FGF21 mRNA induction in skeletal muscle and elevated plasma FGF21 concentrations in human mitochondrial myopathy patients. These findings strengthen the growing appreciation of skeletal muscle as a secretory-endocrine tissue that modulates whole-body energy metabolism (58,61–63). Furthermore, our finding that FGF21 expression is abundant in mouse heart suggests that cardiac FGF21 levels might be elevated in diseases involving defective cardiac mitochondrial function such as Friedreich's ataxia.
In summary, we have used gene expression analysis to explore the unique pathophysiology of a human mitochondrial myopathy caused by Fe-S cluster deficiency. We conclude that chronic Fe-S cluster deficiency in skeletal muscle induces a unique starvation response that is characterized by a predominance of oxidative muscle fibers and enhanced expression of the fatty acid oxidation machinery, despite significant impairments of the TCA cycle and respiratory chain. Fe-S cluster-deficient patient muscles show dramatically increased expression of the ketogenic enzyme HMGCS2 and enhanced secretion of the endocrine hormone FGF21, suggesting that these clinically relevant biomarkers will be useful for evaluating the efficacy of therapeutic treatments for this and other mitochondrial myopathies.
MATERIALS AND METHODS
Patients, tissue biopsies and primary cell cultures
Patient data are summarized in Table 1. Impaired muscle oxidative phosphorylation in ISCU myopathy and mitochondrial myopathy patients was indicated by restricted oxygen extraction from blood during maximal cycle exercise (64). Peak systemic arterio-venous oxygen (a-vO2) difference in the mitochondrial myopathy patients with heteroplasmic mtDNA mutations was 4.5–8.3 ml/dl, compared with an average peak a-vO2 difference of ∼5 ml/dl in the ISCU myopathy patients, and ∼15 ml/dl in healthy controls (64). Skeletal muscle biopsies from vastus lateralis were obtained and prepared as described previously (4). Written informed consent was given by all individuals involved in the study in accordance with the Institutional Review Board of the University of Texas Southwestern Medical Center and the Texas Health Presbyterian Hospital of Dallas. Primary myoblast cells were cultured as described previously (11).
RNA preparation and microarray analysis
RNA from biopsies and cell pellets was prepared using the mirVana™ RNA Isolation Kit (Ambion). RNA quality was verified using an Agilent 2100 Bioanalyzer, and samples were processed by one-cycle target labeling and hybridization with Affymetrix Genechip U133 Plus 2.0 microarrays at the NCI-Frederick Laboratory of Molecular Technology. The R/Bioconductor software was used to identify differentially expressed genes in the data set (65). The robust multi-array average expression measure was applied for background correction, quantile normalization and output of gene expression values in log2 scale. Normalized unscaled standard error and relative log expression plots were generated for quality assessment. We used the LIMMA package to assess differentially expressed genes, and statistical significance was adjusted for multiple testing by controlling the Benjamini and Hochberg's false discovery rate at 5% (66). Functional and pathway analyses were performed using the iProXpress platform (http://pir.georgetown.edu/iproxpress) for gene profiling based on cellular function, pathways and processes (19). The DAVID bioinformatics database was also used for functional classification (67), and Ingenuity Pathway Analysis™ software provided clustering of affected cellular processes. The microarray data are available under the GEO accession GSE48574.
Northern blots, qRT-PCR and statistical analysis
qRT-PCR was performed using SYBR® Green (Applied Biosystems) according to the manufacturer's instructions, following reverse transcription of total RNA into cDNA (Applied Biosystems). qRT-PCR primer sequences are listed in Supplementary Material, Table S3. Relative transcript abundance was calculated using the 2−ΔΔCt method (68), with GAPDH as the internal control. Non-transformed 2−ΔΔCt values were analyzed by paired t-tests to evaluate statistical significance relative to controls. Correct qRT-PCR product size was verified by agarose gel electrophoresis, and melting curve measurements were made after every experiment. Northern blots were performed as reported previously (69). An antisense RNA probe against HMGCS2 mRNA was synthesized using the MAXIScript T7 in vitro transcription kit (Ambion), incorporating α-32P-cytosine triphosphate. The probe template with T7 promoter was generated by PCR using a human liver cDNA library and the following primers: 5′-TAATACGACTCACTATAGGGAGACCATAAGAGAAGGCACCAATCC-3′ and 5′-TCCCCCTGGCCAAAACAGA-3′.
siRNA knockdown of ISCU
A pool of siRNA oligonucleotides directed against ISCU was purchased from Thermo Scientific/Dharmacon (part# L-012837-01-0005); a pool of non-targeting oligonucleotides served as the control. Primary myoblasts were transfected with 100 nm siRNA using the Neon™ Transfection System (Invitrogen), with settings of 1400 V, 20 ms pulse width and 2 pulses per transfection. The cells were transfected every other day for 6 days and were harvested 2 days after the last transfection.
Muscle fiber-type quantification and CD31 histochemistry
Muscle fiber composition was assessed by acid–base myofibrillar ATPase staining (30). Vastus lateralis biopsies from 3 ISCU myopathy patients and 13 healthy control subjects were compared. Immunohistochemistry for the capillary endothelial marker CD31 was performed on a patient muscle biopsy, as well as a biopsy from the healthy heterozygous offspring of this patient, as previously described (32).
Immunofluorescence microscopy
As described in Taivassalo et al. (32), 6 µm frozen vastus lateralis biopsy sections were obtained. Samples were blocked using 0.4% Triton X-100, 4% normal goat serum and 2% ovalbumin in PBS at room temperature. After blocking, a mixture of mouse anti-MTCO1 antibody (1/100, Abcam) and rabbit anti-PGC-1α antibody (1/100, Abcam) was applied for overnight incubation. The next day, samples were washed and incubated with a mixture of secondary antibodies (1/400, Alexa fluoro 546 goat anti-mouse/Alexa fluoro 488 goat anti-rabbit) for 1 h. Sections were washed and mounted with anti-fading media containing DAPI (Vector Laboratories) and analyzed using a confocal microscope (Zeiss LSM710).
For PGC-1α immunohistochemistry, tissue sections were treated with 0.1% H2O2 in PBS for 10 min to block endogenous peroxidase activity, and endogenous biotin was later blocked following the manufacturer's protocol (Avidin/Biotin Blocking Kit, Invitrogen). Samples were blocked and incubated with rabbit anti-PGC-1α antibody (see above) in blocking buffer overnight. Samples were then washed, incubated with biotin-labeled goat anti-rabbit secondary antibody for 1 h. ABC complex (Vector Laboratories) was used to amplify signals, and diaminobenzidine tetrahydrochloride (Sigma) was used as a chromagen. Expression of PGC-1α was analyzed using a bright field microscope (Nikon Eclipse E600).
Western blots
SDS–PAGE and western blotting were performed as described previously (70) using 1.5 mm 4–12% pre-cast bis–Tris gels (Invitrogen). Rabbit polyclonal anti-FECH serum was a kind gift from Dr Harry Dailey. Anti-IRP2, PPAT and ISCU rabbit polyclonal sera were raised against synthetic peptide fragments (20). SDH-B and complex IV, subunit 4 antibodies were from Mitosciences. The rabbit polyclonal HMGCS2 antibody was from Sigma Life Sciences. Mouse monoclonal anti-NTHL1 was from R&D Systems. Mouse monoclonal anti-human TfR1 was from Zymed. PGC-1α antibody for western blotting (H-300) was from Santa Cruz Biotechnology, and the VEGF antibody was from Millipore. RCC1 was a gift from Dr Mary Dasso of NICHD.
Aconitase in-gel assay and electrophoretic mobility shift assay
Aconitase was assayed using a coupled assay following native PAGE separation, as described previously (20). IRP-IRE-binding activity was determined by electrophoretic mobility shift assay using a 32P-labeled ferritin IRE probe, as described previously (20).
FGF21 ELISA and FGF21 secretion experiments
FGF21 protein levels in plasma from human subjects were measured using a commercially available kit (BioVendor). C2C12 cells were propagated in DMEM medium supplemented with 10% FBS, and confluent cultures were differentiated by switching the medium to DMEM containing 5 mm glucose, 2% heat-inactivated horse serum and 0.4 µg/ml dexamethasone for 4 days. FGF21 secretion from C2C12 myotubes cultured on six-well plates was measured following 18 h incubation with metabolic inhibitors dissolved in C2C12 differentiation medium containing 100 µm palmitic acid conjugated to bovine serum albumin (BSA). Myotubes were washed two times with PBS at 37°C, and secreted proteins were collected by incubation of myotubes for 1h at 37°C in 2 ml of secretion medium consisting of a 1:1 mixture of OptiMEM I medium and DMEM medium containing 5 mm glucose, 2 mm glutamine and 110 mg/l sodium pyruvate (Invitrogen). This secretion medium was further modified by the addition of BSA and β-mercaptoethanol at 2 mg/l and 20 mm, respectively. After 1h, secretion medium was collected and placed on ice, the myotubes were lysed with 200 µl of radioimmunoprecipitation assay buffer and the lysates were snap-frozen and stored at −80°C for western blotting. The secretion medium was then centrifuged at 1000g for 5 min, the supernatant was collected and the volume was adjusted to 5 ml with PBS. Five milliliters of methanol was added to this mixture and mixed by inversion. Next, 1.25 ml of chloroform was added and mixed by inversion, and the resulting suspension was centrifuged for 20 min at 4000g, which resulted in two liquid phases with the precipitated protein located at the interface. The upper phase was carefully aspirated, then 5 ml of methanol was added and the solution was mixed by vigorous shaking. Centrifugation at 4000g for 20 min followed by thorough aspiration of the supernatant resulted in a small protein pellet, which was further air-dried for 20 min. Finally, the pellet was re-suspended in 100 µl of 1× western blot sample buffer, incubated at 95°C for 5 min and the solution was clarified by centrifugation at 20 000g for 5 min. Twenty microliters of the supernatant was then subjected to SDS–PAGE and western blotting for FGF21 protein, using a rabbit polyclonal antibody raised against FGF21 (Novus NBP1-59291).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and NIAMS grant #R01 AR050597.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Eric Shoubridge for providing us with the primary human myoblast cells. We would also like to thank Drs Deliang Zhang, Gennadiy Kovtunovych, Carole Sourbier, Elliott Crooke, John Casey, G. Ian Gallicano and Hiroshi Nakai for providing ideas and suggestions that greatly improved the quality of this work.
REFERENCES
- 1.Rotig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. doi:10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 2.Ye H., Jeong S.Y., Ghosh M.C., Kovtunovych G., Silvestri L., Ortillo D., Uchida N., Tisdale J., Camaschella C., Rouault T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. doi:10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cameron J.M., Janer A., Levandovskiy V., Mackay N., Rouault T.A., Tong W.H., Ogilvie I., Shoubridge E.A., Robinson B.H. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet. 2011;89:486–495. doi: 10.1016/j.ajhg.2011.08.011. doi:10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mochel F., Knight M.A., Tong W.H., Hernandez D., Ayyad K., Taivassalo T., Andersen P.M., Singleton A., Rouault T.A., Fischbeck K.H., et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. doi:10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsson A., Lind L., Thornell L.E., Holmberg M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 2008;17:1666–1672. doi: 10.1093/hmg/ddn057. doi:10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- 6.Larsson L.E., Linderholm H., Mueller R., Ringqvist T., Soernaes R. Hereditary metabolic myopathy with paroxysmal myoglobinuria due to abnormal glycolysis. J. Neurol. Neurosurg. Psychiatry. 1964;27:361–380. doi: 10.1136/jnnp.27.5.361. doi:10.1136/jnnp.27.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drugge U., Holmberg M., Holmgren G., Almay B.G., Linderholm H. Hereditary myopathy with lactic acidosis, succinate dehydrogenase and aconitase deficiency in northern Sweden: a genealogical study. J. Med. Genet. 1995;32:344–347. doi: 10.1136/jmg.32.5.344. doi:10.1136/jmg.32.5.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rouault T.A. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 2012;5:155–164. doi: 10.1242/dmm.009019. doi:10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanaker P.S., Toompuu M., Hogan V.E., He L., Tzoulis C., Chrzanowska-Lightowlers Z.M., Taylor R.W., Bindoff L.A. Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy. Biochim. Biophys. Acta. 2010;1802:539–544. doi: 10.1016/j.bbadis.2010.02.010. doi:10.1016/j.bbadis.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 10.Nordin A., Larsson E., Thornell L.E., Holmberg M. Tissue-specific splicing of ISCU results in a skeletal muscle phenotype in myopathy with lactic acidosis, while complete loss of ISCU results in early embryonic death in mice. Hum. Genet. 2011;129:371–378. doi: 10.1007/s00439-010-0931-3. doi:10.1007/s00439-010-0931-3. [DOI] [PubMed] [Google Scholar]
- 11.Crooks D.R., Jeong S.Y., Tong W.H., Ghosh M.C., Olivierre H., Haller R.G., Rouault T.A. Tissue specificity of a human mitochondrial disease: differentiation-enhanced mis-splicing of the Fe-S scaffold gene ISCU renders patient cells more sensitive to oxidative stress in ISCU myopathy. J. Biol. Chem. 2012;287:40119–40130. doi: 10.1074/jbc.M112.418889. doi:10.1074/jbc.M112.418889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linderholm H., Muller R., Ringqvist T., Sornas R. Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Med. Scand. 1969;185:153–166. doi: 10.1111/j.0954-6820.1969.tb07314.x. doi:10.1111/j.0954-6820.1969.tb07314.x. [DOI] [PubMed] [Google Scholar]
- 13.Haller R.G., Henriksson K.G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N.H., Gunder M., Ayyad K., Blomqvist C.G., et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 1991;88:1197–1206. doi: 10.1172/JCI115422. doi:10.1172/JCI115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kollberg G., Tulinius M., Melberg A., Darin N., Andersen O., Holmgren D., Oldfors A., Holme E. Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain. 2009;132:2170–2179. doi: 10.1093/brain/awp152. doi:10.1093/brain/awp152. [DOI] [PubMed] [Google Scholar]
- 15.Kollberg G., Melberg A., Holme E., Oldfors A. Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulphur cluster deficiency myopathy. Neuromuscul. Disord. 2010;21:115–120. doi: 10.1016/j.nmd.2010.11.010. doi:10.1016/j.nmd.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 16.Puccio H., Simon D., Cossee M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001;27:181–186. doi: 10.1038/84818. doi:10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 17.Hall R.E., Henriksson K.G., Lewis S.F., Haller R.G., Kennaway N.G. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 1993;92:2660–2666. doi: 10.1172/JCI116882. doi:10.1172/JCI116882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linderholm H., Essen-Gustavsson B., Thornell L.E. Low succinate dehydrogenase (SDH) activity in a patient with a hereditary myopathy with paroxysmal myoglobinuria. J. Intern. Med. 1990;228:43–52. doi: 10.1111/j.1365-2796.1990.tb00191.x. doi:10.1111/j.1365-2796.1990.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 19.Huang H., Hu Z.Z., Arighi C.N., Wu C.H. Integration of bioinformatics resources for functional analysis of gene expression and proteomic data. Front. Biosci. 2007;12:5071–5088. doi: 10.2741/2449. doi:10.2741/2449. [DOI] [PubMed] [Google Scholar]
- 20.Tong W.H., Rouault T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. doi:10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Schiaffino S., Reggiani C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. doi:10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 22.Mu L., Su H., Wang J., Han Y., Sanders I. Adult human mylohyoid muscle fibers express slow-tonic, alpha-cardiac, and developmental myosin heavy-chain isoforms. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2004;279:749–760. doi: 10.1002/ar.a.20065. doi:10.1002/ar.a.20065. [DOI] [PubMed] [Google Scholar]
- 23.Peuker H., Conjard A., Pette D. Alpha-cardiac-like myosin heavy chain as an intermediate between MHCIIa and MHCI beta in transforming rabbit muscle. Am. J. Physiol. 1998;274:C595–C602. doi: 10.1152/ajpcell.1998.274.3.C595. [DOI] [PubMed] [Google Scholar]
- 24.Rossi A.C., Mammucari C., Argentini C., Reggiani C., Schiaffino S. Two novel/ancient myosins in mammalian skeletal muscles: MYH14/7b and MYH15 are expressed in extraocular muscles and muscle spindles. J. Physiol. 2010;588:353–364. doi: 10.1113/jphysiol.2009.181008. doi:10.1113/jphysiol.2009.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halestrap A.P. The SLC16 gene family – structure, role and regulation in health and disease. Mol. Aspects Med. 2013;34:337–349. doi: 10.1016/j.mam.2012.05.003. doi:10.1016/j.mam.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Fishbein W.N., Merezhinskaya N., Foellmer J.W. Relative distribution of three major lactate transporters in frozen human tissues and their localization in unfixed skeletal muscle. Muscle Nerve. 2002;26:101–112. doi: 10.1002/mus.10168. doi:10.1002/mus.10168. [DOI] [PubMed] [Google Scholar]
- 27.Houtkooper R.H., Canto C., Wanders R.J., Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. doi:10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGarry J.D., Brown N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. doi:10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 29.Fischer H., Gustafsson T., Sundberg C.J., Norrbom J., Ekman M., Johansson O., Jansson E. Fatty acid binding protein 4 in human skeletal muscle. Biochem. Biophys. Res. Commun. 2006;346:125–130. doi: 10.1016/j.bbrc.2006.05.083. doi:10.1016/j.bbrc.2006.05.083. [DOI] [PubMed] [Google Scholar]
- 30.Dubowitz V., Sewry C.A. Adenosine triphosphatase (ATPase) staining. In: Dubowitz V., Sewry C.A., Oldfors A., editors. Muscle Biopsy: A Practical Approach. Philadelphia, PA: Saunders Elsevier; 2007. [Google Scholar]
- 31.Little J.P., Safdar A., Bishop D., Tarnopolsky M.A., Gibala M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1alpha and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300:R1303–R1310. doi: 10.1152/ajpregu.00538.2010. doi:10.1152/ajpregu.00538.2010. [DOI] [PubMed] [Google Scholar]
- 32.Taivassalo T., Ayyad K., Haller R.G. Increased capillaries in mitochondrial myopathy: implications for the regulation of oxygen delivery. Brain. 2012;8:53–61. doi: 10.1093/brain/awr293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenz T., Diabz F., Spiegelman B.M., Moraes C.T. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. doi:10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Fulgham D.L., Widhalm S.R., Martin S., Coffin J.D. FGF-2 dependent angiogenesis is a latent phenotype in basic fibroblast growth factor transgenic mice. Endothelium. 1999;6:185–195. doi: 10.3109/10623329909053409. doi:10.3109/10623329909053409. [DOI] [PubMed] [Google Scholar]
- 35.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. doi:10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Misumi Y., Misumi Y., Miki K., Takatsuki A., Tamura G., Ikehara Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986;261:11398–11403. [PubMed] [Google Scholar]
- 37.Egan B., Zierath J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. doi:10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Suomalainen A., Elo J.M., Pietilainen K.H., Hakonen A.H., Sevastianova K., Korpela M., Isohanni P., Marjavaara S.K., Tyni T., Kiuru-Enari S., et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. doi:10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donoghue P., Doran P., Wynne K., Pedersen K., Dunn M.J., Ohlendieck K. Proteomic profiling of chronic low-frequency stimulated fast muscle. Proteomics. 2007;7:3417–3430. doi: 10.1002/pmic.200700262. doi:10.1002/pmic.200700262. [DOI] [PubMed] [Google Scholar]
- 40.Jarvis J.C., Mokrusch T., Kwende M.M., Sutherland H., Salmons S. Fast-to-slow transformation in stimulated rat muscle. Muscle Nerve. 1996;19:1469–1475. doi: 10.1002/(SICI)1097-4598(199611)19:11<1469::AID-MUS11>3.0.CO;2-O. doi:10.1002/(SICI)1097-4598(199611)19:11<1469::AID-MUS11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 41.Trappe S., Harber M., Creer A., Gallagher P., Slivka D., Minchev K., Whitsett D. Single muscle fiber adaptations with marathon training. J. Appl. Physiol. 2006;101:721–727. doi: 10.1152/japplphysiol.01595.2005. doi:10.1152/japplphysiol.01595.2005. [DOI] [PubMed] [Google Scholar]
- 42.Yan Z., Okutsu M., Akhtar Y.N., Lira V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011;110:264–274. doi: 10.1152/japplphysiol.00993.2010. doi:10.1152/japplphysiol.00993.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gute D., Fraga C., Laughlin M.H., Amann J.F. Regional changes in capillary supply in skeletal muscle of high-intensity endurance-trained rats. J. Appl. Physiol. 1996;81:619–626. doi: 10.1152/jappl.1996.81.2.619. [DOI] [PubMed] [Google Scholar]
- 44.Rasbach K.A., Gupta R.K., Ruas J.L., Wu J., Naseri E., Estall J.L., Spiegelman B.M. PGC-1alpha regulates a HIF2alpha-dependent switch in skeletal muscle fiber types. Proc. Natl Acad. Sci. USA. 2010;107:21866–21871. doi: 10.1073/pnas.1016089107. doi:10.1073/pnas.1016089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vega R.B., Huss J.M., Kelly D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. doi:10.1128/MCB.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fruchart J.C., Duriez P., Staels B. Peroxisome proliferator-activated receptor-alpha activators regulate genes governing lipoprotein metabolism, vascular inflammation and atherosclerosis. Curr. Opin. Lipidol. 1999;10:245–257. doi: 10.1097/00041433-199906000-00007. doi:10.1097/00041433-199906000-00007. [DOI] [PubMed] [Google Scholar]
- 47.Muoio D.M., Koves T.R. Skeletal muscle adaptation to fatty acid depends on coordinated actions of the PPARs and PGC1 alpha: implications for metabolic disease. Appl. Physiol. Nutr. Metab. 2007;32:874–883. doi: 10.1139/H07-083. doi:10.1139/H07-083. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez J.C., Gil-Gomez G., Hegardt F.G., Haro D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994;269:18767–18772. [PubMed] [Google Scholar]
- 49.Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. doi:10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 50.Murphy J.L., Ratnaike T.E., Shang E., Falkous G., Blakely E.L., Alston C.L., Taivassalo T., Haller R.G., Taylor R.W., Turnbull D.M. Cytochrome c oxidase-intermediate fibres: importance in understanding the pathogenesis and treatment of mitochondrial myopathy. Neuromuscul. Disord. 2012;22:690–698. doi: 10.1016/j.nmd.2012.04.003. doi:10.1016/j.nmd.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anitori R., Manning K., Quan F., Weleber R.G., Buist N.R., Shoubridge E.A., Kennaway N.G. Contrasting phenotypes in three patients with novel mutations in mitochondrial tRNA genes. Mol. Genet. Metab. 2005;84:176–188. doi: 10.1016/j.ymgme.2004.10.003. doi:10.1016/j.ymgme.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 52.Lehninger A.L., Nelson D.L., Cox M.M. Principles of Biochemistry. 2nd edn. New York, NY: Worth Publishers, Inc.; 2000. [Google Scholar]
- 53.Angielski S., Lukowicz J. The role of the kidney in the removal of ketone bodies under different acid-base status of the rat. Am. J. Clin. Nutr. 1978;31:1635–1641. doi: 10.1093/ajcn/31.9.1635. [DOI] [PubMed] [Google Scholar]
- 54.Vila-Brau A., De Sousa-Coelho A.L., Mayordomo C., Haro D., Marrero P.F. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J. Biol. Chem. 2011;286:20423–20430. doi: 10.1074/jbc.M111.235044. doi:10.1074/jbc.M111.235044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kharitonenkov A., Shiyanova T.L., Koester A., Ford A.M., Micanovic R., Galbreath E.J., Sandusky G.E., Hammond L.J., Moyers J.S., Owens R.A., et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. doi:10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reitman M.L. FGF21: a missing link in the biology of fasting. Cell. Metab. 2007;5:405–407. doi: 10.1016/j.cmet.2007.05.010. doi:10.1016/j.cmet.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Fisher F.M., Kleiner S., Douris N., Fox E.C., Mepani R.J., Verdeguer F., Wu J., Kharitonenkov A., Flier J.S., Maratos-Flier E., et al. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–281. doi: 10.1101/gad.177857.111. doi:10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyynismaa H., Carroll C.J., Raimundo N., Ahola-Erkkila S., Wenz T., Ruhanen H., Guse K., Hemminki A., Peltola-Mjosund K.E., Tulkki V., et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010;19:3948–3958. doi: 10.1093/hmg/ddq310. doi:10.1093/hmg/ddq310. [DOI] [PubMed] [Google Scholar]
- 59.Yatsuga S., Suomalainen A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum. Mol. Genet. 2011;21:526–535. doi: 10.1093/hmg/ddr482. doi:10.1093/hmg/ddr482. [DOI] [PubMed] [Google Scholar]
- 60.Kim K.H., Jeong Y.T., Oh H., Kim S.H., Cho J.M., Kim Y.N., Kim S.S., Kim do H., Hur K.Y., Kim H.K., et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2012;19:83–92. doi: 10.1038/nm.3014. doi:10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 61.Boström P., Wu J., Jedrychowski M.P., Korde A., Ye L., Lo J.C., Rasbach K.A., Bostrom E.A., Choi J.H., Long J.Z., et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. doi:10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bortoluzzi S., Scannapieco P., Cestaro A., Danieli G.A., Schiaffino S. Computational reconstruction of the human skeletal muscle secretome. Proteins. 2006;62:776–792. doi: 10.1002/prot.20803. doi:10.1002/prot.20803. [DOI] [PubMed] [Google Scholar]
- 63.Pedersen B.K., Febbraio M.A. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012;8:457–465. doi: 10.1038/nrendo.2012.49. doi:10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 64.Taivassalo T., Jensen T.D., Kennaway N., DiMauro S., Vissing J., Haller R.G. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126:413–423. doi: 10.1093/brain/awg028. doi:10.1093/brain/awg028. [DOI] [PubMed] [Google Scholar]
- 65.Hahne F., Huber W., Gentleman R., Falcon S. Bioconductor Case Studies. New York, NY: Springer; 2008. pp. 25–45. [Google Scholar]
- 66.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 67.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. doi:10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 68.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. doi:10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 69.Crooks D.R., Ghosh M.C., Haller R.G., Tong W.H., Rouault T.A. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood. 2010;115:860–869. doi: 10.1182/blood-2009-09-243105. doi:10.1182/blood-2009-09-243105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crooks D.R., Ghosh M.C., Braun-Sommargren M., Rouault T.A., Smith D.R. Manganese targets m-aconitase and activates iron regulatory protein 2 in AF5 GABAergic cells. J. Neurosci. Res. 2007;85:1797–1809. doi: 10.1002/jnr.21321. doi:10.1002/jnr.21321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.