

Abstract
Patients with Axenfeld–Rieger Syndrome (ARS) present various dental abnormalities, including hypodontia, and enamel hypoplasia. ARS is genetically associated with mutations in the PITX2 gene, which encodes one of the earliest transcription factors to initiate tooth development. Thus, Pitx2 has long been considered as an upstream regulator of the transcriptional hierarchy in early tooth development. However, because Pitx2 is also a major regulator of later stages of tooth development, especially during amelogenesis, it is unclear how mutant forms cause ARS dental anomalies. In this report, we outline the transcriptional mechanism that is defective in ARS. We demonstrate that during normal tooth development Pitx2 activates Amelogenin (Amel) expression, whose product is required for enamel formation, and that this regulation is perturbed by missense PITX2 mutations found in ARS patients. We further show that Pitx2-mediated Amel activation is controlled by chromatin-associated factor Hmgn2, and that Hmgn2 prevents Pitx2 from efficiently binding to and activating the Amel promoter. Consistent with a physiological significance to this interaction, we show that K14-Hmgn2 transgenic mice display a severe loss of Amel expression on the labial side of the lower incisors, as well as enamel hypoplasia—consistent with the human ARS phenotype. Collectively, these findings define transcriptional mechanisms involved in normal tooth development and shed light on the molecular underpinnings of the enamel defect observed in ARS patients who carry PITX2 mutations. Moreover, our findings validate the etiology of the enamel defect in a novel mouse model of ARS.
INTRODUCTION
Axenfeld–Rieger Syndrome (ARS) is a rare, autosomal-dominant genetic disease in humans, occurring in about 1:200 000 individuals (1,2). Patients with ARS exhibit a wide spectrum of developmental defects (3,4), including umbilical anomalies, ocular defects and craniofacial abnormalities (5,6). Classic clinical presentations also include various dental defects, often involving hypoplasia of the enamel (6,7). Intriguingly, most of the dental phenotypes associated with ARS include enamel defects that are clinically similar to those observed in patients with Amelogenesis Imperfecta (AI) (8). Approximately 30% of developing enamel is protein, of which 90% is Amelogenin (Amel) (9). The majority of this protein is expressed from the Amelogenin allele on the X chromosome (Amelx), and <10% by that on the Y chromosome (Amely) (10,11). Many enamel defects, such as AI, have been directly associated with abnormal amelogenin expression and/or processing (12,13).
Although ARS patients were first diagnosed almost a century ago, the genetic underpinnings of this syndrome were unknown until the 1990's, when FOXC1 (14,15) and PITX2 (16,17) alleles were found to be closely associated with ARS. FOXC1, a member of the winged helix/forkhead family of transcription factors, plays critical roles in eye development, and multiple studies have implicated mutations in this gene as major causes of ARS-associated anterior eye chamber defects, including iridogoniodysgenesis anomaly and familial glaucoma iridogoniodysplasia (15,18). PITX2 is a bicoid-motif-binding protein and a member of the paired-like homeobox transcription factor family (19). It is also one of the earliest epithelial markers of tooth development (20), and plays fundamental roles in the genetic control of pattern formation and epithelial differentiation in the tooth (19,21,22). Given that Pitx2 has long been considered as a master regulator of the transcriptional hierarchy in early tooth development (23,24), PITX2 mutations are thought to account for many of the tooth defects in ARS patients. Pitx2 is highly expressed in the cervical loop region of the developing tooth epithelium (a stem cell niche) and initiates differentiation. Once the epithelial stem cells start to migrate toward the apical tip of the tooth, Pitx2 expression decreases and is maintained at a relatively low level. Nevertheless, we found that Pitx2 is a major determinant of the later stages of tooth development, especially during amelogenesis (enamel formation).
PITX2 mutations account for 40% of ARS cases, and are associated with most of the dental defects that have been reported for this syndrome (6). Among these are missense mutations that lead to P64L, T68P, R90P, L105V and N108T amino acid substitutions, all of which were identified in clinical reports (6,25–27). ARS patients with these mutations have classical ARS manifestations, including a variety of dental defects: microdontia, hypodontia and enamel hypoplasia.
During tooth organogenesis, a spectrum of proteins other than Pitx2, including signaling molecules and additional transcription factors, contribute to the regulation of amelogenesis (28–30). For example, Dlx2 is a member of the homeobox family of genes and has been shown to play a central role in patterning of the jaw (31), in Dlx-1/Dlx-2 double mutants, the dental epithelial cells are poorly organized and express very little Amel (32). FoxJ1 is a transcription factor of the winged-helix/fork-head family, and is fundamental to cilia development (33). FoxJ1−/− mice present defects in odontogenesis resulting in small maxillary and mandibular incisors and reduced Amel expression (34). Dlx2 and FoxJ1 are both transcriptionally regulated by Pitx2, and their protein products interact physically with each other and with Pitx2, forming a complex in which hierarchical interactions take place to regulate amelogenesis (34,35). Wnt signaling is also required, upstream of these events and specifically for the late stages of tooth development (36,37). In the developing tooth bud, β-catenin is expressed at the same time as Lef-1 and, like Pitx2 and Lef-1, in epithelia (38). β-Catenin and Lef-1 independently interact with PITX2, and synergistically regulate the expression of PITX2 target genes (38,39).
Tooth development depends on the activities of not only transcription factors, but also chromatin-remodeling proteins (40). One candidate for such activity is Hmgn2, a chromatin-associated high-mobility group protein that binds to histones (41,42). Many transcription factors of the homeodomain family bind to Hmgn2 with high affinity, and form inactive complexes with histones on the open chromatin (43). Hmgn2 inhibits homeodomain transcription factors from binding DNA, but recruits transcription factors to chromatin where they are poised to regulate transcription upon interaction with other factors that de-repress the Hmgn2 inactive complex to allow the transcription factors to bind DNA. Hmgn2 is highly expressed in the dental epithelium during early embryogenesis, but its levels decrease similar to Pitx2 as development proceeds.
In the current study, we investigated the functional significance of the Pitx2 transcriptional hierarchy with respect to tooth development, including the roles that the Pitx2 co-factors play in regulating amelogenesis. Although the focus on the molecular-genetic aspects of ARS has increased in recent years, the molecular underpinnings of these particular tooth defects in ARS remain largely unknown. The Pitx2 homeodomain knockout (Pitx2 HD−/−) mice suffer from embryonic lethality at around Day E10.5, rendering them unsuitable for the study of late-stage tooth development (22,44), and prompted us to generate an alternative loss-of-function mouse model to mimic the enamel defects in ARS. Hmgn2 alters Amel expression by interacting with Pitx2 during amelogenesis, and that the overexpression of Hmgn2 in vivo could recapitulate the effects of Pitx2 loss-of-function, with such animals serving as a model for further studies of ARS dental defects.
RESULTS
PITX2 binds to the distal promoter of Amelogenin
Expression of the Amel gene is highly specific, being restricted to the ameloblasts of the dental epithelium. Ideally, a Pitx2 loss-of-function mouse would have served as an in vivo model for the study of such defects. However, our discovery of a temporary delay in Amel expression in Pitx2 HD+/− mice corroborated the hypothesis that Pitx2 plays an important role in regulating Amel expression (Fig. 1). At E18.5, Amel expression is significantly decreased in the Pitx2 HD heterozygous mice lower incisor.
Figure 1.
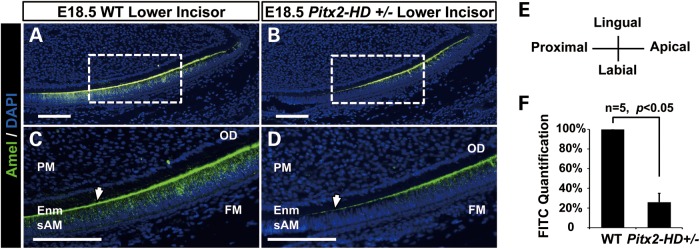
Pitx2 HD+/− mice exhibit delayed Amel expression. Series of sagittal sections of lower incisors from E18.5 wild-type and Pitx2 HD+/− littermate embryos were examined by immunofluorescence staining. Sections were stained for Amel protein, using a Alexa-488 labeled antibody. Selected wild-type (A) and Pitx2 HD+/− (B) sections are shown. (C) and (D) are magnified views of the boxed regions in (A) and (B), respectively. In all sections, 4′,6-diamidino-2-phenylindole (DAPI) staining was used to identify nuclei. Arrowheads indicate sites of decreased Amel expression in the secretory ameloblast region of the mutant mice compared with wild type. FM, follicle mesenchyme; sAM, secretory ameloblast; PM, papilla mesenchyme; Enm, enamel. Scale bars represent 100 μm. (E) The histological orientation of all developmental tooth sections. (F) Quantification of Alexa-488 signal (FITC channel) in a series of stained sections, indicating that Amel expression in Pitx2 HD+/− at E18.5 is reduced.
Our in vitro model for conducting cell-based studies is the LS-8 cell line, which was originally derived from the epithelial layer of the developing enamel organ of mice (45). LS-8 cells have been widely used for in vitro analyses because they express amelogenesis-specific factors, including Pitx2 and Amel, both of which are present at moderate levels (46–48). A reporter system was constructed to pinpoint the transcriptional regulatory events involved in the activation of the Amel promoter. Specifically, the 2.2 kb promoter upstream of the Amel gene was cloned into the luciferase-reporter plasmid pTK-luc. The endogenous activation of this reporter was tested in LS-8 cells, as well as in the odontoblast-like cell line, MDPC-23, a control that does not express epithelium-specific factors. Both the cell lines were also transfected with pTK-luc containing a minimal promoter as additional controls. Assessment of luciferase levels revealed that the 2.2 kb Amel promoter was significantly activated in LS-8 cells but not in MDPC-23 cells, demonstrating the efficiency and specificity of the reporter system (Fig. 2A).
Figure 2.

Pitx2 binds to the distal promoter of Amel. (A) Induction of a luciferase reporter of Amel 2.2 kb promoter activity. Luciferase was induced about 8-fold over endogenous levels in the LS-8 cells, but was not significantly induced in MDPC-23 cells. Structures of Amel and control reporter constructs are illustrated schematically above the plot. (B) Schematic of the Amel 2.2 kb promoter, with a predicted PITX2 binding motif indicated by the vertical arrow. Primers were designed to flank the predicted PITX2 binding site (−2171 to −1927bp; BS primers) and an upstream region that lacks a PITX2 site (−6720 to −6436 bp; NC primers). (C) PCR products from ChIP assays involving immunoprecipitation of endogenous Pitx2 in LS-8 cells. PCR products were resolved in agarose gels. Lanes 1 and 10 contain markers. PCRs for lanes 2–5 contain DNA generated using BS primers; lane 2, PCR using no template; lane 3, PCR using normal rabbit IgG-precipitated chromatin as template; lane 4, PCR using Pitx2 antibody-immunoprecipitated chromatin as template and lane 5, PCR using chromatin as input. PCR reactions represented in lanes 6–9 were performed as for the samples in lanes 2–5, but using the NC control primers instead of the BS primers. (D) Quantitation of real-time PCR performed using the ChIP conditions described in (C). The occupancy of the Amel promoter region is shown as enrichment of Pitx2 binding to chromatin in the Pitx2 antibody-immunoprecipitated DNA compared to binding to the IgG immunoprecipitated DNA.
Sequence analysis of the Amel 2.2 kb promoter revealed a bicoid motif (TAATCC), a potential Pitx2-binding site, 2152 bp upstream of transcriptional start site (Fig. 2B), and chromatin immunoprecipitation (ChIP) assays performed in LS-8 cells demonstrate that endogenous Pitx2 binds to this site on the Amel promoter. A set of primers flanking this Pitx2-binding site (BS primers, Fig. 2B) amplified the Amel promoter when either chromatin (Fig. 2C, lane 5) or Pitx2-immunoprecipitated chromatin (Fig. 2C, lane 4) was used as a template for polymerase chain reactions (PCRs), demonstrating that Pitx2 specifically bound to this bicoid motif within the Amel promoter. A PCR using normal IgG-immunoprecipitated chromatin as a control served as one negative control (Fig. 2C, lane 3), and PCR using the ChIP DNA with primers targeting an upstream region of the Amel promoter that lacks a known Pitx2-binding motif served as another (NC primers, Fig. 2B). The negative result for this second negative control indicates that Pitx2 binding to the identified site is highly specific. Quantitation of binding by a real-time PCR and using the BS primers on chromatin immunoprecipitated with an anti-Pitx2 antibody revealed a 28.7-fold enrichment in amplicon abundance relative to that in the IgG control (Fig. 2D). These results demonstrate that Pitx2 physically interacts with the Amel promoter through the bicoid motif.
PITX2 activates Amel expression
To establish whether the strong binding of Pitx2 protein to the Amel promoter is functionally relevant, we transiently co-transfected LS-8 cells with Myc-tagged PITX2 expression plasmids and the luciferase reporter plasmid containing the 2.2 kb Amel promoter. Three human PITX2 isoforms (PITX2A, PITX2B and PITX2C) differ at their N-termini and differentially regulate transcription (49). Luciferase assays demonstrated that each of the three PITX2 isoforms activated the Amel promoter, 25.5-fold, 11.1-fold and 17.4-fold, respectively (Fig. 3A). Western blotting demonstrated that the exogenous PITX2A, B and C proteins were similarly expressed in LS-8 cells (Fig. 3B). An 86 bp DNA segment (−2124–−2039 bp) that encompasses the Pitx2-binding site identified in the ChIP assays (Fig. 2) was cloned (two tandem copies) into pTK-luc. A control reporter with a similar structure was also constructed; in this case, the Pitx2-binding bicoid motif (TAATCC) was mutated to a non-binding sequence (AGGCCT). In luciferase assays conducted in LS-8 cells co-transfected with PITX2A and the control reporter construct, PITX2 transactivation was reduced to levels comparable with those in the mock control (Fig. 3C), further substantiating the specificity of binding and the requirement for the PITX2-binding site during transactivation of the Amel promoter.
Figure 3.

PITX2 activates Amel expression. (A) Luciferase reporter activity in LS-8 cells co-transfected with PITX2A/B/C expression plasmids and Amel reporter. Transactivation is shown as the mean fold activation compared with activation in the presence of empty expression plasmid (Mock). (B) Whole-cell lysates from (A) were resolved on 10% polyacrylamide gel. Overexpressed PITX2 protein isoforms were detected using antibody against the Myc tag. β-Tubulin is shown as loading control. (C) Activation of luciferase reporter whose expression is driven by a duplicated 86 bp DNA segment derived from the Amel promoter region, encompasses the Pitx2 binding site (see Fig. 2). This construct was transfected into LS-8 cells with or without PITX2A expression plasmid. In parallel, a reporter with a mutated PITX2 binding site (MUT Pitx2-BS) was transfected as control. (D) Amel mRNA isolated from LS-8 cells transiently transfected with PITX2A after 24h of culture in DMEM with 10% fetal bovine serum. Real-time PCR was performed to assess endogenous Amel expression. mRNA levels of PITX2A-overexpressing and mock-transfected cells were normalized to the levels in non-transfected cells, denoted as NC. (E) PITX2 expression in an LS-8 cell line stably transfected with PITX2A. The line was established and cultured in osteogenic medium, and mRNA from cells cultured for 7 days was subjected to real-time PCR, revealing that PITX2 expression was elevated. Non-treated control and empty viral vector control are denoted as NC and Mock, respectively. (F–N) Amel protein in LS-8 cells stably transfected with PITX2A and cultured in osteogenic medium for 0 (F, G and H), 4 (I, J and K) or 7 (L, M and N) days, as assessed by immunofluorescence. DAPI staining was used to identify nuclei. Alexa-555 (red) labels Amel protein in the cytoplasm (K and N). Scale bars represent 50 μm.
To validate the luciferase data, we examined the transfected cells described above (Fig. 3A) for endogenous Amel expression by real-time PCR. Consistent with the findings from the luciferase assays, this experiment revealed significant increases in endogenous Amel expression in the context of PITX2 isoform overexpression (Fig. 3D). In order to extend our findings to a physiological context, we cultured LS-8 cells under amelogenesis-promoting conditions, i.e., in osteogenic differentiation medium (50). An LS-8 cell line was stably transfected with PITX2A using a lentiviral system, thereby ensuring prolonged overexpression of PITX2A in osteogenic culture. Almost all cells remaining after selection with basticidin were infected and expressed PITX2A (data not shown). The average level of PITX2A transcripts were elevated about 170-fold over that in the control group (Fig. 3E). These cells were maintained in the osteogenic differentiation medium until harvested at 1, 4 and 7 days after seeding. The level of endogenous Amel was evaluated by fluorescence immunocytochemistry. From post-seeding days 4–7, the levels of Amel protein were higher in PITX2A-overexpressing (Fig. 3K and N) versus. uninfected (Fig. 3I and L) and mock-virus infected cells (Fig. 3J and M). Collectively, these data reaffirmed that PITX2 strongly activates Amel expression by binding to the 2.2 kb distal Amel promoter.
PITX2 ARS mutants fail to activate the Amel promoter
We next tested whether PITX2 mutations found in ARS patients lead to reduced, or completely abolished, Amel promoter activation. We constructed expression plasmids containing Myc-tagged PITX2A with missense mutations that have been identified in ARS patients, namely those that lead to the P64L (27), T68P (6), R90P (27), L105V (27) and N108T (27) amino acid substitutions, all of which are associated with enamel defects (25,26). Among these, P64L, T68P and R90P occur in the DNA-binding homeodomain, and L105V and N108T in the C-terminus (Fig. 4A). LS-8 cells transiently transfected with any of these five PITX2A mutant constructs showed ectopic expression within 24 h of transfection (Fig. 4B). We next sought to determine whether these proteins are capable of activating endogenous Amel expression. Real-time PCR results demonstrated that cells overexpressing any of the five PITX2A mutants had significantly lower endogenous Amel expression than counterparts transfected with wild-type PITX2A (Fig. 4C). To confirm that the observed decrease in Amel expression was due to defective transcriptional activation by the ARS-associated PITX2 mutants, luciferase assays were conducted; LS-8 cells were transfected with wild-type PITX2A or any of the five PITX2A mutants plus the luciferase reporter of Amel promoter activity. The results indicated that each of the five PITX2 mutants is impaired for transactivation of the Amel promoter, with the effect in the case of the homeodomain mutants (P64L, T68P and R90P) more severe than those of the two C-terminus mutants (Fig. 4D). Taken together, our data demonstrate that ARS-associated mutations of PITX2 are the direct cause of reduced Amel expression, and that this is due to the reduction of Amel promoter transactivation.
Figure 4.

PITX2 ARS mutants reduce activation of the Amel promoter. (A) Schematic illustration of the locations of five ARS missense mutations. Note that the P64L, T68P and R90P amino-acid substitutions are located in the homeodomain. H, homeobox; OAR, otp, aristaless, and rax-homology domain. (B) Levels of PITX2 proteins in LS-8 cells transfected with the ARS mutations. Whole-cell lysates were resolved on a 10% polyacrylamide gel, and PITX2 mutant protein was detected using an antibody against the Myc tag. β-tubulin served as the loading control. (C) Expression of endogenous Amel in PTIX2-transfected LS-8 cells. Real-time PCR revealed that compared to wild type PITX2A, all five ARS mutant forms were impaired in their ability to activate Amel. (D) Activation of the Amel promoter by the ARS PITX2 mutants. The luciferase assay confirmed that the ability of the ARS PITX2 mutant proteins to activate the Amel promoter was impaired. Luciferase activity is shown as mean-fold activation compared to activity in the context of the empty expression plasmid (Mock). All luciferase activities were normalized to β-galactose expression.
Our previous studies have shown that Pitx2 serves as a transcriptional effector of Wnt/β-catenin signaling, and that it does so by interacting with the β-catenin and Lef-1 proteins. When Pitx2 interacts with either β-catenin or Lef-1, the activation of downstream genes is synergistic (39). The luciferase assay demonstrates that this transcription mechanism applies to the Amel gene, with co-transfection of PITX2A with either β-catenin or Lef-1 synergistically enhancing activation of the Amel promoter over the enhancement by either β-catenin or Lef-1 alone (Fig. 5A). Previous studies have also shown that FoxJ1 and Dlx2 physically interact with PITX2 to modulate transcriptional activity, working through the PITX2 C-terminus and homeodomain, respectively, and that both of these binding partners are required for normal tooth and craniofacial development (32,34). Transcription by the Pitx2/FoxJ1/Dlx2 complex depends on Pitx2-mediated activation of FoxJ1 and Dlx2 expression, and one of the downstream targets of the complex is Amel. We have identified this transcriptional interaction as a key to regulating Amel expression (34). In our experiments, both FoxJ1 and Dlx2 were capable of activating the Amel promoter on their own. However, in the presence of PITX2A, the FoxJ1-mediated activation of the Amel promoter was enhanced, whereas the Dlx2-mediated activation was suppressed (Fig. 5A). Considering the importance of PITX2 as an upstream regulator of the expression of not only the tooth development-specific Amel, but also that of FoxJ1 and Dlx2, we speculated that the ARS-mutant forms of PITX2 affect gene expression in a broader spectrum of tissues. We thus tested expression from the Dlx2 and FoxJ1 promoters in cells expressing the PITX2 ARS mutant proteins. Compared with wild-type PITX2A, the ARS homeodomain mutants (P64L, T68P and R90P) again show a significant decrease in the ability to activate the Dlx2 and FoxJ1 promoters. Consistent with a previous report (51), the C-terminus mutants (L105V and N108T) showed only a mild decrease in activation (Fig. 5B, C). FoxJ1 is downstream of Pitx2 and its ability to activate Amel expression independently has been shown previously (34). We thus asked whether FoxJ1 overexpression can rescue Amel activation in the context of the PITX2 mutant proteins. Indeed, FoxJ1 overexpression fully rescued Amel activation in C-terminus mutants L105V and N108T (Fig. 5D). However, it did not in the case of the homeodomain mutant R90P (Fig. 5D).
Figure 5.

PITX2 co-factors modulate Amel promoter activation. (A) Expression plasmids containing the PITX2A, β-catenin, Lef-1, Dlx2 and FoxJ1 cDNAs were co-transfected into LS-8 cells with a luciferase reporter plasmid whose expression is driven by the Amel promoter. Luciferase activity is shown as mean-fold activation compared with that in the presence of empty mock expression plasmid. All luciferase activities were normalized to β-galactose expression. (B, C) Luciferase assay showing that ARS PITX2 mutations are impaired in their abilities to activate (B) the Dlx2 and (C) FoxJ1 promoters. (D) Luciferase reporter activity in the presence of FoxJ1. The transcriptional activity of PITX2 C-terminal mutants (L105V and N108T) is rescued by co-expression of FoxJ1. All luciferase activation values are shown as mean-fold activation compared with the empty mock expression plasmid. All luciferase activities were normalized to β-galactose expression. (E–K) Empty vector, wild type PITX2A and five ARS mutant PITX2A forms were transfected into LS-8 cells, which were fixed and subjected to immunocytochemical staining for Myc-tagged mutant PITX2. Cellular localization of ectopic PITX2A is indicated by Alexa-488 (Green). All cells were stained with DAPI to identify the nuclei. Scale bar represents 50 μm.
We next assessed the possibility that disrupted nuclear localization contributes to the decrease in transactivation by the mutant PITX2 proteins. The transfected LS-8 cells were fixed and immunostained using an Alexa-488 labeled anti-Myc-tag antibody. Like wild-type PITX2A, the P64L, T68P, L105V and N108T mutant forms all localized specifically to the nuclear compartment. In contrast, the PITX2A R90P mutant was predominantly present in the cytoplasm (Fig. 5E–K). These findings are consistent with the dramatic reduction in the ability of FoxJ1 to rescue Amel activation in the context of this mutant (Fig. 5D), because FoxJ1-mediated rescue occurs in the nuclear compartment, at the transcriptional level. Collectively, our evidence strongly suggests that PITX2 can activate Amel independently and in concert with co-factors, and that it contributes to effective modulation of Amel expression as part of a molecular network. Moreover, they suggest that the expression levels of Pitx2 co-factors, particularly FoxJ1, may play a substantial part in determining the severity of the enamel phenotypes in the context of ARS-associated mutations in the PITX2 gene.
The chromatin-remodeling protein Hmgn2 attenuates Amel expression by repressing PITX2
Hmgn2 physically interacts with PITX2A, through the homeodomain and the otp, aristaless domain (OAR) of PITX2, forming an inactive complex at PITX2-binding sites on the promoters of downstream genes (40). We thus asked whether Hmgn2 can regulate PITX2 activation of the Amel promoter. When Hmgn2 and PITX2A were co-transfected into LS-8 cells, Amel activation was repressed to ∼50% of the level measured in the presence of PITX2 alone (Fig. 6A). This repression was fully rescued by co-transfecting shRNAs specific to Hmgn2 (Fig. 6A). The efficiency and specificity of Hmgn2 knockdown were confirmed by blotting lysates from LS-8 cells transfected with the Hmgn2 construct with and without the Hmgn2-shRNA (Fig. 6B). The possibility that loss of transcriptional activity as assessed by luciferase activity might be caused by PITX2 degradation in the context of Hmgn2 overexpression was ruled out, showing that PITX2 expression was constant in all of the cells tested (Fig. 6C). The inhibitory effect of Hmgn2 on Amel expression was validated by a 2-fold increase in endogenous Amel in LS-8 cells transfected with the Hmgn2-shRNA (Fig. 6D).
Figure 6.

Hmgn2 represses PITX2A transactivation of the Amel promoter. (A) Reversible repression of the Amel promoter in the context of Hmgn2. LS-8 cells were co-transfected with Hmgn2 and PITX2A. The resulting repression of Amel luciferase reported activity was reversed by expressing an shRNA specific for Hmgn2 (siHmgn2). Reporter activation values are shown as mean fold activation compared to that obtained by co-expression with the empty expression plasmid. All luciferase activities were normalized to β-galactose expression. (B) Efficiency of shRNA-mediated silencing of ectopic Hmgn2 in LS-8 cells. A non-targeting shRNA (siControl) was tested in parallel, as a negative control. β-tubulin is serves as a loading control. (C) Expression of Pitx2A-myc in whole-cell lysates from (A). Proteins were resolved on a 14% polyacrylamide gel, and overexpressed PITX2A was detected using an antibody against the Myc tag. β-tubulin served as a loading control. (D) Quanitation of Amel expression in LS-8 cells transfected with Hmgn2 shRNA plasmids. Endogenous Amel expression is 2-fold higher than in LS-8 cells transfected with the control shRNA.
During normal tooth development, Pitx2 is expressed before Amel, and its levels remain relatively constant during later stages of development. Hmgn2, on the other hand, though broadly expressed throughout the craniofacial region, declines gradually starting at E16.5. Expression of Hmgn2 throughout the dental epithelia in both incisor and molar teeth at P0 was significantly decreased compared with E16.5 (Fig. 7A–D). We asked whether the down-regulation of Hmgn2 at this specific time point triggers and maintains Amel expression by relieving PITX2 inhibition. We further examined mRNA isolated from mouse maxilla and mandible tissue at different stages of embryogenesis for Hmgn2, Pitx2 and Amel expression, and found a strong negative correlation between the expression of Hmgn2 and Amel (Fig. 7E). This relationship began at E16.5, the stage at which the ameloblasts of the dental epithelium become fully differentiated and first start to secrete Amel. The change in Hmgn2 expression after E16.5 parallels the developmental shift to the secretion of Amel and formation of the enamel layer, indicating that it regulates Pitx2 function both spatially and temporally, and thus that it is an indirect initiator of Amel expression.
Figure 7.

Hmgn2 inhibits Pitx2 binding to the Amel promoter. Expression of Hmgn2 protein in E16.5 and P0 wild type lower incisors and molars were examined by immunofluorescence staining. Hmgn2 protein levels were assessed using an Alexa-555 (Red)-labeled antibody. (A–D) Hmgn2 staining on sections from (A) E16.5 wild type lower incisor, (B) P0 wild type lower incisor, (C) E16.5 wild type lower molars and (D) P0 wild type lower molar. White dotted lines outlined dental epithelia. LI, lower incisor; LM, lower molar; AM, ameloblast; CL, cervical loop; Scale bar represents 250 μm. (E) Levels of Hmgn2, Pitx2 and Amel transcripts as evaluated by real-time PCR, at various embryonic and neonatal time points. In the cases of the Pitx2 and Hmgn2 genes, mRNA levels were normalized to those at E13.5; in the case of Amel, mRNA levels were normalized to those at E16.5 because expression was undetectable prior to this time point. Amel levels increase dramatically after E16.5, and the fold changes were scaled down by 102 to fit the figure. (F) Hmgn2 protein levels in the LS-8 cell line following lentivirus-mediated Dox−inducible overexpression. Hmgn2 was detected using an Hmgn2 antibody, and β-tubulin served as a loading control. (G) ChIP assays performed on non-Dox induced (Dox−) and Dox induced (Dox+) cells, using the Amel promoter BS primer sets, as described in Figure 2. Lane 1 contains markers. Lanes 2 and 3 contain 10% of the amplified PCR product from input chromatin of Dox− or Dox+ cells. Lanes 4 and 5 contain amplified products from DNA fragments immunoprecipitated with an antibody against Pitx2, from Dox+ and Dox− animals, respectively. (H) Occupancy of the Amel promoter by Pitx2. ChIP products were quantitated by real-time PCR, with the amount of anti-Pitx2-immunoprecipitated DNA normalized to the amount of IgG-immunoprecipitated (Mock) DNA. Values shown are the enrichment of reactive DNA in the Dox+ relative to the Dox− samples, with the value of the latter set to 1.0. (±SEM from three independent ChIPs). (I) Levels of endogenous Amel in Dox+ versus Dox− cells, as assessed by real-time PCR. Levels from Dox+ cells were normalized to those in Dox− cells.
We previously showed that Hmgn2 inhibits Pitx2 from binding to DNA (40). To validate the relevance of this mechanism to Amel regulation, and further to elucidate in vivo the early developmental mechanism whereby Hmgn2 might influence PITX2-mediated Amel promoter activation, we established LS-8 cells that can be induced to overexpress Hmgn2, using the Tet-on lenti-virus system. The viral construct contains an Hmgn2 complementary DNA (cDNA) driven by the Tet-on promoter and an IRES-eGFP tag. After doxycycline (Dox)-mediated induction, over 50% of the LS-8 cells expressed eGFP (Supplementary Material, Fig. S1A). The Dox+ cells showed robust Hmgn2 overexpression at both the mRNA (Supplementary Material, Fig. S1B) and protein (Fig. 7F) levels. We also took a quantitative ChIP approach, using both Dox+ and Dox− cells to test the efficiency of Pitx2 binding to the Amel promoter. The DNA fragments immunoprecipitated by a Pitx2 antibody or IgG antibody (mock) were subjected to a real-time PCR, using primers (BS) flanking the Pitx2-binding site on the Amel promoter (Fig. 7G). Quantification of Amel-promoter occupancy by Pitx2, as the relative ratio of enriched Amel promoter DNA from Dox+ or Dox− cells, revealed that Pitx2 binding to the Amel promoter is significantly disrupted in the context of high Hmgn2 expression (Fig. 7H). The downstream effect of this weakened binding was lower Amel expression, as evidenced by a real-time PCR comparing mRNA levels in the two groups of cells (Fig. 7I). These results suggest that Hmgn2 is a potent inhibitor of Pitx2-mediated activation of the Amel promoter, and that it acts by decreasing the binding between the Pitx2 protein and the promoter DNA.
K14-Hmgn2 transgenic mice recapitulate ARS enamel defects
Having validated that PITX2 ARS mutants are defective for transactivation of Amel expression, we wanted to study the effects of loss of Pitx2 function on the formation of dental enamel using an animal model. Pitx2 null mice are unsuitable for this analysis due to the embryonic lethality before amelogenesis. Based on the fact that Hmgn2 inhibits Pitx2 activation of Amel expression, we reasoned that an Hmgn2 transgenic mouse might serve as a good in vivo model of Pitx2 loss of function. We engineered such a transgenic mouse line using the K14 promoter, as it drives expression during the transition, secretory and mature stages of ameloblast differentiation and has been widely used to ensure the epithelial specificity of transgene expression in various studies (52–54). At age of P17, the K14-Hmgn2 transgenic mice had shorter upper and lower incisors compared with those of wild-type mice. Notably, the surface of the all teeth in the wild type mice exhibits yellow pigmentation due to the deposition of iron salt, but the Hmgn2 transgenic teeth had transparent texture and chalky white surface, indicating lack of the enamel deposition and mineralization (Supplementary Material, Fig. S2A–H). However, the K14-Hmgn2 transgenic mice do not display defects in the differentiation of ameloblasts in the lower incisors (P4), as judged by histology and the expression patterns of various differentiation markers, including ameloblastin (Ameb), enamelin (Enam) and dentin sialophosphoprotein (Dspp) (Supplementary Material, Fig. S3). In contrast, amel immunohistochemical staining revealed that the levels in the secretory ameloblast were significantly decreased in P4 incisors and molars compared with wild type (Fig. 8A–D and Supplementary Material, Fig. S2I–P). Quantitative analysis of Amel staining confirmed a substantial decrease of the signal in the ameloblast and enamel layers in the transgenic versus wild-type animals (Fig. 8E). As expected, Hmgn2 was overexpressed in the secretory ameloblast region of the K14-Hmgn2 transgenic mice (Fig. 8F and G). Quantification of fluorescence intensity showed that the overexpressed Hmgn2 was localized mainly in the ameloblast layer rather than the mesenchymal compartment, as expected given the epithelial specificity of the K14 promoter (Fig. 8H). The residual Amel protein in the Hmgn2 transgenic mice tended to aggregate and form a patch-like enamel layer compared with wild type (Fig. 8D and Supplementary Material, Fig. S2I–P). Masson's trichrome staining at the same developmental stage revealed that the enamel layer formed by secreted Amel in the Hmgn2 transgenic mice is severely depleted with respect to both deposition and calcification compared to wild-type (Fig. 8I–N). Notably, the irregular pattern of enamel formation strongly resembles the human enamel hypoplasia observed in ARS patients (55–57). Although the exact changes in the biosynthesis and mineralization of the enamel layer that underlie this phenotype remain to be defined, the K14-Hmgn2 transgenic mouse phenotype is consistent with in vivo down-regulation of Amel expression and enamel hypogenesis. Because Pitx2 activates Dlx2 expression, we examined Dlx2 expression by real-time PCRs with mandibular and maxillary RNAs harvested from of P0 wild-type and K14-Hmgn2 transgenic mice. In accordance with Figure 5B showing the decreased Pitx2 activity leading to transcriptionally repression of Dlx2 in LS-8 cells, Dlx2 was down-regulated ∼20% in the K14-Hmgn2 transgenic craniofacial tissue (Fig. 8O). Together with the observed reduction in Amel expression in the K14-Hmgn2 transgenic mice, these findings support our in vitro data.
Figure 8.

K14-Hmgn2 transgenic mice show enamel defects. Expression of Amel protein in wild-type and K14-Hmgn2 transgenic littermates. Animals were sacrificed at P4 and series of sagittal sections of the lower incisors were examined by immunofluorescence staining. Amel protein levels were assessed using an Alexa-488 (Green)-labeled antibody. (A, B) Amel staining on sections from (A) WT and (B) K14-Hmgn2 transgenic tissue. (C, D) Higher magnifications of boxed regions in (A) and (B), respectively. Arrowheads highlight the differences in Amel protein levels and the patterns of distribution in comparable locations. (E) Quantification of Alexa-488 signal on sections from five individual pairs of heads, showing that Amel levels are significantly decreased in the context of Hmgn2 expression. (F, G) Levels and distribution of Hmgn2 protein, labeled using Alexa-555 (Red), on the labial side of incisor epithelium. In all sections, DAPI staining reveals the nuclei. (H) Quantification of the Alexa-555 signal from five individual pairs of heads, demonstrating the efficiency of K14 promoter-driven Hmgn2 expression. Epithelial specificity was demonstrated by the similarities in the levels of endogenous Hmgn2 expression in the odontoblast layer from both tissues. (I, J) Representative trichrome-stained lower incisors from WT and Hmgn2 transgenic mice, respectively. This method stains the enamel dark red and the dentin blue. (K, L) Magnified views of the left-hand boxes in (I) and (J), respectively, demonstrating that the enamel layer is lost from the secretory segment of the incisor, as indicated by black arrowheads. (M, N) Magnified views of the right-hand boxes in (I) and (J), respectively. White dotted lines indicate that the thickness of the dentin layers is similar in the two genotypes. (O) Dlx2 transcripts from the mandibles and maxillae of P0 WT and K14-Hmgn2 transgenic mice were assessed by real-time PCRs. Dlx2 was down-regulated about 20% in the K14-Hmgn2 transgenic tissue. AM, ameloblast; OD, odontoblast; PM, papilla mesenchyme; Dt, dentin; Enm, enamel. Scale bar represents 100 μm.
DISCUSSION
ARS-associated tooth anomalies include abnormally small teeth (microdontia), missing teeth (hypodontia) and brittle tooth crowns (enamel hypoplasia); patients may present only one or more of these anomalies, in a variety of combinations. It has been documented that the teeth of ARS patients can become brittle and result in tooth loss during early adulthood (7,46). Since the first documentation of dental anomalies in ARS patients decades ago, researchers have discovered many molecular mechanisms and genetic mutations that underlie certain phenotypes—both systemic and localized. However, the etiology of the enamel hypoplasia associated with ARS has not been clearly elucidated. Although this phenotype was long overlooked because it often accompanies microdontia and hypodontia, ARS is the most prominent cause of defective enamel deposition in the tooth crown, which directly leads to brittle teeth and early tooth loss (6). In some cases, enamel hypoplasia is simply caused by loss-of-function mutations in the Amel gene, resulting in Amelogenesis Imperfecta (IA). Although the dental phenotype in ARS patients is similar to that in cases of IA, it is not genetically associated with the Amel gene. Thus, the enamel hypoplasia of ARS must be due to defects in a regulator of Amel expression and or post-translational processing. Amel organizes the crystal pattern of the enamel and regulates its thickness of the enamel. Thus, its abundance is likely critical for normal enamel formation. Both the spatial and temporal patterns of Amel gene expression are under genetic control and regulated by transcription factors. Moreover, it is known that the CCAAT/enhancer-binding protein activates Amel expression, and that Msx2 transcriptionally represses Amel expression (58). In the current study, we focused on the regulation of Amel expression by PITX2, given that this transcriptional factor is specific to the region of Amel expression, i.e., the epithelium.
Pitx2 expression can be detected as early as E8.5 of mouse embryonic development (59). Even then, it is specific to the oral epithelium, and it progressively becomes restricted to the dental placodes, remaining high in the epithelium as it invaginates and, ultimately, in the dental lamina and enamel knot. By E15.5, Pitx2 expression is highest in the undifferentiated cervical loops and pre-ameloblasts. It is also detectable throughout the transitional and secretory ameloblasts, although at lower levels, but is completely lost in the protective ameloblasts (20).
The PITX2 gene was identified as a gene associated with ARS in the mid-1990s, (16). We recently delineated mechanisms underlying the hierarchical expression of major epithelial genes during tooth development (34). One involves direct activation of the Dlx2 promoter by Pitx2, after which Dlx2 binds to the Pitx2 C-terminus and thereby represses its activity. A second involves direct activation of the FoxJ1 promoter by Pitx2, and subsequent binding of FoxJ1 to the Pitx2 homeodomain, which enhances Pitx2 activity. Dlx2 and FoxJ1 also interact with one another physically, synergistically regulating their own promoters and additively regulating the Amel promoter (34). Since Pitx2 plays fundamental roles in the genetic control of tooth development, it was considered a key protein responsible for dental defects observed in ARS patients. Its homeodomain consists of 60 amino acids, including a lysine at position 50 in the third helix, and is characteristic of the bicoid-related proteins (60,61). This homeodomain selectively recognizes the 3′-CC dinucleotide adjacent to the TAAT core of the bicoid motif (62). Indeed, many previous studies showed that PITX2 recognizes this bicoid motif or its 3′ variants on distal promoters of downstream genes, including some that are involved in tooth differentiation, e.g., FoxJ1, Dlx2, Lhx6 and Dact2 (63,64), and others that participate in development of the brain (65), heart (66) and limbs (67). The current study corroborates these findings regarding the bicoid motif-mediated transactivity of PITX2, and extends them by demonstrating that these features also apply to the Amel promoter. Furthermore, we show that three PITX2 isoforms (A, B and C), which differ at their N-termini (3,65), all activate Amel expression, i.e. that PITX2 is a transcriptional activator of Amel.
Mutations in the PITX2 gene have been identified as the underlying causes of a variety of dental anomalies. The spectrum of lesions comprises missense, frame-shifting, splice-site, nonsense and inframe-duplication mutations (4). Previous reports have shown that some of the PITX2 mutations associated with ARS lead to defects in transcriptional activation, due to either disrupted nuclear localization or defective DNA binding (51,68,69). Our analysis of five mutant forms of PITX2 that are caused by missense mutations and cause enamel hypoplasia in ARS patients (P64L, T68P, R90P, L105V and N108T) reveals that each show reduced Amel activation to some degree. This is the first demonstration that PITX2 ARS mutations are the direct causes of Amel insufficiency and enamel defects, and that they do so by abrogating transactivation of the Amel promoter. We have also identified the 19th amino acid of PITX2A (R90) as being crucial to nuclear translocation of the PITX2A protein. In this context, we note that R90 lies within an arginine/lysine-rich motif, KNRRAKWRKR (88–97 amino acids), which may contain a novel nuclear localization signal (70). A previous study of the PITX2 R90C mutant revealed a severe decrease in transactivational activity and normal nuclear localization (51). However, our finding indicates that an arginine to proline (R90P) substitution can abolish the nuclear localization of PITX2A; this could potentially be due to the conformational rigidity of the proline. This novel finding further supports the notion that the molecular basis of tooth anomalies in ARS is an inability of PITX2 to activate genes involved in tooth morphogenesis.
We have also extended our knowledge of normal tooth development by investigating the involvement of the chromatin remodeling protein Hmgn2 during late stages of this process. Hmgn2 belongs to a non-histone family of chromosome-binding proteins that unfold the higher-order chromatin structure (71). It does not have consensus-binding motif on chromatin, but alters the local structure of DNA or chromatin by inducing a conformation that facilitates the binding of specific regulatory factors, including homeobox domain-containing transcription factors (43,72). The expression of Hmgn2 is reduced in adult tissues (73), and in a variety of tissues it correlates with the expression of Pitx2 (19,73). We have shown that when the Wnt signaling pathway is inactive and the levels of nuclear β-catenin are reduced, Hmgn2 binds to and inactivates PITX2 transcriptional activity (40). The inactivation of PITX2 by Hmgn2 would tightly control the transcriptional activity of PITX2. Thus, Hmgn2 is considered a major regulator of the timing of early embryonic development in the mouse (74). During tooth development, Hmgn2 and Amel expression in the dental epithelium are negatively correlated. The divergence starts at E16.5, which coincides with the full differentiation of ameloblast cells in the dental epithelium and secretion of Amel proteins. The change in Hmgn2 expression after E16.5 parallels the developmental shift in Amel secretion, and formation of the enamel layer. Although we have focused on the implications of Amel expression in this study, based on the dynamic expression patterns of Hmgn2 in other tissues, we speculate that it is a more general regulator of Pitx2 function, and active in other developmental processes. Based on the mechanism that we have identified, we hypothesized that increased Hmgn2 expression would lead to Pitx2 loss of function. Indeed, our histological data demonstrated that K14-Hmgn2 transgenic mice suffer from insufficient Amel expression and enamel hypoplasia, mimicking the ARS phenotype in humans. Hence, we have generated a novel model for studying reduced PITX2 activity during amelogenesis. The Hmgn2 transgenic mouse model will likely also be valuable in studying ARS phenotypes in other organs, with its expression driven by appropriate promoters.
Our conclusions are summarized in the schematic models shown in Figure 9. The upper panel depicts normal amelogenesis during tooth development. As embryonic development progresses from E16.5 onward, Hmgn2 levels decrease (red arrowhead), leading to de-repression of Pitx2 and, consequently, activation of Amel expression (green arrowhead). The latter involves both direct transcriptional activation and alteration of the transcriptional hierarchy involving FoxJ1 and Dlx2, which modulate Amel expression and lead to normal enamel deposition and mineralization. Wnt/β-catenin signaling positively regulates this process, synergizing with PITX2 to enhance transcriptional activation. The lower panel illustrates mechanisms that may account for the defective tooth development in ARS patients. In the case of the ARS-associated PITX2 mutants, both the direct activation of Amel expression and indirect modulation through FoxJ1 and Dlx2 are perturbed, leading to a decrease in Amel expression and, ultimately, to a hypoplastic enamel layer. The lower panel also represents tooth development in the K14-Hmgn2 transgenic mice, in which Pitx2 function is inhibited by constitutive high-level Hmgn2 expression in the dental epithelium. Thus, these mice serve as a Pitx2 loss-of-function model and phenocopy the enamel hypoplasia observed in ARS patients.
Figure 9.
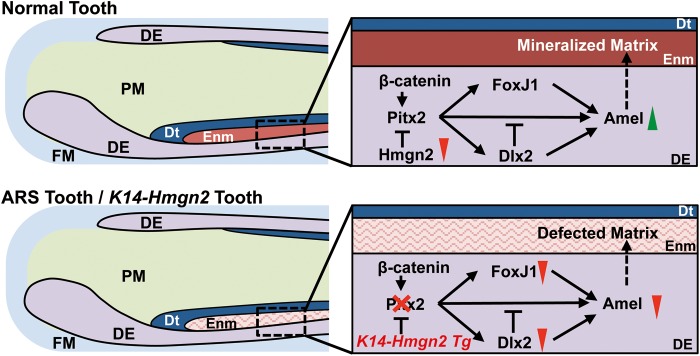
ARS enamel defect model. (Upper panel) Schematic illustration of normal tooth development. During normal amelogenesis, the level of Hmgn2 expression decreases, leading to de-repression of Pitx2 and, consequently, activation of Amel expression, by both direct transcriptional activation and alteration of the transcriptional hierarchy that modulates Amel expression. The outcome is normal enamel deposition and mineralization. (Lower panel) Schematic illustration of the model for tooth development in ARS patients and in the context of Hmgn2 overexpression. When PITX2 loses its transcriptional function due to ARS mutations, both direct and indirect activation of Amel expression are perturbed, leading to decreased enamel deposition, and thus to a hypoplastic enamel layer. The K14-Hmgn2 transgenic mice, in which the high Hmgn2 levels lead to Pitx2 inhibition that mimics Pitx2 loss-of-function, phenocopy of the enamel hypoplasia observed in ARS patients. Red triangles, downregulation of expression; green triangles, upregulation of expression; DE, dental epithelium; FM, follicle mesenchyme; PM, papilla mesenchyme; Dt, dentin; Enm, enamel.
In summary, our data support several conclusions. (1) During normal tooth development, Pitx2 activates Amel expression by both directly activating transcription programs and altering the transcriptional hierarchy that orchestrates Amel expression. (2) ARS-associated PITX2 mutations lead to decreased Amel expression (by both the direct and indirect mechanisms), leading to decreases in Amel deposition and enamel development, and thus to a hypoplastic enamel layer. (3) The K14-Hmgn2 transgenic mouse, which features enamel hypoplasia similar to that in human patients with ARS, is an excellent tool for studying the molecular etiology of ARS in the craniofacial region, as a more viable and subtle substitute for the Pitx2 knockout models which suffer from early lethality. We have uncovered a novel pathway that may provide for innovative treatments for ARS patients and people with tooth developmental defects. Treatments could involve manipulating several parts of the gene network to alleviate the dental problems.
MATERIALS AND METHODS
Mouse strain breeding
All animals were housed in the Program of Animal Resources of the University of Iowa, and were handled in accordance with the principles and procedure of the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the University of Iowa IACUC guidelines. The Pitx2 HD knock-out mouse strain was obtained from Dr James Martin (22). The K14-Hmgn2 transgenic mouse line was generated by inserting the Hmgn2 gene adjacent to the K14 promoter, as previously done for PITX2C (35).
Histology, fluorescent immunohistochemistry
Murine embryos and pups were used for histology and fluorescence immunohistochemistry (FIHC). Samples were fixed in 4% paraformaldehyde, dehydrated and embedded in paraffin. Sections were cut to 7 μm thickness, and for each sample multiple sections were stained by standard hematoxylin and eEosin staining to assess sample quality. Sections to be used for fluorescence immunohistochemistry were rehydrated and treated with 10 mm sodium citrate solution for 15 min at a slow boil for antigen retrieval. Subsequently, sections were incubated with 10% goat serum-phosphate buffer saline-tween (PBST) for 30 min at room temperature, followed by overnight incubation at 4°C with an antibody (diluted 1:500) against one of the following proteins: Amel (Santa Cruz), Hmgn2 (Millipore), Ameb (Santa Cruz), Enml (Santa Cruz) or Dspp (Santa Cruz). After the incubation, the slides were treated with Alexa-488 (FITC channel)- or Alexa-555 (Cy3 channel)-labeled secondary antibody (Invitrogen) at a concentration of 1:500 for 30 min. Each antibody incubation was followed by three to six PBST (phosphate-buffered saline with 0.05% Tween-20) washes. Nuclear counter staining was performed by applying 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting solution after the final wash (Vector Laboratories). Fluorescence signals from specific channels were quantified using imaging software (Nikon), and are presented as normalized mean intensity ± SEM. For trichrome staining (using a modified Masson's protocol), sections were first stained with azocarmine for 1 h, and then with Orange G and Aniline Blue for 2 h, as previously described (75).
Fluorescence immunocytochemistry
Cells were seeded on glass coverslips 24 h prior to fixation. Coverslips were then incubated in ice-cold acetone for 5 min at 4°C. Fixed cells were washed twice with PBST (5 min each). Subsequently, the coverslips were incubated in 10% normal goat serum-PBST for 30 min at room temperature, and then in 1:500 diluted Amel antibody (Santa Cruz) or Myc-tag antibody (Cell signaling) at 4°C overnight. Then cells were rinsed with PBST three times, 10 min each, and were incubated with Alexa-555- or Alexa-488-labeled secondary antibody (Invitrogen) for 30 min at 37°C. Finally, the cells were washed with PBST three times, 10 min each, and counter stained using mounting solution containing DAPI.
Expression, RNAi and luciferase reporter constructs
A pcDNA-3.1-MycHisC (Invitrogen) plasmid was used to express endogenous wild-type PITX2A, PITX2B, PITX2C, FoxJ1, Dlx2, β-catenin S37A, Lef-1 and Hmgn2, as previously described (34,40). ARS mutants PITX2A P64L, T68P, R90P, L105V and N108T were generated by PCR-mediated site-mutagenesis. Luciferase reporters were generated by inserting Dlx2, FoxJ1 or Amel promoter DNA fragments into pTK-Luc vectors, as previously described (34). The Hmgn2 shRNA targets the 5′-TCTGCGAGGTTGTCTGCTA-3′ sequence of the mRNA. This shRNA was cloned into pSilencer 4.1 (Life Technologies), as previously described (40). siControl was supplied by LifeTechnologies and was designed to avoid sequence similarity with all mouse mRNAs. The Pitx2a lentivirus construct was obtained from Open Biosystems. This plasmid contains a Pitx2a cDNA following a CMV promoter, an IRES/eGFP tag and a blasticidin-resistant gene for positive selection. The Hmgn2 Tet-on inducible lentivirus construct was cloned by inserting an Hmgn2 cDNA into the Tet-on virus backbone (ClonTech), which has an IRES/eGFP after the cDNA insertion site. All the cloned constructs were confirmed by DNA sequencing. All plasmids used for transfection were purified by double-banding in CsCI.
Cell culture, transfections, luciferase assays and lentiviral infection
LS-8 and MDPC-23 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 5% bovine growth serum and penicillin/streptomycin, and transfected by electroporation. Cells were fed and seeded in 60 mm dishes 24 h prior to transient transfection. Cells were resuspended in PBS and mixed with 2.5 μg of expression plasmid, 5 μg of reporter plasmid and 0.2 μg of SV-40 β-galactosidase plasmid. Transfection was performed by electroporation at 380 v and 950 mF (Gene Pulser XL, Bio-Rad), or using the Fugene HD (Promega) transfection reagent. Transfected cells were incubated in 60 mm culture dishes, for 24 h unless otherwise indicated, and fed with 10% FBS and DMEM. Following lysis, assays for reporter activity (luciferase assay, Promega) as well as for protein content (Bradford assay, Bio-Rad) were carried out. β-Galactosidase was measured using the Galacto-Light Plus reagents (Tropix Inc.) as an internal normalizer. For each assay, all luciferase activity was normalized to the mean value of the first experimental group, and is shown as mean ± SEM. Lentivirus was produced in HEK293FT cells transfected with lentivirus plasmids using Fugene HD (Promega). Virus was obtained by collecting culture medium after 24 h and filtering out the virus. For lentivirus infection, LS-8 cells were seeded into 6 cm dishes at 20% confluence. Virus was added immediately after seeding and cultured for 1 week; the medium was changed every day. Amel production was enhanced by culturing Pitx2a-overexpressing LS-8 cells in osteogenic differentiation medium (BGJb medium supplemented with 20% horse serum, 10% chick-embryo extract and 0.9 mm ascorbic acid).
Western blot assays
Approximately 25 μg of cell lysate was analyzed on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels. Following electrophoresis, the protein was transferred to polyvinylidene difluoride membrane filters (Millipore), immunoblotted and detected using specific antibodies and ECL reagents from Amersham Biosciences. The following polyclonal antibodies were used to detect the proteins: anti-β-tubulin (Santa Cruz Biotechnology), anti-Myc (Invitrogen) and Hmgn2 (Millipore).
ChIP assay
The ChIP assays were performed as previously described using the ChIP assay kit (Upstate) with the following modifications (34). LS-8 cells were plated in 60-mm dishes and fed 24 h prior to the experiment. Cells were cross-linked by applying 1% formaldehyde for 10 min at 37°C. All PCR reactions were carried out at an annealing temperature of 60°C. Specific primer sets for amplifying the Pitx2-binding site in the Amel promoter were as follows: sense: 5′-GACTGCCTTTTAGTTCCATTCTC-3′ and antisense: 5′-TCTGTGATCCATATTTACACACCTG-3′. All PCR products were evaluated on a 2% agarose gel in TBE for the expected size (244 bp) and were confirmed by sequencing. Controls included PCR including primers but no chromatin, and immunoprecipitation using normal rabbit IgG instead of the specific primary antibody. Additionally, we carried out PCRs with control primers amplifying Amel promoter elements upstream of the putative Pitx2-binding site, as follows: sense: 5′-CAGATCTTATTTGCAGCCTGA-3′ and antisense: 5′-AAAAGACATCTGCCCTCTTCT-3′. The expected product size was 283 bp. The primary antibody used in this assay was polyclonal rabbit Pitx2 antibody (Capra Science). A quantitative real-time PCR was performed using the same primers and annealing temperature. Identical amounts of immunoprecipitated DNA were loaded as template. Relative promoter occupancy was calculated by comparing the abundance of the immunoprecipitated DNA between experimental groups, after normalizing the abundance of the immunoprecipitated DNA with that of the IgG DNA. All products of standard and real-time PCR were confirmed by sequencing.
Real-time PCR assays
Total RNA was isolated from cells or tissue using miRNeasy mini kit (Qiagen). cDNAs were reverse transcribed using oligo (dT) primers according to the manufacturer's instructions (iScript Select cDNA Synthesis Kit, Bio-Rad). cDNA levels were normalized to those obtained using primers to β-actin (5′- GCCTTCCTTCTTGGGTATG-3′ and 5′- ACCACCAGACAGCACTGTG-3′). Primer sequences for Amel, Pitx2, Dlx2 and Hmgn2 are available upon request. All Ct numbers were below 30 cycles. PCR products were examined by melting curve analysis and the sequences were confirmed.
Statistical analysis
All quantified results are presented as mean ± SEM, and with an n-value indicating the number of biological repeats. A two-tailed unpaired Student's t test and either one- or two-way ANOVA were used to determine statistical significance.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the National Institutes of Health, grant DE13941 and DE18885.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Amendt laboratory for helpful discussions and Christine Blaumueller for editorial expertise.
REFERENCES
- 1.Shields M.B., Buckley E., Klintworth G.K., Thresher R. Axenfeld–Rieger syndrome. A spectrum of developmental disorders. Survey Ophthal. 1985;29:387–409. doi: 10.1016/0039-6257(85)90205-x. doi:10.1016/0039-6257(85)90205-X. [DOI] [PubMed] [Google Scholar]
- 2.Amendt B.A. The Molecular Mechanisms of Axenfeld–Rieger Syndrome. Springer, New York, NY, USA; 2005. [Google Scholar]
- 3.Amendt B., Semina E., Alward W. Rieger syndrome: a clinical, molecular, and biochemical analysis. Cell Mol. Life Sci. 2000;57:1652–1666. doi: 10.1007/PL00000647. doi:10.1007/PL00000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lines M.A., Kozlowski K., Walter M.A. Molecular genetics of Axenfeld–Rieger malformations. Hum. Mol. Genet. 2002;11:1177–1187. doi: 10.1093/hmg/11.10.1177. doi:10.1093/hmg/11.10.1177. [DOI] [PubMed] [Google Scholar]
- 5.Fitch N., Kaback M. The Axenfeld syndrome and the Rieger syndrome. J. Med. Genet. 1978;15:30–34. doi: 10.1136/jmg.15.1.30. doi:10.1136/jmg.15.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semina E.V., Reiter R., Leysens N.J., Alward W.L.M., Small K.W., Datson N.A., Siegel-Bartelt J., Bierke-Nelson D., Bitoun P., Zabel B.U. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat. Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. doi:10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 7.O'Dwyer E., Jones D. Dental anomalies in Axenfeld–Rieger syndrome. Int. J. Paediat. Dent. 2005;15:459–463. doi: 10.1111/j.1365-263X.2005.00639.x. doi:10.1111/j.1365-263X.2005.00639.x. [DOI] [PubMed] [Google Scholar]
- 8.Gadhia K., McDonald S., Arkutu N., Malik K. Amelogenesis imperfecta: an introduction. Br. Dent. J. 2012;212:377–379. doi: 10.1038/sj.bdj.2012.314. doi:10.1038/sj.bdj.2012.314. [DOI] [PubMed] [Google Scholar]
- 9.Fincham A., Moradian-Oldak J., Simmer J. The structural biology of the developing dental enamel matrix. J. Struct. Biol. 1999;126:270–299. doi: 10.1006/jsbi.1999.4130. doi:10.1006/jsbi.1999.4130. [DOI] [PubMed] [Google Scholar]
- 10.Snead M.L., Lau E.C., Zeichner-David M., Fincham A.G., Woo S.L., Slavkin H.C. DNA Sequence for cloned cDNA for murine amelogenin reveal the amino acid sequence for enamel-specific protein. Biochem. Biophys. Res. Comm. 1985;129:812–818. doi: 10.1016/0006-291x(85)91964-3. doi:10.1016/0006-291X(85)91964-3. [DOI] [PubMed] [Google Scholar]
- 11.Nakahori Y., Takenaka O., Nakagome Y. A human XY homologous region encodes ‘amelogenin. Genomics. 1991;9:264–269. doi: 10.1016/0888-7543(91)90251-9. doi:10.1016/0888-7543(91)90251-9. [DOI] [PubMed] [Google Scholar]
- 12.Snead M.L. Amelogenin protein exhibits a modular design: implications for form and function. Connect. Tissue Res. 2003;44:47–51. [PubMed] [Google Scholar]
- 13.Dressler S., Meyer-Marcotty P., Weisschuh N., Jablonski-Momeni A., Pieper K., Gramer G., Gramer E. Dental and craniofacial anomalies associated with Axenfeld–Rieger syndrome with PITX2 mutation. Case Rep. Med. 2010 doi: 10.1155/2010/621984. 2010, http://dx.doi.org/10.1155/2010/621984 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mears A.J., Mirzayans F., Gould D.B., Pearce W.G., Walter M.A. Autosomal dominant iridogoniodysgenesis anomaly maps to 6p25. Am. J. Hum. Genet. 1996;59:1321. [PMC free article] [PubMed] [Google Scholar]
- 15.Nishimura D.Y., Swiderski R.E., Alward W.L., Searby C.C., Patil S.R., Bennet S.R., Kanis A.B., Gastier J.M., Stone E.M., Sheffield V.C. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 1998;19:140–147. doi: 10.1038/493. doi:10.1038/493. [DOI] [PubMed] [Google Scholar]
- 16.Murray J.C., Bennett S.R., Kwitek A.E., Small K.W., Schinzel A., Alward W.L., Weber J.L., Bell G.I., Buetow K.H. Linkage of Rieger syndrome to the region of the epidermal growth factor gene on chromosome 4. Nat. Genet. 1992;2:46–49. doi: 10.1038/ng0992-46. doi:10.1038/ng0992-46. [DOI] [PubMed] [Google Scholar]
- 17.Datson N.A., Semina E., Van Staalduinen A., Dauwerse H.G., Meershoek E.J., Heus J.J., Frants R.R., Den Dunnen J., Murray J.C., Van Ommen G. Closing in on the Rieger syndrome gene on 4q25: mapping translocation breakpoints within a 50-kb region. Am. J. Hum. Genet. 1996;59:1297. [PMC free article] [PubMed] [Google Scholar]
- 18.Mirzayans F., Gould D.B., Heon E., Billingsley G.D., Cheung J.C., Mears A.J., Walter M.A. Axenfeld–Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. EJHG. 2000;8:71. doi: 10.1038/sj.ejhg.5200354. doi:10.1038/sj.ejhg.5200354. [DOI] [PubMed] [Google Scholar]
- 19.Hjalt T.A., Semina E.V., Amendt B.A., Murray J.C. The Pitx2 protein in mouse development. Dev. Dyn. 2000;218:195–200. doi: 10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. doi:10.1002/(SICI)1097-0177(200005)218:1<195::AID-DVDY17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 20.Mucchielli M.-L., Mitsiadis T.A., Raffo S., Brunet J.-F., Proust J.-P., Goridis C. Mouse Otlx2/RIEG expression in the odontogenic epithelium precedes tooth initiation and requires mesenchyme-derived signals for its maintenance. Dev. Biol. 1997;189:275–284. doi: 10.1006/dbio.1997.8672. doi:10.1006/dbio.1997.8672. [DOI] [PubMed] [Google Scholar]
- 21.Lin C.R., Kioussi C., O'Connell S., Briata P., Szeto D., Liu F., Izpisúa-Belmonte J.C., Rosenfeld M.G. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401:279–281. doi: 10.1038/45803. doi:10.1038/45803. [DOI] [PubMed] [Google Scholar]
- 22.Lu M.-F., Pressman C., Dyer R., Johnson R.L., Martin J.F. Function of Rieger syndrome gene in left–right asymmetry and craniofacial development. Nature. 1999;401:276–278. doi: 10.1038/45797. doi:10.1038/45797. [DOI] [PubMed] [Google Scholar]
- 23.Pispa J., Thesleff I. Mechanisms of ectodermal organogenesis. Dev. Biol. 2003;262:195–205. doi: 10.1016/s0012-1606(03)00325-7. doi:10.1016/S0012-1606(03)00325-7. [DOI] [PubMed] [Google Scholar]
- 24.Tucker A., Sharpe P. The cutting-edge of mammalian development; how the embryo makes teeth. Nat. Rev. Genet. 2004;5:499–508. doi: 10.1038/nrg1380. doi:10.1038/nrg1380. [DOI] [PubMed] [Google Scholar]
- 25.Weisschuh N., Dressler P., Schuettauf F., Wolf C., Wissinger B., Gramer E. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld–Rieger malformations. Invest. Ophthal. Vis. Sci. 2006;47:3846–3852. doi: 10.1167/iovs.06-0343. doi:10.1167/ioversus06-0343. [DOI] [PubMed] [Google Scholar]
- 26.Perveen R., Lloyd I.C., Clayton–Smith J., Churchill A., van Heyningen V., Hanson I., Taylor D., McKeown C., Super M., Kerr B. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations. Invest. Ophthal. Vis. Sci. 2000;41:2456–2460. [PubMed] [Google Scholar]
- 27.Phillips J.C. Four novel mutations in the PITX2 gene in patients with Axenfeld–Rieger syndrome. Ophthal. Res. 2002;34:324–326. doi: 10.1159/000065602. doi:10.1159/000065602. [DOI] [PubMed] [Google Scholar]
- 28.Gibson C.W. The amelogenin proteins and enamel development in humans and mice. J. Oral Biosci. 2011;53:248–256. doi: 10.2330/joralbiosci.53.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright J.T., Hart T.C., Hart P.S., Simmons D., Suggs C., Daley B., Simmer J., Hu J., Bartlett J.D., Li Y., et al. Human and mouse enamel phenotypes resulting from mutation or altered expression of AMEL, ENAM, MMP20 and KLK4. Cells Tissues Organs. 2009;189:224–229. doi: 10.1159/000151378. doi:10.1159/000151378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson C.W. Regulation of amelogenin gene expression. Crit. Rev. Eukaryot. Gene Expr. 1999;9:45–57. [PubMed] [Google Scholar]
- 31.Thomas B.L., Liu J.K., Rubenstein J., Sharpe P.T. Independent regulation of Dlx2 expression in the epithelium and mesenchyme of the first branchial arch. Development. 2000;127:217–224. doi: 10.1242/dev.127.2.217. [DOI] [PubMed] [Google Scholar]
- 32.Lézot F., Thomas B., Greene S.R., Hotton D., Yuan Z.A., Castaneda B., Bolaños A., Depew M., Sharpe P., Gibson C.W. Physiological implications of DLX homeoproteins in enamel formation. J. Cell. Physiol. 2008;216:688–697. doi: 10.1002/jcp.21448. doi:10.1002/jcp.21448. [DOI] [PubMed] [Google Scholar]
- 33.Blatt E.N., Yan X.H., Wuerffel M.K., Hamilos D.L., Brody S.L. Forkhead transcription factor HFH-4 expression is temporally related to ciliogenesis. Am. J. Respir. Cell Mol. Biol. 1999;21:168. doi: 10.1165/ajrcmb.21.2.3691. doi:10.1165/ajrcmb.21.2.3691. [DOI] [PubMed] [Google Scholar]
- 34.Venugopalan S.R., Li X., Amen M.A., Florez S., Gutierrez D., Cao H., Wang J., Amendt B.A. Hierarchical interactions of homeodomain and forkhead transcription factors in regulating odontogenic gene expression. J. Biol. Chem. 2011;286:21372–21383. doi: 10.1074/jbc.M111.252031. doi:10.1074/jbc.M111.252031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venugopalan S.R., Amen M.A., Wang J., Wong L., Cavender A.C., D'souza R.N., Akerlund M., Brody S.L., Hjalt T.A., Amendt B.A. Novel expression and transcriptional regulation of FoxJ1 during oro-facial morphogenesis. Hum. Mol. Genet. 2008;17:3643–3654. doi: 10.1093/hmg/ddn258. doi:10.1093/hmg/ddn258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu F., Chu E.Y., Watt B., Zhang Y., Gallant N.M., Andl T., Yang S.H., Lu M.-M., Piccolo S., Schmidt-Ullrich R. Wnt/β-catenin signaling directs multiple stages of tooth morphogenesis. Dev. Biol. 2008;313:210–224. doi: 10.1016/j.ydbio.2007.10.016. doi:10.1016/j.ydbio.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J., Lan Y., Baek J.-A., Gao Y., Jiang R. Wnt/beta-catenin signaling plays an essential role in activation of odontogenic mesenchyme during early tooth development. Dev. Biol. 2009;334:174–185. doi: 10.1016/j.ydbio.2009.07.015. doi:10.1016/j.ydbio.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amen M., Liu X., Vadlamudi U., Elizondo G., Diamond E., Engelhardt J.F., Amendt B.A. PITX2 And β-catenin interactions regulate Lef-1 isoform expression. Mol. Cell. Biol. 2007;27:7560–7573. doi: 10.1128/MCB.00315-07. doi:10.1128/MCB.00315-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vadlamudi U., Espinoza H.M., Ganga M., Martin D.M., Liu X., Engelhardt J.F., Amendt B.A. PITX2, β-catenin, and LEF-1 interact to synergistically regulate the LEF-1 promoter. J. Cell Sci. 2005;118:1129–1137. doi: 10.1242/jcs.01706. doi:10.1242/jcs.01706. [DOI] [PubMed] [Google Scholar]
- 40.Amen M., Espinoza H.M., Cox C., Liang X., Wang J., Link T.M.E., Brennan R.G., Martin J.F., Amendt B.A. Chromatin-associated HMG-17 is a major regulator of homeodomain transcription factor activity modulated by Wnt/beta-catenin signaling. Nucleic Acids Res. 2008;36:462–476. doi: 10.1093/nar/gkm1047. doi:10.1093/nar/gkm1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phair R.D., Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–608. doi: 10.1038/35007077. doi:10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 42.Catez F., Yang H., Tracey K.J., Reeves R., Misteli T., Bustin M. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. doi:10.1128/MCB.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hock R., Furusawa T., Ueda T., Bustin M. HMG Chromosomal proteins in development and disease. Trends Cell Biol. 2007;17:72–79. doi: 10.1016/j.tcb.2006.12.001. doi:10.1016/j.tcb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin C.R., Kioussi C., O'Connell S., Briata P., Szeto D., Liu F., Izpisua-Belmonte J.C., Rosenfeld M.G. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401:279–282. doi: 10.1038/45803. doi:10.1038/45803. [DOI] [PubMed] [Google Scholar]
- 45.Chen L., Couwenhoven R., Hsu D., Luo W., Snead M. Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch. Oral Biol. 1992;37:771–778. doi: 10.1016/0003-9969(92)90110-t. doi:10.1016/0003-9969(92)90110-T. [DOI] [PubMed] [Google Scholar]
- 46.Dhamija S., Liu Y., Yamada Y., Snead M.L., Krebsbach P.H. Cloning and characterization of the murine ameloblastin promoter. J. Biol. Chem. 1999;274:20738–20743. doi: 10.1074/jbc.274.29.20738. doi:10.1074/jbc.274.29.20738. [DOI] [PubMed] [Google Scholar]
- 47.Green P.D., Hjalt T.A., Kirk D.E., Sutherland L.B., Thomas B.L., Sharpe P.T., Snead M.L., Murray J.C., Russo A.F., Amendt B.A. Antagonistic regulation of Dlx2 expression by PITX2 and Msx2: implications for tooth development. Gene Expr. 2001;9:265–281. doi: 10.3727/000000001783992515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jheon A.H., Mostowfi P., Snead M.L., Ihrie R.A., Sone E., Pramparo T., Attardi L.D., Klein O.D. PERP Regulates enamel formation via effects on cell–cell adhesion and gene expression. J. Cell Sci. 2011;124:745–754. doi: 10.1242/jcs.078071. doi:10.1242/jcs.078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cox C.J., Espinoza H.M., McWilliams B., Chappell K., Morton L., Hjalt T.A., Semina E.V., Amendt B.A. Differential regulation of gene expression by PITX2 isoforms. J. Biol. Chem. 2002;277:25001–25010. doi: 10.1074/jbc.M201737200. doi:10.1074/jbc.M201737200. [DOI] [PubMed] [Google Scholar]
- 50.Thesleff I. Differentiation of odontogenic tissues in organ culture. Eur. J. Oral Sci. 1976;84:353–356. doi: 10.1111/j.1600-0722.1976.tb00503.x. doi:10.1111/j.1600-0722.1976.tb00503.x. [DOI] [PubMed] [Google Scholar]
- 51.Footz T., Idrees F., Acharya M., Kozlowski K., Walter M.A. Analysis of mutations of the PITX2 transcription factor found in patients with Axenfeld–Rieger syndrome. Invest. Ophthal. Vis. Sci. 2009;50:2599–2606. doi: 10.1167/iovs.08-3251. doi:10.1167/ioversus08-3251. [DOI] [PubMed] [Google Scholar]
- 52.Plikus M.V., Zeichner-David M., Mayer J.A., Reyna J., Bringas P., Thewissen J., Snead M.L., Chai Y., Chuong C.M. Morphoregulation of teeth: modulating the number, size, shape and differentiation by tuning Bmp activity. Evol. Dev. 2005;7:440–457. doi: 10.1111/j.1525-142X.2005.05048.x. doi:10.1111/j.1525-142X.2005.05048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andl C.D., Mizushima T., Nakagawa H., Oyama K., Harada H., Chruma K., Herlyn M., Rustgi A.K. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J. Biol. Chem. 2003;278:1824–1830. doi: 10.1074/jbc.M209148200. doi:10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 54.Andl T., Ahn K., Kairo A., Chu E.Y., Wine-Lee L., Reddy S.T., Croft N.J., Cebra-Thomas J.A., Metzger D., Chambon P. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development. 2004;131:2257–2268. doi: 10.1242/dev.01125. doi:10.1242/dev.01125. [DOI] [PubMed] [Google Scholar]
- 55.Drum M., Kaiser-Kupfer M., Guckes A., Roberts M. Oral manifestations of the Rieger syndrome: report of case. J. Am. Dent. Assoc. 1985;110:343–346. doi: 10.14219/jada.archive.1985.0324. [DOI] [PubMed] [Google Scholar]
- 56.Idrees F., Bloch-Zupan A., Free S.L., Vaideanu D., Thompson P.J., Ashley P., Brice G., Rutland P., Bitner-Glindzicz M., Khaw P.T. A novel homeobox mutation in the PITX2 gene in a family with Axenfeld–Rieger syndrome associated with brain, ocular, and dental phenotypes. Am. J. Med. Genet. Part B: Neuropsyc. Genet. 2006;141:184–191. doi: 10.1002/ajmg.b.30237. doi:10.1002/ajmg.b.30237. [DOI] [PubMed] [Google Scholar]
- 57.Brooks J.K., Coccaro P.J., Zarbin M.A. The Rieger anomaly concomitant with multiple dental, craniofacial, and somatic midline anomalies and short stature. Oral Surg. Oral Med. Oral Path. 1989;68:717–724. doi: 10.1016/0030-4220(89)90161-8. doi:10.1016/0030-4220(89)90161-8. [DOI] [PubMed] [Google Scholar]
- 58.Bei M., Stowell S., Maas R. Msx2 controls ameloblast terminal differentiation. Dev. Dyn. 2004;231:758–765. doi: 10.1002/dvdy.20182. doi:10.1002/dvdy.20182. [DOI] [PubMed] [Google Scholar]
- 59.St Amand T.R., Zhang Y., Semina E.V., Zhao X., Hu Y., Nguyen L., Murray J.C., Chen Y. Antagonistic signals between BMP4 and FGF8 define the expression of Pitx1 and Pitx2 in mouse tooth-forming anlage. Dev. Biol. 2000;217:323–332. doi: 10.1006/dbio.1999.9547. doi:10.1006/dbio.1999.9547. [DOI] [PubMed] [Google Scholar]
- 60.Simeone A., Acampora D., Mallamaci A., Stornaiuolo A., D'Apice M.R., Nigro V., Boncinelli E. A vertebrate gene related to orthodenticle contains a homeodomain of the bicoid class and demarcates anterior neuroectoderm in the gastrulating mouse embryo. EMBO J. 1993;12:2735. doi: 10.1002/j.1460-2075.1993.tb05935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanes S.D., Brent R. DNA Specificity of the bicoid activator protein is determined by homeodomain recognition helix residue 9. Cell. 1989;57:1275–1283. doi: 10.1016/0092-8674(89)90063-9. doi:10.1016/0092-8674(89)90063-9. [DOI] [PubMed] [Google Scholar]
- 62.Gehring W.J., Qian Y.Q., Billeter M., Furukubo-Tokunaga K., Schier A.F., Resendez-Perez D., Affolter M., Otting G., Wüthrich K. Homeodomain-DNA recognition. Cell. 1994;78:211. doi: 10.1016/0092-8674(94)90292-5. doi:10.1016/0092-8674(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Z., Gutierrez D., Li X., Bidlack F., Cao H., Wang J., Andrade K., Margolis H.C., Amendt B.A. The LIM homeodomain transcription factor LHX6: a transcriptional repressor that interacts with pituitary homeobox 2 (pitx2) to regulate odontogenesis. J. Biol. Chem. 2013;288:2485–2500. doi: 10.1074/jbc.M112.402933. doi:10.1074/jbc.M112.402933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li X., Florez S., Wang J., Cao H., Amendt B.A. Dact2 represses PITX2 transcriptional activation and cell proliferation through Wnt/beta-catenin signaling during odontogenesis. PloS One. 2013;8:e54868. doi: 10.1371/journal.pone.0054868. doi:10.1371/journal.pone.0054868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smidt M.P., Cox J.J., Schaick H.S.v., Coolen M., Schepers J., Kleij A.M., Burbach J.P.H. Analysis of three Ptx2 splice variants on transcriptional activity and differential expression pattern in the brain. J. Neurochem. 2000;75:1818–1825. doi: 10.1046/j.1471-4159.2000.0751818.x. doi:10.1046/j.1471-4159.2000.0751818.x. [DOI] [PubMed] [Google Scholar]
- 66.Toro R., Saadi I., Kuburas A., Nemer M., Russo A.F. Cell-specific activation of the atrial natriuretic factor promoter by PITX2 and MEF2A. J. Biol. Chem. 2004;279:52087–52094. doi: 10.1074/jbc.M404802200. doi:10.1074/jbc.M404802200. [DOI] [PubMed] [Google Scholar]
- 67.L'Honoré A., Ouimette J.-F., Lavertu-Jolin M., Drouin J. Pitx2 defines alternate pathways acting through myoD during limb and somitic myogenesis. Development. 2010;137:3847–3856. doi: 10.1242/dev.053421. doi:10.1242/dev.053421. [DOI] [PubMed] [Google Scholar]
- 68.Amendt B.A., Sutherland L.B., Semina E.V., Russo A.F. The molecular basis of Rieger syndrome: analysis of Pitx2 homeodomain protein activities. J. Biol. Chem. 1998;273:20066–20072. doi: 10.1074/jbc.273.32.20066. doi:10.1074/jbc.273.32.20066. [DOI] [PubMed] [Google Scholar]
- 69.Kozlowski K., Walter M.A. Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum. Mol. Genet. 2000;9:2131–2139. doi: 10.1093/hmg/9.14.2131. doi:10.1093/hmg/9.14.2131. [DOI] [PubMed] [Google Scholar]
- 70.Dingwall C., Laskey R.A. Nuclear targeting sequences—a consensus? Trends Biochem. Sci. 1991;16:478. doi: 10.1016/0968-0004(91)90184-w. doi:10.1016/0968-0004(91)90184-W. [DOI] [PubMed] [Google Scholar]
- 71.Herrera J.E., Sakaguchi K., Bergel M., Trieschmann L., Nakatani Y., Bustin M. Specific acetylation of chromosomal protein HMG-17 by PCAF alters its interaction with nucleosomes. Mol. Cell. Biol. 1999;19:3466–3473. doi: 10.1128/mcb.19.5.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shirakawa H., Herrera J.E., Bustin M., Postnikov Y. Targeting of high mobility group-14/-17 proteins in chromatin is independent of DNA sequence. J. Biol. Chem. 2000;275:37937–37944. doi: 10.1074/jbc.M000989200. doi:10.1074/jbc.M000989200. [DOI] [PubMed] [Google Scholar]
- 73.Lehtonen S., Olkkonen V.M., Stapleton M., Zerial M., Lehtonen E. HMG-17, a chromosomal non-histone protein, shows developmental regulation during organogenesis. Internat. J. Dev. Biol. 1998;42:775. [PubMed] [Google Scholar]
- 74.Mohamed O.A., Bustin M., Clarke H.J. High-mobility group proteins 14 and 17 maintain the timing of early embryonic development in the mouse. Dev. Biol. 2001;229:237–249. doi: 10.1006/dbio.2000.9942. doi:10.1006/dbio.2000.9942. [DOI] [PubMed] [Google Scholar]
- 75.Cao H., Wang J., Li X., Florez S., Huang Z., Venugopalan S.R., Elangovan S., Skobe Z., Margolis H.C., Martin J.F., et al. MicroRNAs play a critical role in tooth development. J. Dent. Res. 2010;89:779–784. doi: 10.1177/0022034510369304. doi:10.1177/0022034510369304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.