

Abstract
As a continuation of our efforts directed towards the development of anti-diabetic agents from natural sources, piplartine was isolated from Piper chaba, and was found to inhibit recombinant human ALR2 with an IC50 of 160 µM. To improve the efficacy, a series of analogues have been synthesized by modification of the styryl/aromatic and heterocyclic ring functionalities of this natural product lead. All the derivatives were tested for their ALR2 inhibitory activity, and results indicated that adducts 3c, 3e and 2j prepared by the Michael addition of piplartine with indole derivatives displayed potent ARI activity, while the other compounds displayed varying degrees of inhibition. The active compounds were also capable of preventing sorbitol accumulation in human red blood cells.
Keywords: Piper chaba, Piplartine, Chemical modification, Aldose reductase inhibition, Michael addition, Baylis–Hillman reaction
1. Introduction
According to the latest WHO estimates, approximately 200 million people worldwide are suffering from diabetes. This will increase to at least 350 million by the year 2025, which could have a severe impact on human health [1]. Prolonged exposure to uncontrolled chronic hyperglycaemia in diabetes which can lead to various complications of cardiovascular, renal, neurological and visual systems is the main cause of morbidity and mortality in diabetic patients. Although mechanisms leading to diabetic complications are not completely understood, many biochemical pathways associated with hyperglycaemia have been implicated [2]. Among these, the polyol pathway has been extensively studied. Aldose reductase (ALR2) is the rate-limiting enzyme of the polyol pathway and reduces excess glucose to sorbitol. Among human aldo-keto reductases (AKRs), ALR2 is unique in its ability to catalyse the NADPH-dependent conversion of glucose to sorbitol (Schemes 1 and 2). Under euglycaemic conditions, ALR2 plays a minor role in glucose metabolism. During diabetes, its contribution is significantly enhanced and it converts excess glucose to sorbitol in insulin independent tissues like nerve, lens, retina and kidney [3,4]. Osmotic stress due to accumulation of high concentrations of sorbitol is postulated to be a major factor in the development of diabetic complications such as neuropathy, nephropathy, retinopathy and cataract. The inhibitory effects of various synthetically and naturally derived compounds on ALR2 in vitro and in different animal models suggest that ALR2 could be a potential target in controlling diabetic complications.
Scheme 1.
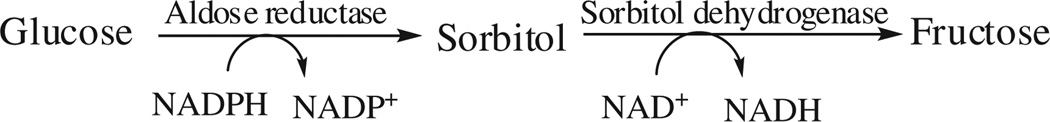
Polyol pathway.
Scheme 2.

Miachael addition reaction of piplartine with various indoles.
Two chemical classes of ALR2 inhibitors (ARI) have also been tested under Phase II clinical trials (Fig. 1). While carboxylic acid inhibitors (such as zopolrestat, ponalrestat and tolerestat) have shown poor tissue permeability and were not very potent in vivo, spiroimide (spirohydantoin) inhibitors (like sorbinil) penetrate tissues more efficiently but many have caused skin reactions and liver toxicity [5–7]. Although strict glycaemic control is expected to control or prevent diabetic complications; most individuals with diabetes rarely achieve consistent euglycaemia. Hence, agents that can substantially delay or prevent the onset and development of diabetic complications, irrespective of glycaemic control, would offer many advantages. In principle, ARI can be included in this category.
Fig. 1.

Some known ALR2 inhibitors.
The past decade has witnessed an increasing interest in search of plant based lead compounds for the development of new pharmaceuticals [8]. Hence, libraries designed and synthesized around the basic structure of such compounds have better chance of displaying desirable biological and pharmacological properties. During the past ten years, great deal of our efforts has been devoted to the phytochemical investigations of the medicinal plants of the genus Piper of Piperaceae family, which has resulted in a series of structurally intriguing alkaloids with potential biological activities [9]. As part of our continuing efforts, we have focused our attention on piperaceous amides, a well established family of natural products, which have attracted global attention for several decades because of their wide range of biological activities [10]. In our preliminary investigation of the piper species for its ALR2 inhibition property (Table 1), the chloroform extract of Piper chaba was found to have significant activity. Therefore, activity guided separation resulted in the isolation of several known compounds (see Supporting information) including piplartine (1), which showed moderate activity against ALR2. On careful review at specific structural similarities of apparently diverse ARI and piplartine (1), it was noticed that the imide functionality (–CO–NH–CO–) is common in most of the known standard drugs (Fig.1) including piplartine, and we also predicted that imide functionality may be able to contribute the activity of ARI. Therefore, this drug-like small molecule natural product is an ideal structural template for the synthesis of a series of analogues in order to explore their structure–activity relationships (SAR), thus affording the information for further lead optimization of this class of compounds as potential ARI. Our work in this area has focused primarily on identifying derivatives with significantly improved efficacy. We describe here our synthetic approaches to piplartine analogues together with their in vitro biological activity. We also report herein the SAR of this compound class as inhibitors of ALR2 and docking studies.
Table 1.
IC50 values for extracts of both Piper chaba and P. nigrum.
| S. No | Piper species | Extract | IC50 (µg/mL)a |
|---|---|---|---|
| 1 | P. chaba | Hexane | 63 |
| 2 | P. chaba | Chloroform | 22.5 |
| 3 | P. chaba | Methanol | 344 |
| 4 | P. chaba | aq. Methanol | 84 |
| 5 | P. nigrum | Hexane | 168 |
| 6 | P. nigrum | Chloroform | 80 |
| 7 | P. nigrum | Methanol | 390 |
Data are average of four experimental values.
2. Results and discussion
2.1. Chemistry
In view of the fact that the imide functionality is essential for ARI activity, we focused on the synthesis of new analogues, by exploiting the other functional moieties (aromatic/styryl and heterocyclic rings) (Fig. 2). In this context, we have designed analogues of piplartine by introducing various substituents around the aromatic/styryl and heterocyclic moieties and keeping the imide moiety intact (Fig. 2). Each structural moiety can be modified independently in order to facilitate the systematic refinement of the search for increasingly effective analogues of piplartine. It is also envisioned that analogues with the variations in the aromatic or cinnamyl ring as head group region and modification in heterocyclic unit may be rewarding.
Fig. 2.

Key structural moieties of piplartine and design of piplartine derivatives.
2.1.1. Michael addition reaction of piplartine with indole
Our initial medicinal chemistry efforts around the lead molecule, piplartine (1) demonstrated by Michael addition reaction with various substituted indoles (Scheme 3). As shown in (Fig. 2), piplartine contains two double bonds (trans and cis) which act as Michael acceptors and indoles act as Michael donors. The Michael adducts formed by this reaction, 3-substituted indoles, have been received much attention from organic chemists because of their wide range of applications in cancer therapy [11,12]. In accordance with the importance of compounds possessing this skeleton, a number of methods have been developed for its construction [13]. Initially, we have investigated the Michael addition reaction of piplartine with 3.0 equiv of 5-nitro indole was carried out in presence of variety of Lewis and Bronsted acids (Table S1, see Supporting information). To our delight, molecular iodine provided the highest yields (Table S1, see Supporting information). By using the solvent acetonitrile (Table S2, see Supporting information), the adduct (4) was observed, while methyl ester of 3,4,5-trimethoxycinnamicacid (5) was formed when methanol was used as a solvent, and in both cases the products were isolated. The best outcome was obtained with 10 mol % of iodine in dichloroethane at reflux conditions. Iodine-catalysed Michael addition reaction has been reported on α,β-unsaturated esters, sulphones, nitro olefins [14]. The role of iodine in this reaction can be attributed to its mild Lewis acid ability, which enhances both the nucleophilicity of the indole and electrophilicity of the piplartine via enol forms. The catalytic activity of Lewis acids like iodine mainly relies on their coordinating character to assemble both Michael donors and acceptors on their coordination surface. Having optimized these reaction conditions, a wide range of indoles were investigated for the Michael addition reaction and the results are summarized in Table S3 (see Supporting information). The reaction resulted in mono-adducts via trans double bond and di-adducts via trans and cis double bonds. All mono-adducts from trans were isolated in good yields except for the entries 4, 5 (Table S3, see Supporting information). The effect of temperature and volume of solvent also played an important role in this reaction. At 30–80 °C, reaction resulted in high yield of mono-adduct, and low yields of di-adduct (observed in entry 9, Table S3, see Supporting information). While at 50–100 °C, the reaction yielded both mono- and di-adducts equally, at 85–120 °C high yields of di-adduct and low yield of mono-adduct were formed. High volume of solvent leads to formation of mono-adduct with high yield whereas low volume of solvent leads to formation of di-adduct with high yield and medium volume of solvent leads to equal quantities of both mono- and di-adducts.
Scheme 3.

Synthesis of piplartine.
The order of reactivity of 5-substiuted indoles, 1-substituted indoles and 2-substituted indoles were observed to be OMe < Me < I = Br < F < NO2, Benzyl < Me and Phenyl < Me respectively. From the above data, it can be inferred that, reactivity of indole with piplartine increases with increase in the electron donating group at 1st and 2nd position and electron withdrawing group at 5th position. Among all the Michael donors (substituted indoles) 5-NO2 indole was highly reactive towards the Michael acceptor (piplartine) and all products (2i and 3i) were isolated (Table S3, Entry 9, see Supporting information) after reaction for 3–48 h, achieving 80% of completion. The enantio and diastereo-selectivity was determined using HPLC. The mono-adduct (2g) formed a racemic mixture with the ratio of 1:1, as analysed by HPLC (column: Chiral pak IA 250 × 4.6 mm, 5 µm, Flow rate: 1.0 mL/min, 225 nm, PDA detector) elution with 15% isopropanol in hexane, di-adduct (3e) formed as diastereomers with the ratio of 1:1, as analysed by HPLC (column: YMC silica 150 × 4.6 mm, Flow rate: 1.0 mL/min) elution with 4% isopropanol in hexane.
2.1.2. Modification of the aromatic ring/styryl ring
To facilitate the modification on the phenyl/styryl ring, we initially developed a new synthetic strategy for piplartine. As outlined in Scheme 4, synthesis started from the trimethoxy cinnamaldehyde (6), which was subjected to the C2-Wittig olefination reaction to yield trimethoxy ethyl cinnamate (7) [15]. Hydrolysis of 7 in the presence of LiOH followed by condensation with 9 in the presence of n-BuLi/pivalyl chloride in THF afforded the piplartine in good yields (65%) [16]. The key intermediate 9 was prepared in a two step sequence starting from homoallylamine 10, which is condensed with acryl chloride to give 11, subsequent ring closing metathesis (RCM) reaction of 11 using Grubbs II catalyst [17].
Scheme 4.
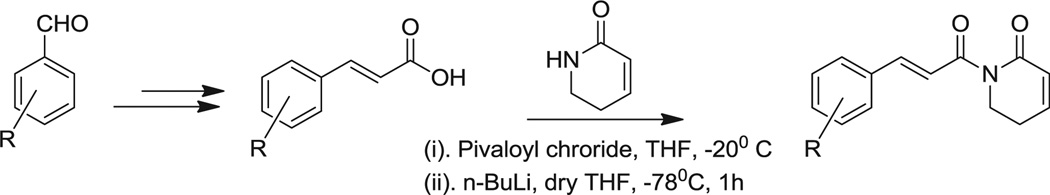
Synthesis of piplartine analogues.
Upon completion of synthesis of natural product lead, the synthetic route was utilized for the preparation of analogues with the purpose of revealing SAR (Scheme 5). In choosing the constituents of the library, we aimed to probe both size and nature of the binding pocket into which aryl side chain projects. Inspection of the literature reveals that the biology of compounds prepared using this process has not been investigated. Thus, with the retention of the other structural moieties, twenty analogues (12–31) were prepared according to the general synthetic approach illustrated in Scheme 5. While the preparation of 12–23 was straightforward, the synthesis of 24–27 involved the condensation of corresponding benzoic acid derivatives with key intermediate 9. Compounds 28 & 29 were prepared with lipophilic heterocyclic azole rings which may improve the solubility and bioavailability of the target compounds. To assess the importance of the chain, we also prepared 30 and 31, which are homologues of the piplartine by following the known procedure.
Scheme 5.

2.1.3. Baylis–Hillman reaction of piplartine (modification of heterocyclic ring)
The rationale of this study was to evaluate the influence of various substitutions around the heterocyclic ring and their substitution pattern on the ARI activity of the overall lead molecule. To develop compound libraries based on this fused heterocyclic scaffold, we considered a Baylis–Hillman reaction [18], which can introduce aromatic rings around the heterocyclic moiety. The basic skeleton of piplartine consists of trimethoxycinnamyl (TMC) moiety, attached to a six membered heterocyclic moiety which contains a double bond at the 3rd position. Thus, the target compounds 32a–32j were prepared through one step reaction of piplartine with wide variety of aldehydes using DABCO in catalytic amount at room temperature in 1,4-dioxane (Scheme 6).
Scheme 6.

In addition to these compounds, we also prepared the few miscellaneous derivatives through the condensation of trimethoxy cinnamic acid with the corresponding amines afforded the piplartine derivatives (33–43) in good yields (Scheme 7) [19]. The crude products were purified by flash chromatography using a SP-1 (Biotage) purification system with dichloromethane and methanol mixture (1%–50% gradient condition). Purity and identity were verified for all compounds by HPLC/MS and 1H NMR. The purity and NMR data of final compounds are available in the Supporting information. All purified products were dissolved in DMSO to a standard concentration of 10 mM for biological testing.
Scheme 7.

2.2. Biological activity
The major objective of this program has been to identify highly potent, and efficacious ALR2 inhibitors for treatment of chronic diabetic complications. We have previously reported ARI activity contained in a few spice/dietary sources including Piper using in vitro, and ex vivo models [20]. This work has been further extended to test extracts of Piper. nigrum, Piper. longum, and P. chaba, for their inhibitory effects on ALR2. Among them extracts of P. chaba showed significant activity towards ALR2 (Table 1), and piplartine was isolated as the lead compound, which prompted us to generate the derivatives and screen for the ARI activity. All the analogues including piplartine were initially tested for their potency against human ALR2 using the drug sorbinil as a positive control and results from these experiments are listed in (Table 2). Compounds showing 50% inhibition (IC50) at concentrations of less than 10 µM were considered to be active and were further evaluated for their potential on sorbitol accumulation in human red blood cells (RBC) under high glucose conditions. It is interesting to note that compared to the parent compound piplartine, most of the derivatives synthesized displayed potent ARI activity, higher than the standard, sorbinil. Among the test compounds, piplartine derivatives prepared from the Michael addition reaction with indoles, displayed best activity. From these, adducts 3c and 3e exhibited the highest ARI activity with an IC50 value of 4 µM followed by 2j, 2g and 3d with IC50 values of 8, 15 and 40 µM, respectively (Fig. 3; Table 2). Of these, the molecules 3c, 3e and 2j showed a significantly higher inhibition of ALR2 when compared to well known ARIs, quercetin and sorbinil, which inhibited the human recombinant ALR2 with an IC50 value of 40 and 8 µM, respectively.
Table 2.
IC50a values for piplartine and its analogues.
| Compound | IC50 (µM) | Compound | IC50 (µM) | Compound | IC50 (µM) |
|---|---|---|---|---|---|
| 1 | 160 | 4 | 72 | 32b | >500 |
| 2a | >500 | 5 | >500 | 32c | >500 |
| 3a | 80 | 12 | >500 | 32d | >500 |
| 2b | 100 | 13 | 400 | 32e | >500 |
| 3b | >500 | 14 | >500 | 32f | >500 |
| 2c | 8 | 15 | 350 | 32g | >500 |
| 3c | 4 | 16 | >500 | 32h | >500 |
| 2db | – | 17 | >500 | 32i | >500 |
| 3d | 40 | 18 | 350 | 32j | >500 |
| 2eb | – | 19 | 300 | 33 | >500 |
| 3e | 4 | 20 | >500 | 34 | >500 |
| 2f | >500 | 21 | 350 | 35 | >500 |
| 3f | >500 | 22 | >500 | 36 | >500 |
| 2g | 15 | 23 | 50 | 37 | >500 |
| 3g | >500 | 24 | 350 | 38 | >500 |
| 2h | >500 | 25 | >500 | 39 | >500 |
| 3h | >500 | 26 | 400 | 40 | >500 |
| 2i | >500 | 27 | 300 | 41 | >500 |
| 3i | 100 | 28 | >500 | 42 | >500 |
| 2j | 8 | 29 | 500 | 43 | >500 |
| 3j | >500 | 30 | 500 | Sorbinil | 8 |
| 2k | 100 | 31 | 150 | Quercetin | 40 |
| 3k | 100 | 32a | >500 |
Data are average of four experimental values.
2d & 2e were not formed during the reaction.
Fig. 3.

Inhibition of human recombinant ALR2 by 3c, 3e, 2j compounds. Representative inhibition plots for 3c, 3e and 2j against recombinant human ALR2 in vitro. IC50 value was calculated by linear regression analysis of log inhibitor concentration versus percentage of inhibition. ALR2 activity in the absence of inhibitors was considered as 100%. Data were presented in Table 2 are average of four experimental values.
In addition to lens, activation of ALR2 in RBC, retina, nerve and kidney cells, leads to the accumulation of sorbitol [21]. We have also found a direct correlation between erythrocyte ALR2 and sorbitol levels [22]. Therefore, we assessed accumulation of sorbitol in RBC under high glucose conditions (ex vivo) to understand the significance of in vitro inhibition of ALR2 by these compounds, particularly their effect on osmotic stress. In vitro incubation of RBC with 55 mM glucose resulted in the accumulation of sorbitol three to four folds higher than the control (Fig. 4). Incubation of RBC in the presence of piplartine analogues under high glucose conditions lead to reduction in the accumulation of intracellular sorbitol. Though degree of inhibition varied according to the IC50 values of different analogues, on average there was 40–50% reduction with the concentrations equal to their IC50 value of the analogues. These results indicate the significance of ARI potential of these analogues in terms of preventing the accumulation of intracellular sorbitol. Hence, we believe that these compounds, particularly, 3c, 3e and 2j might be useful for the treatment and/or prevention of diabetic complications.
Fig. 4.

Effect of 3c, 3e and 2j on sorbitol accumulation in RBC under high glucose conditions. In panel 3c the bars represent sorbitol levels in RBC under normal glucose concentration (5.5 mM) (control RBC; bar 1) and under high glucose (55 mM) conditions in the absence (bar 2) and presence of 1.4, 5.0,10 and 20 µM 3c compound (bars 3–6, respectively). In panel 3e the bars represent sorbitol levels in RBC under normal (5.5 mM) glucose concentration (control RBC; bar 1) and under high glucose (55 mM) conditions in the absence (bar 2) and presence of 2, 4 and 10 µM 3e compound (bars 3–5, respectively). In panel 2j the bars represent sorbitol levels in RBC under normal (5.5 mM) glucose concentration (control RBC; bar 1) and under high glucose (55 mM) conditions in the absence (bar 2) and presence of 4, 8 and 15 µM 2j compound (bars 3–5, respectively). Data are mean ± SE (n = 6).
2.2.1. Influence on the Michael adducts
As summarized in Table 2, Michael adducts obtained by addition of indoles to piplartine significantly enhanced the inhibition of ALR2, and different substitutions at indoles also affected ARI activity. Among all adducts, di-adducts showed more potent activity than mono-adducts. It is noteworthy to point out that the hydrolysis products 4, 5 were inactive towards the enzyme inhibition. Interestingly, activity was significantly enhanced (40-folds) when Michael adducts were prepared from N-benzyl indoles (3c, IC50 = 4 µm) and N-phenyl indoles (3e, 4 µm) than N-methyl indoles. Similar results were observed with the compound 3e in which the 2nd position of the indole ring was substituted with phenyl group (3e, IC50 = 4 µm), displayed more potent ALR2 inhibition activity than with methyl group substitution (3d, IC50 = 40 µM). This may be predicted due to the orientation of the inhibitors in the active site, the benzyl and phenyl side chains reach more deeply into the active site of the enzyme than the methyl groups. In contrast to the strong substitution effects seen on the nitrogen, substituents on the aromatic ring of indole framework have little effect. Thus, methoxy group substitution on the indole ring exhibited considerable activity (2j, IC50 = 8 µm), when compared to the methyl and nitro substitutions. Among the halogens, bromine showed moderate activity (2g, IC50 = 15 µm) compared to the iodine and chlorine groups. From these results, it can be concluded that the adduct that possess an N-benzyl group and hydrophobic groups like phenyl and electron donating groups like methoxy groups on the indole ring are indispensable for significant activity.
2.2.2. Influence of structural variations at phenyl/styryl ring
Piplartine analogues with different substituents in the aromatic ring (12–31) were screened for their ARI activity, and the results indicated that a modification on the aromatic/styryl ring does not produce the significant increase in activity. Replacement of the aromatic ring with the heterocyclic rings (21–23, 28 and 29) also did not enhance the activity of the piplartine. Similarly, compounds 24–27, in which the aromatic ring is directly attached to the heterocyclic ring also did not affect ARI. Further, homologation of the chain between the aromatic ring and heterocyclic rings further led to the drastic decrease in the activity of piplartine analogues (compounds 30 and 31). Based on these observations, we conclude that double bond and methoxy substitutions at aromatic ring preferably at 3rd, 4th and 5th –positions are an essential characteristic for piplartine to impart the ARI activity.
2.2.3. Influence of structural variations at heterocyclic ring
The activity results shown in Table 2 indicated that derivatives (32a–32j) prepared through Baylis–Hillman reaction were not active against the ALR2. This indicates the ARI mechanism of piplartine regarding its reactive site to the target would be on the piperidone ring, which may be considered as the “warhead” of the molecule. It is thus postulated that piplartine may act like maleimide derivatives as a Michael acceptor [23], reacting at C-3 of the heterocyclic ring with a nucleophilic site of the target (enzyme). Thus, the C-3 position was replaced by the large phenyl group in the derivatives (32a–32j), and the resulting activity decreases significantly presumably because of decreased affinity to the target. Further, it is also observed that replacement of the piperidone ring with the heterocyclic rings did not improve the activity of the piplartine.
2.3. Molecular docking
To rationalize and identify the structural features of the active molecules, we performed molecular docking studies with piplartine and its analogues into the active site of ALR2 [24]. The 0.93 A crystal structure of human ALR2 (PDB No 1PWM) was typed using the charmM force field and amino acid residues were corrected for physiological pH, and water molecules were removed. Fidarestat, a known ALR2 inhibitor, and piplartine analogues were minimized, and docked into the active site of ALR2. High-scoring complexes were identified based on the DOCK score and GOLD SCORE fitness, and the visualized protein–ligand binding conformation (Table S4, see Supporting information). The active site of ALR2 consists of two major pockets, the so-called “anionic” pocket and the “specificity” pocket [25] and the top ranked structures were identified from molecular docking. The ligands occupied active site of ALR2 and extended towards the hydrophobic cleft or specificity pocket. Interestingly 3c and 2j revealed similar binding patterns and formed hydrogen bonds with active site residues Trp-20, Tyr-48, His-110, Trp-111, Cys-298, Leu-300, Leu-301, and nicotinamide ring, which are involved in catalysis (Fig. 5A and B). Apart from active site, binding of these inhibitors was also extended towards the flexible hydrophobic pocket through the indole linked to the N-phenyl moiety. The indole attached to the N-phenyl moiety of 3c interacted with Phe-122 and Trp-219 forming the hydrophobic interactions and hydrogen bonds with Trp-20 Pro-218, Trp-219 and Leu-300. These inhibitors did not cause opening of the hydrophobic cleft because there was no hydrophobic interaction with Trp-111. Trp-111 indole moiety was closed by the side chain of Leu-300 and the inhibition is known as closed type conformation.
Fig. 5.

Stereo views of ALR2 docked with 3c (A) and interactions of 3c with active site and specificity pocket (B). Panel A: 3c docked into active site of ALR2 and depicts its interaction with residues Trp-20, Tyr-48, His-110, Trp-111, Cys-298, Leu-300, Leu-301, and nicotinamide ring. Panel B: Interaction of amino acids in active site and specificity pocket forming the hydrophobic interactions (Phe-112 and Leu-301) and hydrogen bond (Pro-218).
The compound 3e formed hydrogen bonds with Trp-20, Val-47 and the nicotinamide ring (substrate binding site) and also formed hydrogen bonds with Leu-300, Leu-301 and Ser-302 (specificity pocket) (Fig. 6A), and the trimethoxyphenyl and indole containing phenyl moiety hydrophobic regions particularly in pi–pi stacking with Trp-219 and Phe-122. Another phenyl containing indole moiety completely occupied the active site and interacted through extensive hydrophobic attractions (Trp-20, Val-47, Tyr-48, Trp-79, His-110 and NADP) (Fig. 5B). In the specificity pocket formation of hydrogen bonds with Leu-300 was pulled upon towards amide moiety of 3e and also because of pi stacking with Phe-122, Leu-300 was away from Trp-111 thereby opening of specificity pocket. This type of inhibition is known as open type conformation which is similar to that of zopolrestat. As compared to 3c and 2j, 3e occupies both the anionic and specificity pockets with complete opposite orientation resulting in increased active site interactions and binding energy (Fig. 6A and B). We also calculated binding energies in the active site of ALR2 (Table S5, see Supporting information), and found that 3e has the highest binding energy among all piplartine analogues probably due to formation of more stable non covalent interactions with the (hydrogen bonds and hydrophobic interactions) active site of ALR2 over the sister compounds (3c and 2j). We compared the binding pattern of the 3c and 3e with fidarestat in the active site of ALR2. Fidarestat occupied only limited space in the active site pocket, but unlike 3c, 3e and 2j, it did not extend into the hydrophobic cleft (Figs. 7 and 8). The compounds 3e and 2j also extended more deeply into the active site than fidarestat, indicating the strong affinity and massive interactions of enzyme (Fig. 8). We plotted the GOLD SCORE against in vitro IC50 values of piplartine analogues and there is evidence of a correlation, considering 40 mm to be a cutoff for activity, in that we found with 2j, 3c, 3d and 3e which fall in this cutoff among the many analogues (Fig. 9).
Fig. 6.

Stereo view of ALR2 docked with 3e (A) and interactions of 3e with active site and specificity pocket (B). Panel A: 3e docked into active site of ALR2 and depicts its hydrogen bond interaction with residues Trp-20, Val-47 and nicotinamide ring (substrate binding site) and also forming hydrogen bonds with Leu-300, Leu-301 and Ser-302 (specificity pocket). Panel B: Massive hydrophobic interaction of 3e indole containing phenyl moiety with active site amino acids (Trp-20, Val-47, Tyr-48, Trp-79, His-110 and NADP+) and specificity pocket forming the hydrophobic interactions (Phe-122) and hydrogen bond (Leu-301).
Fig. 7.

Stereo view of the docked structures of ALR2 active site in presence of fidarestat, 3c and 3e.
Fig. 8.

Stereo view of the 3e overlaid with fidarestat in the active site of ALR2 depicting the binding into the deep groove of anionic pocket as compared to the fidarestat.
Fig. 9.

Prediction of binding affinity to ALR2 with GOLD SCORE fitness and measured IC50 in µM.
3. Conclusion
This study has demonstrated the synthesis, biological activity, and SAR of a series of piplartine derivatives bearing different substituents around the piplartine. Among the derivatives, 3c, 3e and 2j showed better potency towards the ALR2 inhibition as compared to well known inhibitor sorbinil. Further, these compounds inhibited the sorbitol accumulation in human RBC under ex vivo high glucose conditions thereby suggesting their membrane permeability and their potential to inhibit osmotic stress in insulin independent cells like eye lens, retina, neuron and nephrons. This has laid a solid foundation for further lead optimization of this class of compounds by a systematic chemical modification including the synthesis of water-soluble compounds to improve their overall pharmaceutical properties.
4. Experimental section
4.1. General
Optical rotations were measured using a JASCO DIP 300 digital polarimeter and at 1 mL cell at 25 °C. IR spectra were recorded on a Nicolet-740 spectrometer with KBr pellets. The NMR spectra were recorded on a Bruker FT-300 MHz spectrometer at 300 MHz for 1H and 75 MHz for 13C, respectively, using TMS as internal standard. The chemical shifts are expressed as δ values in parts per million (ppm) and the coupling constants (J) are given in hertz (Hz). Mass spectra were performed on a LC-MS/MS (Agilent Technologies 6510) Q-TOF Mass spectrometer. Column chromatography was performed with silica gel (100–200 mesh, Qing-dao Marine Chemical, Inc., Qingdao, China). Preparative HPLC was performed on a Dionex P680 equipped with PDA detector and Shimadzu PRC-ODS (K) column: Zorbax SB (C18, 9.4 × 50 mm, 5 µm), Analytical TLC was performed on precoated Merck plates (60 F254, 0.2 mm) with the solvent system EtOAc–hexane (50:50), and compounds were viewed under a UV lamp and sprayed with 10% H2SO4, followed by heating. Analytical HPLC used to determine the purity of target compounds was conducted on Silica column (250 mm × 4.6 mm, 5 µm) using two solvent systems, (a) 25% IPA in Hexane and (b) 25% ethanol in hexane, flow rate 1.0 mL/min and UV detection at 254 nm for a 25 min run. The results are expressed as retention times in minutes, and the relative content is expressed as a percentage. All the reagents were obtained from commercial vendors in appropriate grades and were used without further purification.
4.2. Plant material
The roots of the plants P. chaba, P. nigrum and P. longum were collected from Indian Medicines and Pharmaceuticals Limited (IMPCL), Uttarakhand, India. It was authenticated by Dr. K. Madhava Chetty, and a voucher specimen of P. chaba was deposited in the herbarium of the Botany Department, Sri Venkateswara University, Tirupati, Andhra Pradesh, India.
4.3. Extraction and isolation
The roots of P. chaba (3 kg) were shade dried, powdered, and extracted with hexane at boiling temperature for 72 h. The resulting hexane extract was evaporated to dryness under reduced pressure, affording syrupy residue (100 g), which exhibited the ARI activity. Active hexane extract (100 g) was subjected to column chromatography (silica gel, 100–200 mesh, eluting with hexane/ EtOAc mixtures of increasing polarity) to give three major fractions (F1–F3). Fraction F1 was purified by repeated flash chromatography on silica gel (100–200 mesh) by eluting with EtOAc/hexane (05:95) to yield 1 g of pipataline (1a), 0.7 g of pellitorine (1b), 6 g of piplartine (1), 0.1 g of piperanal (1c), 0.02 g of methyl ester of 4-hydroxy-3-methoxycinnamicacid (1d). Repeated-chromatography of the fraction F2 on silica gel with the elution of EtOAc/hexane (30:70) gave 0.4 g of piperine (1e), 0.06 g of trichostachine (1f), 0.054 g of 4,5-dihydropiperlongumine (1g) and 0.05 g of guineensine (1h), respectively. Purification of the fraction F3 was achieved by the flash chromatography with EtOAc/hexane (40:60) to give 0.030 g of flavone (1i), 0.020 g of flavone (1j), 0.050 g of brachystamide B (1k), 0.030 g of sesamin (1l) (see Supporting information for structures).
4.4. Typical experimental procedure for the Michael reaction of piplartine
To a mixture of piplartine (1 mmol) and Indole (3 mmol), Iodine (10 mol %) was added. The contents were refluxed in dichloroethane (5 mL) for an appropriate time (12–48 h) and reaction was monitored by thin-layer chromatography (TLC). After complete conversion, the solvent was evaporated, and the product was washed with hypo solution (10 mL), and then extracted with chloroform. The combined organic layers was dried over anhydrous sodium sulphate and evaporated under reduced pressure, purified by silica-gel column chromatography to afford pure product mono-adduct (2a–2k) and di-adduct (3a–3k).
4.5. Spectral chemical and physical properties of piplartine, hydrolysis products (4, 5), Michael adducts (2a–2k) and (3a–3k)
4.5.1. 5,6-Dihydro-1-((E)-3-(3,4,5-trimethoxyphenyl)acryloyl) pyridin-2(1H)-one (1)
As white needles; mp 124 °C, IR (KBr) νmax: 1660, 1670 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.44–2.52 (2H, m), 3.85 (3H, s), 3.89 (6H, s), 4.04 (2H, t, J = 6.61 Hz), 6.03 (1H, td, J = 9.6, 1.7 Hz), 6.78 (2H, s), 6.92 (1H, m), 7.41 (1H, d, J = 15.48 Hz), 7.64 (1H, d, J = 15.48 Hz). 13C NMR (75 MHz, CDCl3): δ 24.3, 41.5, 56.2 (2), 61.0, 105.5 (2), 121.0, 125.5, 130.5, 139.5, 139.9, 143.2, 145.5, 153.5, 165.5, 169.5; HRESIMS m/z 318.1349 [M+ + H], calcd for C17H19NO5 318.1336.
4.5.2. 3-(3,4,5-Trimethoxyphenyl)-3-(2-methyl-1H-indol-3-yl) propanoic acid (4)
As light indigo solid, mp: 159 °C; IR (KBr) νmax: 746, 1125, 1237, 1393, 1458, 1590, 1681, 2934 and 3394 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.37 (3H, s), 3.16–3.32 (2H, m), 3.75 (3H, s), 3.81 (3H, s) 4.74 (1H, t J = 7.93 Hz), 6.58 (2H, s), 7.02 (1H, t, J = 7.55 Hz), 7.10 (1H, t, J = 7.55 Hz), 7.26–7.30 (1H, d, J = 7.90 Hz), 7.48–7.52 (1H, d, J = 7.90 Hz), 7.82 (1H, br s). 13C NMR (75 MHz, CDCl3):δ 29.7, 37.9, 39.6 55.8, 60.7 (2), 104.6 (2), 110.4, 113.1, 119.0, 119.2, 120.8, 124.9, 134.5, 137.2, 137.5, 139.1, 153.0 (2), 185.5; ESIMS m/z C21H23NO5 [M+ + Cl]− 369.0.
4.5.3. (E)-Methyl 3-(3,4,5-trimethoxyphenyl)acrylate (5)
White solid, mp: 110 °C; 1H NMR (300 MHz, CDCl3): δ ppm 3.80 (3H, s), 3.86 (3H, s), 3.89 (6H, s), 6.30 (1H, d J = 15.86 Hz), 6.72 (2H, s), 7.57 (1H, d, J = 15.86 Hz). 13C NMR (75 MHz, CDCl3): δ 51.7, 56.4 (2), 60.6, 105.6 (2), 116.9, 129.0, 144.9, 153.8 (2), 167.0; ESIMS m/z C13H16O5 [M+ + H] 253.0.
4.5.4. 1-(3-(1H-Indol-3-yl)-3-(3,4,5-trimethoxyphenyl)propanoyl)-5,6-dihydropyridin-2(1H)-one (2a)
As a pale yellow solid, mp: 110 °C; IR (KBr) νmax: 812, 866, 929, 1037, 1157, 1200, 1248, 1344, 1444, 1496, 1536, 1610, 1653, 1740, 2857, 2921 and 3414 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.16 (2H, m), 3.74 (2H, m), 3.78 (9H, s), 3.84−3.94 (2H, m), 4.85 (1H, t, J= 7.5 Hz), 5.95 (1H, td, J = 9.6, 1.7 Hz), 6.59 (2H, s), 6.79–6.87 (1H, m), 7.02–7.09 (2H, m), 7.12–7.20 (1H, m), 7.33 (1H, d, J = 8.1 Hz), 7.55 (1H, d, J = 7.9 Hz), 8.12 (NH, br s). 13C NMR (100 MHz, CDCl3): δ 24.5, 39.6, 41.2, 44.5, 56.0 (2), 60.6, 104.7 (2), 111.0, 118.9, 119.5 (2), 121.3, 122.0 (2), 125.6, 126.6, 136.4, 139.9, 145.4 (2), 152.9, 165.9, 175.0; HRESIMS m/z 457.1745 [M+ + Na] (calcd. for C25H26N2O5 457.1734). Anal. HPLC [tR (min) (content)]: 9.50 (97.6%) from solvent a or 9.27 (95.5%) from solvent b.
4.5.5. 1-(3-(1H-Indol-3-yl)-3-(3,4,5-trimethoxyphenyl)propanoyl)-4-(1H-indol-3-yl)piperidin-2-one (3a)
As a pale yellow solid, mp: 165 °C; IR (KBr) νmax: 588, 663, 747, 818, 909, 1005, 1124, 1175, 1235, 1333, 1389, 1422, 1458, 1504, 1591, 1687, 2362, 2852, 2923 and 3361 cm–1. 1H NMR (300 MHz, CDCl3): δ ppm 1.73–2.09 (2H, m), 2.14 (1H, m), 2.58–2.75 (1H, m), 2.86–2.97 (1H, dd, J = 17.1, 5.6 Hz), 3.34–3.69 (2H, m), 3.76 (3H, s), 3.79 (6H, s), 3.85–4.03 (2H, m), 4.85 (1H, t, J = 7.7 Hz), 6.59–6.62 (2H, s), 6.69 (1H, d, J = 2.0 Hz), 6.85 (1H, d, J = 1.8 Hz), 7.04–7.14 (2H, m), 7.14–7.23 (2H, m), 7.36 (2H, m), 7.55 (2H, m), 8.02–8.13 (2H, NH, br t, J = 8.8 Hz). 13C NMR (75 MHz, CDCl3): δ 28.6, 30.0, 39.2, 40.8, 43.9, 44.7, 55.9 (2), 60.9, 105.2 (2), 110.4, 110.6, 112.0, 112.2, 118.6, 118.9, 119.1 (2), 119.3, 120.8 (2), 126.3, 127.5, 130.5, 132.1, 135.2, 135.3, 139.9, 152.8 (2), 173.4, 176.2; HRESIMS m/z 574.2320 [M+ + Na] (calcd. for C33H33N3O5 574.2312). Anal. HPLC [tR (min) (content)]: 7.49 (95.9%) from solvent a or 9.25 (95.7%) from solvent b.
4.5.6. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(1-methyl-1H-indol-3-yl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2b)
As a pale yellow solid, mp: 165°C; IR (KBr) νmax: 770, 1001, 1125, 1188, 1278, 1317, 1460, 1504, 1646, 2852 and 2923 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.10–2.23 (2H, m), 3.65–3.73 (2H, m), 3.74 (3H, s), 3.78 (3H, s), 3.80 (6H, s), 3.80–3.94 (2H, m), 4.83 (1H, t, J = 7.3 Hz), 5.92–5.98 (1H, td J = 9.8,1.5 Hz), 6.59 (2H, s), 6.78–6.87 (1H, m), 6.88 (1H, br s) 7.02–7.08 (1H, m), 7.26 (1H, d, J = 8.2 Hz), 7.53–7.57 (1H, d, J = 8.1 Hz), 7.15–7.22 (1H, m). 13C NMR (75 MHz, CDCl3): δ 24.5, 29.6, 32.6, 41.2, 45.0, 56.0 (2), 60.7, 104.9 (2), 109.0, 117.5, 118.8 (2), 119.4, 121.6, 125.5 (2), 126.1, 127.0, 136.2, 137.1, 139.9, 145.4, 152.4 (2), 165.5, 174.9. HRESIMS m/z 471.1901 [M+ + Na] (calcd. for C26H28N2O5 471.1890). Anal. HPLC [tR (min) (content)]: 8.99 (94.2%) from solvent a or 9.06 (96.8%) from solvent b.
4.5.7. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(1-methyl-1H-indol-3-yl) propanoyl)-4-(1-methyl-1H-indol-3-yl)piperidin-2-one (3b)
As a pale yellow solid, mp: 183 °C; IR (KBr) νmax: 665, 743, 819, 1011, 1126, 1175, 1233, 1326, 1375, 1422, 1464, 1505, 1590, 1689, 2852, and 2927 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.67–1.88 (1H, m), 1.97–2.10 (1H, m), 2.52–2.71 (1H, m), 2.74–2.96 (1H, m), 2.23–3.58 (2H, m), 3.63–3.97 (18H, m), 4.77 (1H, m), 6.52–6.63 (2H, m), 6.66–6.80 (1H, m), 6.82–6.95 (1H, m), 6.97–7.11 (2H, m), 7.13–7.33 (4H, m), 7.40–7.58 (2H, m).13C NMR (75 MHz, CDCl3): δ 29.7, 29.4, 30.0, 32.7, 40.1, 41.4, 43.4, 45.5, 56.2 (2), 60.2,105.6 (2), 109.0, 109.0, 116.6, 117.6, 118.7, 118.9, 119.0 (2), 119.1, 119.7, 121.7 (2), 122.1, 124.6, 126.1, 126.5, 126.5, 127.2, 136.5, 137.2, 139.9, 153.0 (2), 172.2, 175.5. HRESIMS m/z 602.2617 [M+ + Na] (calcd. for C35H37N3O5 602.2625). Anal. HPLC [tR(min) (content)]: 7.48 (96.4%) from solvent a or 6.62 (97.9%) from solvent b.
4.5.8. 1-(3-(1-Benzyl-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2c)
As a pale indigo solid, mp: 95 °C; IR (KBr) νmax: 819, 1124, 1460, 1638, 2055, 2362, 2851, 2922, 1444, 1496, 1536, 1610, 1653, 1740, 2857, 2921 and 3414 cm–1. 1H NMR (300 MHz, CDCl3): δ ppm 2.06–2.20 (2H, m), 3.64–3.74 (2H, m), 3.77 (6H, s), 3.78 (3H, s), 3.81–3.93 (2H, m), 4.86 (1H, t, J = 7.5 Hz), 5.29 (2H, s), 5.90–5.96 (1H, td, J = 9.6, 1.7 Hz), 6.57 (2H, s), 6.76–6.84 (1H, m), 6.99–7.09 (4H, m), 7.10–7.16 (1H, m), 7.19–7.31(H, m), 7.50–7.57 (1H, d, J = 7.9 Hz). 13C NMR (75 MHz, CDCl3): δ 29.6, 41.3, 45.3, 49.9, 56.0 (2), 60.0, 104.9 (2), 109.6, 118.2, 119.1, 119.6, 121.9, 125.4, 125.6, 126.5 (2), 127.4, 127.5, 128.6 (2), 136.2, 136.8, 137.7, 139.9, 145.3 (2), 152.9 (2), 165.5, 174.9. HRESIMS m/z 547.2225 [M+ + Na] (calcd. for C32H32N2O5 547.2203). Anal. HPLC [tR (min) (content)]: 4.28 (97.8%) from solvent a or 9.32 (96.7%) from solvent b.
4.5.9. 1-(3-(1-Benzyl-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-4-(1-benzyl-1H-indol-3-yl)piperidin-2-one (3c)
As a pale indigo solid, mp: 110 °C; IR (KBr) νmax: 744, 1008, 1124, 1176, 1236, 1328, 1460, 1502, 1640, 2063, 2924 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.64–1.81 (1H, m), 1.96–2.18 (1H, m), 2.48–2.65 (1H, m), 2.80–2.91 (1H, m), 3.231–3.49 (2H, m), 3.58–3.71 (2H, m), 3.75 (10H, br s), 4.79 (1H, t, J = 7.7 Hz), 5.21 (2H, s), 5.29 (2H, s), 6.54 (2H, br s), 6.93–7.13 (9H, m), 7.13–7.31 (9H, m), 7.44–7.52 (2H, d, J = 7.5 Hz). 13C NMR (75 MHz, CDCl3): δ 29.4, 29.8, 30.2, 39.9, 41.6, 43.7, 45.7, 50.0, 56.0 (2), 60.0, 105.0 (2), 109.6, 110.0, 117.5, 118.1, 118.9, 119.4 (2), 119.8 (2), 122.1, 122.3, 124.0, 125.5, 126.5 (2), 126.7 (2), 127.6, 127.7 (2), 128.8 (2), 136.5, 136.9, 137.4, 137.8, 139.4, 153.0 (2), 172.8, 175.6. HRESIMS m/z 732.3435 [M+ + H] (calcd. for C47H45N3O5 732.3432). Anal. HPLC [tR (min) (content)]: 8.31 (97.8%) from solvent a or 9.94 (97.1%) from solvent b.
4.5.10. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(2-methyl-1H-indol-3-yl) propanoyl)-4-(2-methyl-1H-indol-3-yl)piperidin-2-one (3d)
As a pale indigo solid, mp: 110 °C; IR (KBr) νmax: 600, 674, 744, 838, 921, 1006, 1125, 1175, 1244, 1330, 1424, 1459, 1505, 1590, 1691, 2361, 2932, 3396 and 3738 cm−1. 1H NMR (300 MHz, CDCl3):δ ppm 1.66–1.86 (2H, m), 2.26 (3H, d, J = 6.4 Hz), 2.41 (3H, d, J = 3.5 Hz), 2.46–2.66 (1H, m), 2.83–3.17 (1H, m), 3.18–3.57 (2H, m), 3.73 (6H, s), 3.79 (3H, s), 3.81–3.91 (1H, m), 4.01–4.21 (1H, m), 4.89 (1H, m), 6.566 (2H, s), 6.99–7.15 (4H, m), 7.18–7.33 (3H, m), 7.62 (1H, m), 7.99 (NH, br s), 8.06 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 28.5, 30.0, 31.2, 38.6, 39.2, 40.6, 43.9, 44.7, 55.9 (2), 60.6, 104.7 (2), 110.4, 110.5, 112.0, 112.5, 118.5, 119.1, 119.2, 119.3, 120.6, 126.4, 127.5, 130.5, 132.2, 135.2, 135.3, 136.0, 140.0, 140.1, 152.8 (2), 173.4, 176.1. HRESIMS m/z 602.2620 [M+ + Na] (calcd. for C35H37N3O5 602.2625). Anal. HPLC [tR (min) (content)]: 6.11 (95.2%) from solvent a or 9.70 (95.3%) from solvent b.
4.5.11. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(2-phenyl-1H-indol-3-yl) propanoyl)-4-(2-phenyl-1H-indol-3-yl)piperidin-2-one (3e)
As a pale white solid, mp: 150 °C; IR (KBr) νmax: 861, 609, 666, 700, 744, 770, 836, 921, 1006, 1125, 1175, 1242, 1320, 1422, 1455, 1501, 1592, 1687, 2845, 2928, 3056 and 3394 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.50–1.74 (1H, m), 1.78–1.96 (1H, m), 2.30–2.61 (1H, m), 2.77–3.04 (1H, m), 3.05–3.34 (2H, m), 3.43–3.65 (1H, m), 3.69 (6H, s), 3.79 (3H, s), 3.85–4.28 (2H, m), 5.04 (1H, m), 6.61 (1H, s), 6.62 (1H, s), 7.01–7.12 (1H, m), 7.12–7.23 (2H, m), 7.27–7.34 (1H, m), 7.34–7.49 (10H, m), 7.50–7.58 (2H, d, J = 7.9 Hz), 7.68–7.81 (1H, m), 7.97 (1H, s), 8.10 (NH, br s), 8.11 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 29.0, 29.6, 31.3, 37.6, 40.8, 44.4, 55.9 (2), 60.7, 104.7 (2), 105.9, 106.6, 110.9, 111.1, 111.3, 112.0, 113.2, 113.4, 119.6, 119.8, 120.5, 121.3, 122.0, 122.2, 124.9, 125.9, 126.3, 128.0, 128.1, 128.2, 128.7, 128.8, 130.3, 131.7, 132.7, 134.9, 136.1, 137.6, 140.1, 152.9 (2), 160.1, 173.2. HRESIMS m/z 742.2685 [M+ + K] (calcd. for C45H41N3O5 742.2678). Anal. HPLC [tR (min) (content)]: 4.63 (98.8%) from solvent a or 3.71 (95.1%) from solvent b.
4.5.12. 1-(3-(5-Iodo-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2f)
As a pale white solid, mp: 180 °C; IR (KBr) νmax: 590, 656, 784, 814, 878, 909, 1000, 1133, 1179, 1231, 1301, 1328, 1360, 1387, 1458, 1504, 1588, 1684, 2361, 2828, 2926, 2994, 3055, 3400 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.16–2.25 (2H, m), 3.61–3.79 (2H, m), 3.80 (9H, s), 3.81–3.95 (2H, m), 4.78 (1H, t, J = 7.7 Hz), 5.95–6.01 (1H, td, J = 9.8, 1.7 Hz), 6.56 (2H, s), 6.82–6.90 (1H, m), 7.02 (1H, br d, J = 2.2 Hz), 7.09–7.14 (1H, d, J = 8.4 Hz), 7.38–7.43 (1H, dd, J = 8.4, 1.7 Hz), 7.87 (1H, d, J = 1.3 Hz), 8.09 (NH, br s). 13C NMR (100 MHz, CDCl3): δ 24.7, 39.4, 41.4, 45.1, 55.9 (2), 61.0, 105.0 (2), 113.2, 118.3, 122.2, 125.7, 128.1, 129.2, 130.3, 130.5, 135.5, 136.2, 139.4, 145.4, 152.8 (2), 165.5, 174.8. HRESIMS m/z 599.0456 [M+ + K] (calcd. for C25H25IN2O5 599.0440). Anal. HPLC [tR (min) (content)]: 10.27 (94.9%) from solvent a or 9.31 (96.0%) from solvent b.
4.5.13. 1-(3-(5-Iodo-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-4-(5-iodo-1H-indol-3-yl)piperidin-2-one (3f)
As a pale white solid, mp 201 °C; IR (KBr) νmax: 664, 770, 877, 1002, 1124, 1175, 1222, 1307, 1387, 1421, 1458, 1505, 1591, 1686, 2361, 2851, 2923 and 3419 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.75–1.90 (1H, m), 2.08–2.20 (1H, m), 2.58–2.74 (1H, m), 2.88–3.00 (1H, dd, J = 17.3, 5.8 Hz), 3.30–3.40 (1H, m), 3.48–3.76 (3H, m), 3.82 (6H, s), 3.83 (3H, s), 3.84–3.98 (1H, m), 4.77 (1H, t, J= 7.5 Hz), 6.55 (1H, s), 6.57 (1H, s), 6.64 (1H, d, J = 1.7 Hz), 7.03 (1H, d, J = 2.0 Hz), 7.11–7.19 (2H, m), 7.39–7.49 (2H, m), 7.83–7.90 (2H, m), 8.10 (NH, br s), 8.18 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 29.5, 30.7, 39.6, 43.1, 45.4, 49.4, 56.1 (2), 60.9, 104.9 (2), 113.1, 113.3, 117.6, 118.2, 121.0, 122.3, 127.7, 128.3, 129.2, 130.4, 130.7, 135.5, 139.3, 139.6, 143.8, 144.3, 145.3, 145.8, 153.1 (2), 173.0, 175.4. HRESIMS m/z 826.0244 [M+ + Na] (calcd. for C33H31I2N3O5 826.0245). Anal. HPLC [tR (min) (content)]: 10.39 (97.6%) from solvent a or 8.39 (95.7%) from solvent b.
4.5.14. 1-(3-(5-Bromo-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2g)
As a pale white solid, mp: 178 °C; IR (KBr) νmax: 667, 770, 884, 1000, 1124, 1176, 1223, 1323, 1422, 1460, 1505, 1592, 1687, 2852, 2923, and 3430 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.18 (2H, m), 3.61–3.75 (2H, m), 3.79 (6H, s), 3.81 (3H, s), 3.84–3.94 (2H, m), 4.78 (1H, t, J = 7.5 Hz), 5.94–6.00 (1H, td, J = 9.8, 1.7 Hz), 6.56 (2H, s), 6.80–6.89 (1H, m), 7.07 (1H, d, J = 2.0 Hz), 7.02–7.23 (2H, m), 7.65 (1H, br d, J = 1.5 Hz), 8.10 (NH, br s). 13C NMR (75 MHz, CDCl3): d 24.5, 39.2, 41.3, 45.2, 56.2 (2), 60.7, 105.0 (2), 112.5, 112.7, 118.8, 121.9, 122.6, 125.0, 125.2, 125.6, 128.4, 135.0, 139.3, 145.5, 153.0 (2), 165.6, 174.6. HRESIMS m/z 535.0839 [M+ + Na] (calcd. for C25H25BrN2O5 535.0859). Anal. HPLC [tR (min) (content)]: 9.13 (95.1%) from solvent a or 6.64 (95.7%) from solvent b.
4.5.15. 1-(3-(5-Bromo-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-4-(5-bromo-1H-indol-3-yl)piperidin-2-one (3g)
As a pale white solid, mp: 210 °C; IR (KBr) νmax: 593, 636, 671, 794, 858, 882, 926, 1000, 1048, 1122, 1176, 1241, 1292, 1322, 1363, 1422, 1458, 1506, 1592, 1669, 1700, 2361, 2929, 3355 and 3737 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.74–1.89 (1H, m), 2.07–2.22 (1H, m), 2.59–2.74 (1H, m), 2.83–2.98 (1H, m), 3.27–3.40 (1H, m), 3.54–3.73 (1H, m), 3.77 (6H, s), 3.80 (3H, s), 3.82–3.98 (2H, m), 4.78 (1H, t, J = 7.3 Hz), 6.56 (2H, s), 6.68 (1H, d, J = 2.2 Hz), 6.91 (1H, d, J = 2.4 Hz), 7.19–7.30 (5H, m), 7.62–7.69 (2H, m), 8.19 (NH, br s), 8.24 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 29.6, 31.5, 39.6, 41.1, 42.9, 45.4, 56.1 (2), 60.8, 105.0 (2), 112.7, 112.8, 117.4, 117.8, 118.5, 118.6, 121.1, 121.4, 121.9, 122.5, 122.7, 125.0, 125.2, 125.3, 127.7, 128.4, 135.0, 139.9, 153.0 (2), 173.0, 175.4. HRESIMS m/z 732.0524 [M+ + Na] (calcd. for C33H31Br2N3O5 732.0523). Anal. HPLC [tR (min) (content)]: 10.39 (98.8%) from solvent a or 10.20 (97.9%) from solvent b.
4.5.16. 1-(3-(5-Fluoro-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2h)
As a pale white solid, mp: 170 °C; IR (KBr) νmax: 721, 772, 820, 1004, 1125, 1177, 1220, 1305, 1383, 1462, 1587, 1690, 2852, 2923 and 3367 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.14–2.26 (2H, m), 3.64–3.76 (2H, m), 3.79 (6H, s), 3.80 (3H, s), 3.81–3.91 (2H, m), 4.77 (1H, t, J = 7.3 Hz), 5.93–5.99 (1H, td, J = 9.6, 1.7 Hz), 6.55 (2H, s), 6.80–6.95 (2H, m), 7.12 (1H, d, J = 2.0 Hz), 7.13–7.18 (1H, m), 7.20–7.25 (1H, m), 8.05 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 24.5, 39.6, 41.2, 45.0, 56.2 (2), 60.7, 104.2, 105.0 (2), 110.3, 110.7, 111.5, 111.7, 119.2, 119.3, 123.1, 125.6, 132.9, 139.5, 145.5, 153.0 (2), 165.6, 174.6. HRESIMS m/z 491.1380 [M+ + K] (calcd. for C25H25FN2O5 599.1379). Anal. HPLC [tR (min) (content)]: 8.37 (95.0%) from solvent a or 8.12 (96.8%) from solvent b.
4.5.17. 1-(3-(5-Fluoro-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl) propanoyl)-4-(5-fluoro-1H-indol-3-yl)piperidin-2-one (3h)
As a pale white solid, mp: 180 °C; IR (KBr) νmax: 812, 866, 929, 1037, 1157, 1200, 1248, 1344, 1444, 1496, 1536, 1610, 1653, 1740, 2857, 2921 and 3414 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.76–1.93 (1H, m), 2.09–2.21 (1H, m), 2.60–2.74 (1H, m), 2.83–2.99 (1H, m), 3.27–3.42 (1H, m), 3.54–3.75 (2H, m), 3.80 (6H, s), 3.81 (3H, s), 3.83–3.98 (1H, m), 4.77 (1H, t, J = 7.17 Hz), 6.56 (1H, s), 6.57 (1H, s), 6.88–7.01 (2H, m), 7.10–7.16 (1H, m), 7.16–7.22 (2H, m), 7.23–7.25 (1H, m), 7.27–7.32 (2H, m), 8.11 (NH, br d, J = 7.5 Hz), 8.18 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 28.5, 29.5, 39.8, 41.2, 42.9, 45.3, 56.2 (2), 60.7, 103.5, 105.0 (2), 110.3, 110.7, 110.9, 111.7, 112.1, 117.8, 118.1, 119.0, 121.8, 123.1, 126.3, 127.1, 133.0, 136.3, 139.5, 153.0 (2), 156.1, 159.2, 173.1, 175.5. HRESIMS m/z 610.2127 [M+ + Na] (calcd. for C33H31F2N3O5 610.2124). Anal. HPLC [tR (min) (content)]: 9.84 (95.3%) from solvent a or 7.52 (96.1%) from solvent b.
4.5.18. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(5-nitro-1H-indol-3-yl) propanoyl)-5,6-dihydropyridin-2(1H)-one (2i)
As yellow solid, mp: 194 °C; IR (KBr) νmax: 605, 657, 741, 775, 819, 899, 1015, 1125, 1180, 1232, 1331, 1385, 1424, 1463, 1514, 1589, 1623, 1684, 2358, 2837, 2933, and 3366 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.22–2.33 (2H, m), 3.67–3.78 (2H, m), 3.80 (3H, s), 3.82 (6H, s), 3.84–3.98 (2H, m), 4.89 (1H, t, J = 7.3 Hz), 5.96–6.02 (1H, td, J = 9.8, 1.5 Hz), 6.58 (2H, s), 6.85–6.95 (1H, m), 7.24 (1H, br d, J = 1.8 Hz), 7.36 (1H, d, J = 9.0 Hz), 8.05–8.10 (1H, dd, J = 9.0, 2.0 Hz), 8.50 (NH, br s), 8.52 (1H, br d, J = 2.0 Hz). 13C NMR (75 MHz, CDCl3): δ 24.5, 39.2, 41.1, 45.9, 56.2 (2), 60.7, 104.7 (2), 111.1, 116.6, 117.8, 121.7, 124.4, 125.6, 126.1, 136.5, 139.1, 139.5, 141.5, 145.7, 153.1 (2), 165.6, 174.2. HRESIMS m/z 480.1779 [M+ + H] (calcd. for C25H25N3O7 480.1765). Anal. HPLC [tR (min) (content)]: 4.87 (97.4%) from solvent a or 9.34 (96.6%) from solvent b.
4.5.19. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(5-nitro-1H-indol-3-yl) propanoyl)-4-(5-nitro-1H-indol-3-yl)piperidin-2-one (3i)
As yellow solid, mp: 220 °C; IR (KBr) νmax: 656, 770, 819, 902, 1002, 1124, 1177, 1223, 1330, 1384, 1424, 1463, 1512, 1589, 1624, 1687, 2852, 2924 and 3342 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.95–2.08 (1H, m), 2.19–2.32 (1H, m), 2.63–2.81 (1H, m), 2.93–2.05 (1H, m), 3.50–3.60 (2H, m), 3.61–3.68 (2H, m), 3.79 (6H, s), 3.84 (3H, s), 4.02–4.15 (1H, m), 4.89 (1H, m), 6.58 (1H, s), 6.59 (1H, s), 6.76 (1H, br d, J = 2.2 Hz), 7.32–7.46 (3H, m), 8.07–8.17 (2H, m), 8.46–8.52 (3H, m), 8.69 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 28.5, 29.5, 39.8, 41.2, 42.9, 45.3, 56.2 (2), 60.7, 103.5, 105.0 (2), 110.3, 110.7, 110.9, 111.7, 112.1, 117.8, 118.1, 119.0, 121.8, 123.1, 126.3, 127.1, 133.0, 136.3, 139.5, 153.0 (2), 156.1, 159.2, 173.1, 175.5. HRESIMS m/z 664.2024 [M+ + Na] (calcd. for C33H31N5O9 664.2014). Anal. HPLC [tR (min) (content)]: 8.23 (97.8%) from solvent a or 9.72 (96.4%) from solvent b.
4.5.20. 1-(3-(5-Methoxy-1H-indol-3-yl)-3-(3,4,5-trimethoxyphenyl)propanoyl)-5,6-dihydropyridin-2(1H)-one (2j)
As a pale white solid, mp: 90 °C; IR (KBr) νmax: 668, 768, 820, 1034, 1123, 1216, 1284, 1458, 1586, 1683, 2853, 2923 and 3468 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.10–2.20 (2H, m), 3.67–3.77 (2H, m), 3.77–3.81 (12H, br s), 3.84–3.91 (2H, m), 4.79 (1H, t, J = 7.5 Hz), 5.92–5.98 (1H, td, J = 9.6, 1.7 Hz), 6.59 (2H, s), 6.81 (1H, m), 6.83 (1H, d, J = 2.26 Hz), 6.98 (1H, d, J = 2.26 Hz), 7.03 (1H, d, J = 1.7 Hz), 7.19–7.24 (1H, d, J = 8.4 Hz), 7.93 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 24.5, 39.8, 41.3, 44.9, 55.8, 56.0 (2), 60.8, 101.4, 105.0 (2), 111.6, 112.2, 118.7, 122.2, 125.6, 127.1, 131.6, 136.2, 139.7, 145.5, 152.8 (2), 153.9, 165.6, 175.5. HRESIMS m/z 465.2042 [M+ + H] (calcd. for C26H28N2O6 465.2020). Anal. HPLC [tR (min) (content)]: 16.73 (95.3%) from solvent a or 3.71 (96.2%) from solvent b.
4.5.21. 1-(3-(3,4,5-Trimethoxyphenyl)-3-(5-methyl-1H-indol-3-yl) propanoyl)-4-(5-methoxy-1H-indol-3-yl)piperidin-2-one (3j)
As a pale white solid, mp: 101 °C; IR (KBr) νmax: 765, 1123, 1216, 1459, 1638, 2851, 2923 and 3438 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.78–1.89 (1H, m), 2.09–2.19 (1H, m), 2.58–2.70 (1H, m), 2.87–2.97 (1H, dd, J = 17.3, 5.2 Hz), 3.30–3.42 (1H, m), 3.45–3.58 (1H, m), 3.60–3.73 (2H, m), 3.78 (6H, s), 3.80 (3H, s), 3.81 (3H, s), 3.86 (3H, s), 3.87–3.98 (1H, m), 4.79 (1H, t, J = 8.1 Hz), 6.60 (1H, s), 6.61 (1H, s), 6.69 (1H, br d, J = 2.4 Hz), 6.81–6.87 (12H, m), 6.95–7.00 (2H, dd, J = 7.5, 2.2 Hz), 7.10 (1H, dd, J = 2.2 Hz), 7.21–7.29 (2H, m), 7.95 (2NH, br s). 13C NMR (75 MHz, CDCl3): δ 28.7, 29.5, 40.0, 41.2, 43.0, 45.3, 55.8, 56.0, 56.2 (2), 60.8, 100.9, 101.6, 105.1 (2), 111.7, 112.0, 112.4, 115.9, 117.7, 118.5, 120.8, 122.3, 126.4, 127.2, 127.4, 131.8, 139.9, 149.9, 152.9 (2), 153.9, 154.0, 173.3, 175.8. HRESIMS m/z 612.2680 [M+ + H] (calcd for C35H37N3O7 612.2704). Anal. HPLC [tR (min) (content)]: 6.52 (94.9%) from solvent a or 7.57 (96.4%) from solvent b.
4.5.22. (5-MethylIndoleMs)1-(3-(3,4,5-trimethoxyphenyl)-3-(5-methyl-1H-indol-3-yl)propanoyl)-5,6-dihydropyridin-2(1H)-one (2k)
As a pale white solid, mp: 152 °C; IR (KBr) νmax: 594, 647, 675, 814, 847, 908, 1000, 1025, 1134, 1180, 1233, 1302, 1328, 1360, 1388, 1410, 1464, 1505, 1589, 1625, 1685, 2362, 2829, 2928, 2996, 3053, 3114, and 3385 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 2.10–2.20 (2H, m), 2.41 (3H, s), 3.64–3.77 (2H, m), 3.79 (9H, s), 3.81–3.93 (2H, m), 4.81 (1H, t, J = 7.5 Hz), 5.91–5.99 (1H, d, J = 9.6 Hz), 6.59 (2H, s), 6.79–6.87 (1H, m), 7.01 (2H, br s), 7.22 (1H, d, J = 8.3 Hz), 7.34 (1H, s), 7.94 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 24.5, 29.6, 39.6, 41.2, 45.0, 56.0 (2), 60.7, 104.9 (2), 110.7, 118.5, 118.9, 121.5, 123.7, 125.6, 126.9, 128.5, 134.7, 136.2, 139.8, 145.4, 152.9 (2), 165.4, 175.0. HRESIMS m/z 487.1634 [M+ + K] (calcd for C26H28N2O5 487.1630). Anal. HPLC [tR (min) (content)]: 15.83 (96.2%) from solvent a or 10.82 (97.4%) from solvent b.
4.5.23. (5-MethylIndoleDs)1-(3-(3,4,5-trimethoxyphenyl)-3-(5-methyl-1H-indol-3-yl)propanoyl)-4-(5-methyl-1H-indol-3-yl) piperidin-2-one (3k)
As a pale white solid, mp: 180 °C; IR (KBr) νmax: 771, 1125, 1219, 1419, 1457, 1507, 1636, 2850, 2921, 2996, 3053, 3114, and 3457 cm−1. 1H NMR (300 MHz, CDCl3): δ ppm 1.76–1.93 (1H, m), 2.00–2.10 (1H, m), 2.41 (3H, s), 2.45 (3H, s), 2.58–2.71 (1H, m), 2.87–2.97 (1H, dd, J = 17.3, 5.2 Hz), 3.31–3.40 (1H, m), 3.46–3.60 (1H, m), 3.60–3.75 (2H, m), 3.78 (3H, s), 3.81 (6H, s), 3.84–3.98 (1H, m), 4.82 (1H, t, J = 7.5 Hz), 6.60 (1H, s), 6.61 (1H, s), 6.80 (1H, br s), 6.96–7.10 (3H, m), 7.20–7.25 (2H, m), 7.28–7.37 (2H, m), 7.91 (NH, br s), 7.96 (NH, br s). 13C NMR (75 MHz, CDCl3): δ 21.5, 28.7, 29.6, 39.7, 41.2, 43.1, 45.4, 49.4, 56.1 (2), 60.7, 105.0 (2), 110.7, 117.2, 117.6, 118.4, 118.9, 119.1, 120.1, 120.3, 121.5, 121.6, 123.4, 123.7, 124.0, 126.2, 128.6, 128.8, 134.8 (2), 140.0, 152.9 (2), 165.6, 175.8. HRESIMS m/z 618.2381 [M+ + K] (calcd. for C35H37N3O5 618.2365). Anal. HPLC [tR (min) (content)]: 5.92 (96.2%) from solvent a or 10.97 (95.9%) from solvent b.
4.6. General procedure for synthesis of piplartine
4.6.1. (i). (E)-Ethyl 3-(3,4,5-trimethoxyphenyl)acrylate (7)
To a stirred solution of trimethoxy benzaldehyde (6) (1 g, 5.10 mmol) benzene (10 mL) was added 1.2 eq of phosphine ester (2.12 g, 6.12 mmol). Resultant mixture was allowed to stir at room temperature for 3 h, and reaction was monitored by TLC. The solvent was removed under vacuum to give the crude product, which was purified by flash column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (9:1) as an eluent to give pale yellow solid in 97% yield. 1H NMR (300 MHz CDCl3): δ 7.58–7.48 (1H, d, J = 15.8 Hz), 6.73–6.65 (1H, d, J = 15.8 Hz), 6.69 (2H, s), 4.20 (2H, t, J = 21.3 Hz), 3.89 (6H, s), 3.84 (3H, s), 1.29 (3H, t, J = 14.3 Hz); 13C NMR (75 MHz, CDCl3): δ 164.8, 153.5 (2C), 142.7, 139.8, 130.8, 116.5, 105.3 (2C), 62.1, 60.6, 56.1 (2C), 15.3.
4.6.2. (ii). (E)-3-(3,4,5-Trimethoxyphenyl)acrylic acid (8)
To a solution of 7 (0.500 g, 1.87 mmol) in THF/H2O (2:1,10 mL) was added LiOH (0.172 g, 7.51 mmol) at room temperature and the resultant mixture was allowed to stir for 12 h. After completion (monitored by TLC), the reaction was neutralized with 1NHCl and extracted with ethylacetate (2 × 5 mL). Organic layer was dried over anhydrous Na2SO4 and the solvent was removed under vacuum. The crude product was purified by flash column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (6:4) as an eluent to give pale yellow solid in 95% yield. 1H NMR (300 MHz, CDCl3): δ 7.61–7.49 (1H, d, J = 15.6 Hz), 6.70 (2H, s), 6.59 (1H, d, J = 15.6 Hz), 3.88 (6H, s), 3.83 (3H, s); 13C NMR (75 MHz, CDCl3): 168.1, 153.3 (2C), 141.9, 139.8, 130.6, 117.6, 105.2 (2C), 60.4, 55.9 (2C).
4.6.3. (iii). N-(But-3-enyl)acrylamide (11)
To a stirred solution of homoallylamine (10) (1 g, 14.08 mmol) in DCM (20 mL) was added Et3N (2.95 mL, 21.12 mmol) at 0 °C. To this reaction mixture, 1.2 eq of acryloylchloride (1.6 mL, 16.90 mmol) was added and the resultant mixture was allowed to stir at room temperature for 3 h. After completion of the reaction, diluted with water, and then extracted into DCM (2 × 10 mL). The solvent was removed under vacuum to give the crude product. The crude product was purified by flash column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (7:3) as an eluent to give pale yellow liquid in 80% yield. 1H NMR (300 MHz, CDCl3): δ 7.27 (1H, s), 6.25 (1H, d, J = 18.3 Hz), 6.10 (1H, d, J = 10.1 Hz), 5.84–5.67 (1H, m), 5.60 (1H, d, J = 10.1 Hz), 5.15–5.02 (2H, m), 3.39 (2H, m), 2.30 (2H, m); 13C NMR (75 MHz, CDCl3): 164.6, 135.3, 131.0, 126.2, 117.5, 39.3, 32.0.
4.6.4. 5,6-Dihydropyridin-2(1H)-one (9)
To a solution of 11 (0.125 g, 1 mmol) in anhydrous DCM (160 mL) was added Grubbs-II catalyst (42 mg, 5 mol %) and stirred at reflux for 6 h. Then it was stirred for an additional 1 h at room temperature in open air to deactivate the catalyst. The reaction mixture was filtered through celite, concentrated, and the residue was purified by column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (2:8) as an eluent to give brown liquid 70% yield. 1H NMR (300 MHz, CDCl3): δ 7.25 (1H, s), 6.51 (1H, m), 5.90 (1H, m), 3.98 (2H, m), 2.46 (2H, m); 13C NMR (75 MHz, CDCl3): 166.5, 140.9, 124.6, 42.3, 25.1.
4.6.5. Synthesis of piplartine (1)
(i) To a solution of trimethoxy cinnamic acid (8) (0.1 g, 0.420 mmol) in freshly distilled THF (2 mL) was added triethyl-amine 0.05 mL To this reaction mixture pivaloyl chloride (0.06 mL, 0.33 mmol) was added at −20 °C and reaction was allowed to stir for 45 min. After completion of the reaction (monitored by TLC), this mixture was used for the next step without purification and work up.
(ii) To a solution of 9 (0.048 g, 0.504 mmol) in freshly distilled THF (2 mL), was added 1.2 eq of n-BuLi (0.31 mL) at −78 °C under inert atmosphere conditions and the reaction was allowed to stir for 45 min and then added anhydride prepared from the above step. The resultant reaction mixture was allowed to stir for 1 h, and the reaction mixture was quenched with saturated NH4Cl (1 mL) then extracted in to ethylacetate (5 mL). The organic layer was separated washed with sat. NaCl (2 × 3 mL) and dried over anhydrous Na2SO4. The combined organic layers were evaporated to dryness to give crude product which was purified by flash column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (6:4) as eluent to give pale yellow solid piplartine 65% yield. 1H NMR (300 MHz, CDCl3): δ 8.07 (1H, d, J = 15.4 Hz), 7.36 (1H, d, J = 15.4 Hz), 7.01 (2H, s), 6.93 (1H, m), 6.02 (1H, td, J = 9.2 Hz, 3.2 Hz), 4.04 (2H, t, J = 12.8 Hz), 3.91 (6H, s), 3.89 (3H, s), 2.49 (2H, m); 13C NMR (75 MHz, CDCl3): δ 168.9, 165.6, 153.4 (2C), 145.5, 141.0, 139.6, 130.1, 123.4, 118.1, 105.7 (2C), 60.6, 56.0 (2C), 41.2, 25.1.
4.7. General procedure for the synthesis of compounds 12–31
(i) To a stirred solution of trimethoxy cinnamic acid (1 eq) in dry THF (3 mL) was added Et3N (1.5 eq) at 0 °C. To this reaction mixture pivaloyl chloride (1.1 eq) was added and allowed to stir for 45 min, reaction progress was monitored by TLC. After completion of the reaction, this mixture was used for the next step without purification.
(ii)To a stirred solution of 9 (1.1 eq) in freshly distilled THF (2 mL), was added 1.1 eq of n-BuLi under inert atmosphere at −78 °C. After being stirred for 45 min, the mixed anhydride prepared from above procedure was added and the resultant reaction mixture was allowed to stir for 1 h. After completion of the reaction, mixture was quenched with saturated NH4Cl (1 mL) then extracted in to ethyl-acetate (5 mL). The organic layer was separated washed with sat. NaCl and dried over anhydrous Na2SO4. The combined organic layers were evaporated to dryness to give crude product which was purified by flash column chromatography on silica gel (60–120 mesh) using hexane/EtOAc (7:3) as eluent to give the desired products.
4.7.1. 1-Cinnamoyl-5,6-dihydropyridin-2(1H)-one (12)
Yield 62%; brown liquid; 1H NMR (300 MHz, CDCl3): δ 7.76–7.66 (1H, d, J = 15.6 Hz), 7.61–7.44 (3H, m), 7.39–6.26 (3H, m), 6.90 (1H, m), 6.01 (1H, td, J = 9.6 Hz, J = 1.5 Hz), 4.02 (2H, t, J = 12.6 Hz), 2.42 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 168.5, 166.3, 145.0, 143.7, 135.1, 130.0, 128.7 (2C), 128.3 (2C), 126.1, 121.8, 41.5, 24.8; ESIMS: m/z 228 [M + H]+, HRESIMS m/z [M + Na]+ 250.0849 (calcd for C14H13NO2, 250.0838). Anal. HPLC [tR (min) (content)]: 5.72 (99.0%) from solvent a or 5.88 (97.5%) from solvent b.
4.7.2. (E)-1-(3-(3-(Trifluoromethyl)phenyl)acryloyl)-5,6-dihydropyridin (13)
Yield 63%; pale yellow solid, mp 137 °C; 1H NMR (300 MHz, CDCl3): δ 7.81–7.72 (2H, m), 7.66 (1H, s), 7.63–7.46 (3H, m), 6.93 (1H, m), 6.04 (1H, td, J = 9.6 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.8 Hz), 2.46 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.2, 165.4, 145.2, 141.2, 136.0, 131.1, 129.3, 126.3, 126.3, 126.0, 125.0, 125.0, 123.8, 41. 24.9; ESIMS: m/z 318 [M + Na]+, HRMS (ESI): m/z 318.0710 [M + Na]+ (calcd. for C15H12NO2F3, 318.0712). Anal. HPLC [tR (min) (content)]: 5.95 (97.0%) from solvent a or 5.80 (95.9%) from solvent b.
4.7.3. (E)-1-(3-(2,5-Dimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (14)
Yield 67%; pale yellow liquid; 8.01 (1H, d, J = 15.6 Hz), 7.51 (1H, d, J = 15.6 Hz), 7.09 (1H, m), 6.93–6.75 (3H, m), 6.01 (1H, d, J = 9.6 Hz), 4.03 (2H, t, J = 12.8 Hz), 3.85 (3H, s), 3.76 (3H, s), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.6, 165.0, 153.7, 153.0, 144.2, 138.9, 126.5, 125.0, 122.3, 117.2, 113.0, 112.4, 56.1, 55.5, 41.5, 25.0; ESIMS: m/z 288 [M + H]+. Anal. HPLC [tR (min) (content)]: 6.65 (95.8%) from solvent a or 6.93 (94.6%) from solvent b.
4.7.4. (E)-1-(3-(4-Bromophenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (15)
Yield 62%; pale yellow solid, mp: 93 °C; 1H NMR (300 MHz, CDCl3): δ 7.66–7.58 (1H, d, J = 15.8 Hz), 7.53–7.40 (5H, m), 6.95–6.87 (1H, m), 6.01 (1H, td, J = 9.0 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.8 Hz), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3):δ 168.5, 165.6, 145.4, 142.0, 134.0, 131.9 (2C), 129.6 (2C), 125.8, 124.2, 122.5, 41.5, 24.7; ESIMS: m/z 329 [M + Na]+, HRESIMS m/z [M + Na]+ 329.9931 (calcd for C20H17NO3, 329.9944). Anal. HPLC [tR (min) (content)]: 5.69 (95.0%) from solvent a or 5.84 (97.6%) from solvent b.
4.7.5. (E)-1-(3-(Benzo[d][1,3]dioxol-5-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (16)
Yield 65%; yellow liquid; 1H NMR (300 MHz, CDCl3): δ 7.64 (1H, d, J = 15.1 Hz), 7.34 (1H, d, J = 15.1 Hz), 7.08 (2H, m), 6.93–6.86(1H, m), 6.77 (1H, d, J = 8.3 Hz), 6.01 (1H, td, J = 9.0 Hz,J = 1.5 Hz), 6.00 (2H, s), 4.01 (2H, t, J = 12.8 Hz), 2.46 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.5, 165.3, 154.9, 148.3, 144.7, 143.7, 129.7, 126.2, 124.6, 119.8, 108.4, 106.9, 101.3, 41.4, 24.9; ESIMS: m/z 294 [M + Na]+, HRESIMS m/z [M + Na]+ 294.0713 (calcd for C15H13NO4, 294.0737). Anal. HPLC [tR (min) (content)]: 6.36 (99.3%) from solvent a or 6.89 (96.6%) from solvent b.
4.7.6. (E)-1-(3-(4-Chlorophenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (17)
Yield 62%; pale yellow liquid; 1H NMR (300 MHz, CDCl3): δ 7.62 (1H, d, J = 15.4 Hz), 7.54–7.40 (3H, m), 7.30 (2H, d, J = 8.1 Hz), 6.90 (1H, m), 5.98 (1H, d, J = 9.2 Hz), 4.01 (2H, t, J = 12.4 Hz), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.2, 165.2, 144.2, 142.1, 135.9, 133.7, 129.5 (2C), 128.9 (2C), 126.4, 122.4, 41.5, 24.9; ESIMS: m/z 284 [M + Na]+, HRMS (ESI): m/z 284.0449 [M + Na]+ (calcd. for C14H12NO2Cl, 284.0448). Anal. HPLC [tR (min) (content)]: 5.80 (98.2%) from solvent a or 5.85 (97.3%) from solvent b.
4.7.7. (E)-1-(3-(Naphthalen-2-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (18)
Yield 60%; white solid, mp 95 °C; 1H NMR (300 MHz, CDCl3): δ 7.93 (2H, d, J = 15.8 Hz), 7.82 (5H, m), 7.59 (1H, d, J = 15.8 Hz), 7.50 (2H, m), 6.92 (1H, m), 6.05 (1H, td J = 9.8 Hz J = 1.5 Hz), 4.06 (2H, t, J = 12.8 Hz), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): d 168.4, 165.2, 144.6, 143.9, 134.2, 133.4, 132.8, 129.9, 128.6, 128.5, 127.7, 126.2, 126.4, 126.3, 124.1, 122.0, 41.5, 24.9; ESIMS: m/z 300 [M + Na]+, HRMS (ESI): m/z 300.0999 [M + Na]+ (calcd. for C18H15NO2, 300.0995). Anal. HPLC [tR (min) (content)]: 5.74 (99.9%) from solvent a or 5.91 (97.3%) from solvent b.
4.7.8. (E)-1-(3-Mesitylacryloyl)-5,6-dihydropyridin-2(1H)-one (19)
Yield 66%; white solid, mp 105 °C; 1H NMR (300 MHz, CDCl3); δ 7.89 (1H, d, J = 15.8 Hz), 7.15 (1H, d, J = 15.8 Hz), 6.92 (3H, m), 5.98 (1H, td, J = 9.8 Hz, J = 1.5 Hz), 4.02 (2H, t, J = 12.8 Hz), 2.44 (2H, m), 2.40 (6H, s), 2.26 (3H, s). 13C NMR (75 MHz, CDCl3): δ 168.6, 165.1, 144.4, 142.0, 137.7, 137.1, 131.6, 129.2 (2C), 126.5, 126.3, 113.8, 41.5, 24.9, 21.3 (2C), 21.2; ESIMS: m/z 292 [M + Na]+, HRESIMS m/z [M + Na]+ 292.1318 (calcd for C17H19NO2, 292.1308). Anal. HPLC [tR (min) (content)]: 5.14 (96.6%) from solvent a or 5.66 (97.1%) from solvent b.
4.7.9. (E)-1-(3-(3-Phenoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (20)
Yield 65%; pale yellow solid, mp 103 °C; 1H NMR (300 MHz, CDCl3): δ 7.67–7.58 (1H, d, J = 15.6 Hz), 7.47–7.39 (1H, d, J = 15.6 Hz), 7.35–7.25 (4H, m), 7.19 (1H, s), 7.09 (1H, t, J = 7.3 Hz), 7.01 (4H, m), 6.01 (1H, td, J = 9.6 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 13.0 Hz), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.4, 165.2, 157.6, 156.9, 144.8, 142.8, 137.0, 130.0, 129.8 (2C), 126.0, 123.4, 123.0, 122.6, 120.2, 119.0 (2C), 118.6, 41.5, 24.8; ESIMS: m/z 342 [M + Na]+, HRESIMS m/z [M + Na]+ 342.1101 (calcd for C20H17NO3, 342.1117). Anal. HPLC [tR (min) (content)]: 7.47 (98.3%) from solvent a or 5.56 (98.1%) from solvent b.
4.7.10. (E)-1-(3-(Pyridin-2-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (21)
Yield 64%; brown liquid; 1H NMR (300 MHz, CDCl3): δ 8.62 (1H, d, J = 4.1 Hz), 7.82 (1H, d, J = 15.2 Hz), 7.71–7.60 (3H, m), 7.47(1H, d, J = 7.9 Hz), 6.90 (1H, m), 6.03 (1H, td, J = 9.8 Hz, J = 1.7 Hz), 3.98 (2H, t, J = 12.8 Hz), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168, 165.9, 153.8, 150.0, 144.6, 141.8, 136.2, 126.1 (2C), 123.7, 123.5, 41.4, 24.9; ESIMS: m/z 229 [M + H]+, HRESIMS m/z [M + H]+ 229.0958 (calcd for C13H12N2O2, 229.0972). Anal. HPLC [tR (min) (content)]: 11.60 (95.3%) from solvent a or 10.59 (95.0%) from solvent b.
4.7.11. (E)-1-(3-(Thiophen-2-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (22)
Yield 60%; pale yellow liquid; 1H NMR (300 MHz, CDCl3): δ 7.81 (1H, d, J = 15.2 Hz), 7.37–7.24 (3H, m), 7.02 (1H, m), 6.90 (1H, m), 6.01 (1H, td, J = 9.8 Hz, J = 1.8 Hz), 4.01 (2H, t, J = 12.4 Hz), 2.44 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.0, 165.0, 144.4, 140.7, 136.3, 130.6, 128.1, 127.1, 126.3, 120.7, 41.4, 24.9; ESIMS: m/z 234 [M + H]+. Anal. HPLC [tR (min) (content)]: 5.60 (96.3%) from solvent a or 6.33 (95.5%) from solvent b.
4.7.12. (E)-1-(3-(Furan-2-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (23)
Yield 60%; liquid; 1H NMR (300 MHz, CDCl3): δ 7.62 (2H, d, J = 17.9 Hz), 7.40 (1H, s), 7.24 (1H, d, J = 14.3 Hz), 6.90 (1H, m), 6.65 (1H, s), 6.01 (1H, td, J = 9.6 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.8 Hz), 2.46 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.3, 165.2, 144.6, 144.5, 144.1, 133.9, 126.3, 123.5, 121.6, 108.0, 41.4, 24.9; ESIMS: m/z 234 [M + H]+.
4.7.13. 1-(4-Nitrobenzoyl)-5,6-dihydropyridin-2(1H)-one (24)
Yield 60%; pale yellow oil; 1H NMR (300 MHz, CDCl3); δ 8.25 (2H, d, J = 9.0 Hz), 7.63 (2H, d, J = 8.0 Hz), 7.01 (1H, m), 5.96 (1H, td, J = 9.8 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.4 Hz), 2.53 (2H, m).13C NMR (75 MHz, CDCl3): δ 171.3, 164.2, 149.2, 145.7, 142.0, 128.7 (2C), 125.2, 123.3 (2C), 42.9, 24.9; ESIMS: m/z 247 [M + H]+, HRMS (ESI): m/z 269.0539 [M + Na]+ (calcd. for C12H10N2O4, 269.0532). Anal. HPLC [tR (min) (content)]: 6.65 (97.6%) from solvent a or 9.01 (95.6%) from solvent b.
4.7.14. 1-(4-Fluorobenzoyl)-5,6-dihydropyridin-2(1H)-one (25)
Yield 62%, brown oil; 1H NMR (300 MHz, CDCl3): δ 7.61–7.53 (2H m), 7.09–6.90 (3H, m), 5.93 (1H, td, J = 9.6 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.4 Hz), 2.54 (2H, m).13C NMR (75 MHz, CDCl3): δ 172.2, 166.5, 165.0, 144.8, 132.1, 131.2, 131.0, 125.8, 115.3, 114.8, 43.4, 25.14; ESIMS: m/z 242 [M + Na]+, HRMS (ESI): m/z 242.0591 [M + Na]+ (calcd. for C12H10NO2F, 242.0587). Anal. HPLC [tR (min) (content)]: 7.32 (99.2%) from solvent b.
4.7.15. 1-(2-Chlorobenzoyl)-5,6-dihydropyridin-2(1H)-one (26)
Yield 60%; oil; 1H NMR (300 MHz, CDCl3): δ 7.24 (4H, s), 6.92–6.85 (1H, m), 5.94 (1H, td, J = 9.8 Hz, J = 1.5 Hz), 4.15 (2H, t, J = 12.8 Hz), 2.51 (2H, m). 13C NMR (75 MHz, CDCl3):δ 168.8, 163.4, 145.1, 137.6, 130.2, 129.5, 129.3, 128.4, 126.7, 125.6, 41.7, 24.9; ESIMS: m/z 236 [M + H]+, HRESIMS m/z [M + Na]+ 258.0309 (calcd for C12H10NO2Cl, 258.0292). Anal. HPLC [tR (min) (content)]: 6.17 (98.8%) from solvent a or 6.79 (95.5%) from solvent b.
4.7.16. 1-(2-Iodobenzoyl)-5,6-dihydropyridin-2(1H)-one (27)
Yield 58%; brown oil; 1H NMR (300 MHz, CDCl3): δ 7.74 (1H, d, J = 7.5 Hz), 7.42–7.33 (1H, m), 7.24–7.15 (1H, m), 7.05 (1H, m), 6.94 (1H, m), 5.92 (1H, td, J = 9.8 Hz, J = 1.5 Hz), 4.01 (2H, t, J = 12.8 Hz), 2.54 (2H, m). 13C NMR (75 MHz, CDCl3): 165.8, 163.4, 145.0, 143.7, 138.8, 130.2, 127.9, 127.7, 125.7, 91.2, 41.6, 24.7; ESIMS: m/z 327 [M]+, HRESIMS m/z [M + Na]+ 349.9629 (calcd for C12H10N2OCl, 349.9648). Anal. HPLC [tR (min) (content)]: 6.27 (95.4%) from solvent a or 6.89 (96.6%) from solvent b.
4.7.17. (E)-1-(3-(1,3-Diphenyl-1H-pyrazol-4-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (28)
Yield 60%; oil; 1H NMR (300 MHz, CDCl3): δ 8.33 (1H, s), 7.85–7.65 (5H, m), 7.54–7.24 (7H, m), 6.90 (1H, m), 6.01 (1H, td, J = 9.8 Hz, J = 1.8 Hz), 4.01 (2H, t, J = 12.48 Hz), 2.46 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.4, 165.3, 153.5, 144.7, 139.7, 134.7, 132.4, 129.4 (2C), 128.8 (2C), 128.7 (2C), 128.4, 126.9, 126.3, 126.2, 121.0, 119.3 (2C), 118.5, 41.4, 24.9; ESIMS: m/z 392 [M + Na]+, HRMS (ESI): m/z 370.1553 [M + H]+ (calcd. for C23H20N3O2, 370.1550). Anal. HPLC [tR (min) (content)]: 5.16 (97.5%) from solvent a.
4.7.18. (E)-1-(3-(3-Methyl-1-phenyl-1H-pyrazol-4-yl)acryloyl)-5,6-dihydropyridin-2(1H)-one (29)
Yield 64%; brown oil; 1H NMR (300 MHz, CDCl3); δ 8.10 (1H, s), 7.68 (3H, m), 7.40 (2H, m), 7.34 (1H, d, J = 15.6 Hz), 7.24 (1H, m), 6.91 (1H, m), 6.01 (1H, td, J = 9.6 Hz, J = 1.7 Hz), 4.02 (2H, t, J = 12.4 Hz), 2.43 (2H, m), 2.49–2.45 (3H, s): 13C NMR (75 MHz, CDCl3): δ 168.5, 165.2, 150.5, 144.4, 139.6, 134.3, 129.4 (2C), 129.3, 126.7, 126.5 (2C), 120.0, 119.0, 118.9, 41.4, 25.0, 13.4; ESIMS: m/z 330 [M + Na]+, HRMS (ESI): m/z 308.1395 [M + H]+ (calcd. for C18H18N3O2, 308.1393). Anal. HPLC [tR (min) (content)]: 7.29 (95.8%) from solvent b.
4.7.19. 1-((2E,4E)-5-(Benzo[d][1,3]dioxol-5-yl)penta-2,4-dienoyl)-5,6-dihydropyridin-2(1H)-one (30)
Yield 58%; yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.57 (1H, m), 7.25 (1H, s), 6.98 (1H, m), 6.81 (4H, m), 5.98 (2H, s), 4.01 (2H, t, J = 12.8 Hz), 2.43 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.2, 164.9, 148.5, 148.3, 144.3, 144.1, 140.0, 131.0, 126.5, 125.80, 124.3, 122.8, 108.5, 106.1, 101.2, 41.3, 25.0; ESIMS: m/z 298 [M + H]+, HRMS (ESI): m/z 320.0892 [M + Na]+ (calcd. for C17H15NO4, 308.1393). Anal. HPLC [tR (min) (content)]: 6.28 (97.7%) from solvent a or 6.91 (96.2%) from solvent b.
4.7.20. 1-((2E,4E,6E)-7-(3,4,5-Trimethoxyphenyl)hepta-2,4,6-trienoyl)-5,6-dihydropyridin-2(1H)-one (31)
Yield 60%; yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.95 (1H, m), 7.62–7.16 (2H, m), 7.14–6.78 (2H, m), 6.79–6.18 (4H, m), 5.98 (1H, m), 4.01 (2H, t, J = 12.4 Hz), 3.90 (6H, s), 3.84 (3H, s), 2.46 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.2, 164.9, 153.59, 144.3, 143.85, 140.4, 140.3, 138.5, 136.9, 136.3, 131.3, 127.8, 124.7, 104.7, 60.7, 56.1, 41.4, 24.9; ESIMS: m/z 392 [M + Na]+.
4.8. General procedure for the preparation of Baylis–Hillman adducts of piplartine (32a–32j)
To a solution of piplartine (0.157 mmol) and aldehyde (0.189 mmol) in 1,4-dioxane (2 mL) 2 eq of DABCO was added at room temperature and the reaction allowed to stir for 72 h. Progress of the reaction was monitored by TLC. Upon completion, the reaction mixture was partitioned with tert-butyl methyl ether (5 mL) and water (3 mL). The organic phase was washed with brine (2 × 3 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel, (60–120 mesh) using hexane/EtOAc (6:4) as eluent to give the desired product.
4.8.1. (E)-3-((4-Chlorophenyl)(hydroxy)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32a)
Yield 60%; yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.64–7.54 (1H, d, J = 15.4 Hz), 7.33–7.23 (5H, m), 6.76 (3H, m), 5.62 (1H, s), 3.96 (2H, t, J = 7.5 Hz), 3.90 (6H, s), 3.83 (3H, s), 2.41 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.3, 166.1, 153.4 (2C), 144.2, 140.9 (2C), 139.9, 138.1, 133.5, 130.4, 128.5 (2C), 128.0 (2C), 120.8, 105.7 (2C), 71.4, 60.7, 56.0 (2C), 41.5, 24.5; ESIMS: m/z 457 [M]+, HRESIMS m/z [M + Na]+ 480.1319 (calcd for C24H24NO6Cl, 480.1315). Anal. HPLC [tR (min) (content)]: 6.53 (97.2%) from solvent a or 7.57 (94.3%) from solvent b.
4.8.2. (E)-3-((4-Fluorophenyl)(hydroxy)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32b)
Yield 55%; pale yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.59(1H, d, J = 15.4 Hz), 7.32 (3H, m), 7.01 (2H, m), 6.72 (2H, s), 6.67 (1H, t, J = 7.7 Hz), 5.60 (1H, s), 3.94 (2H, m), 3.89 (6H, s), 3.84 (3H, s), 2.48 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.3, 166.2, 160.6, 153.4 (2C), 144.2, 140.7, 140.4, 138.4, 137.1, 130.4, 128.3, 128.2, 120.8, 115.3, 115.1, 106.5 (2C), 71.6, 60.8, 56.0 (2C), 41.5, 24.5; ESIMS: m/z 442 [M + H]+, HRESIMS m/z [M + Na]+ 464.1492 (calcd for C24H24NO6F, 464.1485). Anal. HPLC [tR (min) (content)]: 6.73 (96.0%) from solvent a or 8.02 (95.8%) from solvent b.
4.8.3. (E)-3-(Hydroxy(2-nitrophenyl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32c)
Yield 65%; pale yellow solid, mp 137 °C; 1H NMR (300 MHz, CDCl3): δ 7.93(1H, d, J = 9.0 Hz), 7.87(1H, d, J = 7.7 Hz), 7.72 (2H, m), 7.50 (1H, m), 7.37 (1H, d, J = 15.6 Hz) 6.78 (2H, s), 6.59 (1H, t, J = 8.1 Hz), 6.27 (1H, br s), 3.98 (2H, m), 3.92 (6H, s), 3.87 (3H, s), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.3, 166.2, 160.6, 153.3 (2C), 144.2, 140.7, 140.4, 138.4, 137.1, 130.4, 128.3, 128.2, 120.8, 115.3, 115.1, 105.7 (2C), 71.4, 60.6, 56.0 (2C), 41.5, 24.5; ESIMS: m/z 469 [M + H]+, HRESIMS m/z 469.1628 [M + H]+ (calcd for C24H24N2O8, 469.1610). Anal. HPLC [tR (min) (content)]: 7.72 (99.3%) from solvent a or 9.48 (95.4%) from solvent b.
4.8.4. (E)-3-(Hydroxy(4-nitrophenyl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32d)
Yield 63%; yellow solid, mp 135 °C; 1H NMR (300 MHz, CDCl3): δ 8.20 (2H, d, J = 8.6 Hz), 7.63 (1H, d, J = 15.6 Hz), 7.60 (2H, d, J = 8.8 Hz), 7.28 (1H, d, J = 14.1 Hz), 6.73 (3H, m), 5.71 (1H, brs), 4.01 (2H, m), 3.91 (6H, s), 3.86 (3H, s), 2.50 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.0, 165.7, 153.0 (2C), 148.1, 147.1, 144.2, 141.5 (2C), 136.9, 129.9, 126.9 (2C), 123.2 (2C), 120.2, 105.3 (2C), 71.4, 60.4, 55.7 (2C), 41.2, 24.1; ESIMS: m/z 469 [M + H]+, HRESIMS m/z 469.1628 [M + H]+ (calcd for C24H24N2O8, 469.1610). Anal. HPLC [tR (min) (content)]: 8.00 (97.7%) from solvent a or 9.00 (97.2%) from solvent b.
4.8.5. (E)-4-(Hydroxy(2-oxo-1-(3-(3,4,5-trimethoxyphenyl) acryloyl)-1,2,5,6-tetrahydropyridin-3-yl)methyl)benzonitrile (32e)
Yield 60%; pale yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.59–7.43 (5H, m), 7.23 (1H, d, J = 15.4 Hz), 6.74 (1H, m), 6.72 (2H, s), 5.64 (1H, s), 3.89 (2H, m), 3.87 (6H, s), 3.83 (3H, s), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.2, 165.6, 153.2 (2C), 147.2, 144.0, 141.3 (2C), 137.5, 131.8 (2C), 130.2, 127.2 (2C), 120.7, 118.2, 111.3, 105.6 (2C), 70.8, 60.5, 55.9 (2C), 41.4, 24.3; ESIMS: m/z 471 [M + Na]+, HRESIMS m/z [M + Na]+ 471.1538 (calcd for C25H24N2O6, 471.1532). Anal. HPLC [tR (min) (content)]: 9.01 (95.1%) from solvent a or 10.33 (96.8%) from solvent b.
4.8.6. (E)-3-(Hydroxy(pyridin-2-yl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32f)
58%; oil; 1H NMR (300 MHz, CDCl3): δ 8.49 (1H, J = 4.7 Hz) 7.70–7.53 (3H, m), 7.33 (1H, d, J = 15.4 Hz), 7.20 (1H, t, J = 11.7 Hz), 7.02 (1H, t, J = 7.3 Hz), 6.74 (2H, s), 5.72 (1H, s), 4.10 (2H, m), 3.91 (6H, s), 3.85 (3H, s), 2.51 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.4, 166.1, 160.2, 153.4 (2C), 148.1, 143.9, 141.1, 137.7, 136.8 (2C), 130.6, 122.5, 121.7, 121.1, 105.7 (2C), 70.5, 60.7, 55.8 (2C), 41.6, 24.6; ESIMS: m/z 425 [M + H]+. Anal. HPLC [tR (min) (content)]: 6.24 (98.2%) from solvent a or 14.8 (97.1%) from solvent b.
4.8.7. (E)-3-(Hydroxy(thiophen-2-yl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32g)
Yield 58%; yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.60 (1H, d, J = 15.4 Hz), 7.33 (1H, d, J = 15.4 Hz), 7.21 (1H, dd, J = 4.9 Hz, J = 1.1 Hz), 6.94 (2H, m), 6.84 (1H, t, J = 7.9 Hz), 6.74 (2H, s), 5.83 (1H, s), 4.01 (2H, m), 3.89 (6H, s), 3.82 (3H, s), 2.51 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.4, 165.9, 153.4 (2C), 146.0, 144.0, 140.9 (2C), 137.2, 130.4, 136.9, 125.5 (2C), 120.9, 105.6 (2C), 71.0, 60.9, 56.7 (2C), 41.4, 24.0; ESIMS: m/z 429 [M]+; HRMS (ESI): m/z 452.1143 [M + Na]+ (calcd for C22H23NO6, 452.1138) Anal. HPLC [tR (min) (content)]: 6.11 (95.0%) from solvent a or 8.16 (93.9%) from solvent b.
4.8.8. (E)-3-(Hydroxy(naphthalen-2-yl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32h)
Yield 63%; pale brown solid, mp 143 °C; 1H NMR (300 MHz, CDCl3): δ 7.84 (1H, s), 7.86 (3H,m), 7.58 (1H, d, J = 15.4 Hz), 7.42 (3H, m), 7.32 (1H, d, J = 15.4 Hz), 6.73 (2H, s), 6.67 (1H, t, J = 7.7 Hz), 5.80 (1H, s), 3.98 (2H, m), 3.86 (6H, s), 3.83 (3H, s), 2.42 (2H, m). 13C NMR (75 MHz, CDCl3): δ 167.9, 165.9, 153.0 (2C), 143.6, 140.6, 139.9, 138.4, 137.9, 132.9, 132.5, 130.0, 127.7, 127.6, 127.2, 125.7, 125.5, 125.2, 124.2, 120.5, 105.3 (2C), 71.5, 60.2, 55.6 (2C), 41.1, 24.1; ESIMS: m/z 496 [M + Na]+, HRESIMS m/z [M + Na]+ 496.1730 (calcd for C28H27NO6, 496.1736). Anal. HPLC [tR (min) (content)]: 6.24 (95.8%) from solvent a or 7.68 (95.8%) from solvent b.
4.8.9. (E)-3-(Hydroxy(p-tolyl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32i)
Yield 62%; pale yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.61 –7.55 (1H, d, J = 15.4 Hz), 7.32 (1H, d, J = 15.4 Hz), 7.24 (2H, d, J = 7.9 Hz), 7.11 (2H, d, J = 7.9 Hz), 6.75 (2H, s), 6.66 (1H, t, J = 8.1 Hz), 5.59 (1H, s), 3.99 (2H, m), 3.89 (6H, s), 3.84 (3H, s), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.8, 166.7, 153.8 (2C), 144.4, 141.1, 140.7, 139.9, 138.8, 137.5, 130.9, 129.4 (2C), 127.0 (2C), 121.4, 106.1 (2C), 72.3, 61.3, 56.5 (2C), 42.0, 24.9, 21.6; HRESIMS m/z 460.1732 [M + Na]+ (calcd for C25H27NO6, 460.1730). Anal. HPLC [tR (min) (content)]: 6.04 (95.9%) from solvent a or 7.38 (95.2%) from solvent b.
4.8.10. (E)-3-(Hydroxy(phenyl)methyl)-1-(3-(3,4,5-trimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (32j)
Yield 56%; pale brown solid, mp: 137 °C; 1H NMR (300 MHz, CDCl3): δ 7.59 (1H, d, J = 15.4 Hz), 7.36–7.20 (6H, m), 6.73 (2H, s), 6.67 (1H, t, J = 7.3 Hz), 5.62 (1H, s), 3.94 (2H, m), 3.89 (6H, s), 3.83 (3H, s), 2.45 (2H, m). 13C NMR (75 MHz, CDCl3): δ 168.3, 166.2, 153.3 (2C), 143.9, 141.4, 140.8, 140.2, 138.3, 130.4, 128.2 (2C), 127.1, 126.6 (2C), 120.9, 105.6 (2C), 71.8, 60.6, 56.0 (2C), 41.5, 24.4; ESIMS: m/z 423 [M]+, HRMS (ESI): m/z 446.1563 [M + Na]+ (calcd for C24H25NO6, 446.1574). Anal. HPLC [tR (min) (content)]: 6.73 (98.2%) from solvent a or 7.95 (94.8%) from solvent b.
4.9. General procedure for the preparation of 33–43
To a stirred solution of trimethoxy cinnamic acid (0.210 mmol) in DCM (3 mL) was added Et3N (0.315 mmol) and pivaloyl chloride (0.231 mmol) at 0 °C and the reaction mixture was allowed to stir for 45 min. Subsequently, amines (0.315 mmol) were added and the resultant mixture was further allowed to stir for 2 h at room temperature. Upon completion, the reaction mixture was quenched with sat. NaHCO3 (0.5 mL) and diluted with water (2 mL), then extracted in to DCM (5 mL). The organic phase was washed with brine (2 × 3 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel, (60–120 mesh) using hexane/EtOAc (7:3) as an eluent to give the desired product.
4.9.1. (E)-1-(Piperidin-1-yl)-3-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (33)
Yield 80%; pale yellow solid, mp 98 °C; 1H NMR (300 MHz, CDCl3): δ 7.51 (1H, d, J = 15.6 Hz), 6.72 (1H, d, J = 15.6 Hz), 6.69 (2H, s), 3.87 (6H, s), 3.82 (3H, s), 3.61 (4H, br m), 1.7–1.67 (2H, m), 1.63 (4H, m). 13C NMR (75 MHz, CDCl3): δ 164.8, 153.5 (2C), 142.7, 139.8, 130.8, 116.5, 105.3 (2C), 60.6, 56.1 (2C), 46.9, 43.5, 27.3, 24.2 (2C); ESIMS: m/z 306 [M + H]+, HRESIMS m/z [M + H]+ 306.1714 (calcd for C17H23NO4, 306.170). Anal. HPLC [tR (min) (content)]: 7.99 (95.1%) from solvent a or 7.33 (98.9%) from solvent b.
4.9.2. (E)-1-(3-(3,4,5-Trimethoxyphenyl)acryloyl)pyrrolidin-2-one (34)
Yield 70%; white solid, mp 165 °C; 1H NMR (300 MHz, CDCl3): δ 7.78 (1H, d, J = 15.1 Hz), 7.69 (1H, d, J = 15.1 Hz), 6.80 (2H, s), 3.97 (2H, m), 3.91 (6H, s), 3.84 (3H, s) 2.63 (2H, t, J = 15.8 Hz), 2.08 (2H, m). 13C NMR (75 MHz, CDCl3): δ 174.8, 165.6, 153.4 (2C), 145.5, 140.0, 130.1, 118.1, 105.7 (2C), 60.6, 56.0 (2C), 45.7, 33.8, 17.2; ESIMS: m/z 306 [M + H]+, HRESIMS m/z [M + H]+ 306.1350 (calcd. for C16H19NO5, 306.1336). Anal. HPLC [tR(min) (content)]: 10.25 (95.0%) from solvent a or 8.00 (94.0%) from solvent b.
4.9.3. (E)-3-(3-(3,4,5-Trimethoxyphenyl)acryloyl)benzo[d]oxazol-2(3H)-one (35)
Yield 85%; yellow solid, mp 95 °C; 1H NMR (300 MHz, CDCl3): δ 8.12 (1H, m), 7.83 (2H, s), 7.21–7.10 (3H, m), 6.81 (2H, s), 3.90 (6H, s), 3.85 (3H, s). 13C NMR (75 MHz, CDCl3):δ 164.0, 153.4 (2C), 151.3, 148.4. 146.3, 142.3, 130.6, 128.3, 125.6, 124.6, 123.8, 122.4, 116.2, 106.0 (2C), 60.79, 50.0 (2C); ESIMS: m/z 378 [M + Na]+, HRESIMS m/ z [M + Na]+ 378.0937 (calcd for C19H17NO6, 378.0948). Anal. HPLC [tR (min) (content)]: 4.34 (97.2%) from solvent a.
4.9.4. (E)-N-Ethanethioyl-3-(3,4,5-trimethoxyphenyl)acrylamide (36)
Yield 65%; pale yellow solid, mp 133 °C; 1H NMR (300 MHz, CDCl3): δ 7.79 (1H, d, J = 15.8 Hz), 6.73 (2H, s), 6.31 (1H, d, J = 15.8 Hz), 3.90 (6H, s), 3.85 (3H, s), 1.26 (3H, s). 13C NMR (75 MHz, CDCl3): δ 172.2 (2C), 153.5 (2C), 146.9, 140.6, 129.4, 116.6, 105.7 (2C), 60.7, 56.0 (2C), 29.1; ESIMS: m/z 318 [M + H]+.
4.9.5. (E)-2-(3-(3,4,5-Trimethoxyphenyl)acryloyl)benzo[d] isothiazol-3(2H)-one (37)
Yield 60%; yellow solid, mp 218 °C; 1H NMR (300 MHz, CDCl3): δ 8.15–7.71 (4H, m), 7.45 (1H, m), 7.05 (2H, s) 6.99 (1H, d, J = 15.6 Hz), 3.90 (3H, s), 3.83 (6H, s). 13C NMR (75 MHz, CDCl3): δ 164.7, 163.5, 153.4 (2C), 147.9, 145.1, 141.3, 134.2, 131.4, 129.9, 127.8, 125.8 (2C), 120.5, 106.1 (2C), 60.8, 56.1 (2C); ESIMS: m/z 394 [M + Na]+, HRESIMS m/z [M + Na]+ 394.0737 (calcd for C19H17NO5S, 394.072). Anal. HPLC [tR (min) (content)]: 5.92 (96.6%) from solvent a.
4.9.6. (E)-1-(Pyrrolidin-1-yl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (38)
Yield 85%; pale yellow solid, mp 155 °C; 1H NMR (300 MHz, CDCl3): δ 7.61–7.49 (1H, d, J = 15.6 Hz), 6.70 (2H, s), 6.59 (1H, d, J = 15.6 Hz), 3.88 (6H, s), 3.83 (3H, s), 3.59 (4H, m), 2.01 (4H, m). 13C NMR (75 MHz, CDCl3): δ 164.1, 153.3 (2C), 141.9, 139.8, 130.6, 117.6, 105.2 (2C), 60.4, 55.9 (2C), 46.3, 45.8, 26.1, 24.2; ESIMS: m/z 292 [M + H]+, HRESIMS m/z [M + H]+ 292.1547 (calcd for C16H21NO4, 292.1543) Anal. HPLC [tR (min) (content)]: 13.3 (95.2%) from solvent a or 9.33 (96.9%) from solvent b.
4.9.7. (E)-1-(3-(3,4,5-Trimethoxyphenyl)acryloyl)piperidin-2-one (39)
Yield 60%; white solid, mp 129 °C; 1H NMR (300 MHz, CDCl3): δ 7.40 (1H, d, J = 15.1 Hz), 6.52 (1H, d, J = 15.1 Hz), 6.55 (2H, s), 3.82 (3H, s), 3.75 (8H, s), 2.10 (2H, m), 1.45 (4H, m). 13C NMR (75 MHz, CDCl3): δ 165.7 (2C), 154.0, 153.3, 143.0, 139.7, 130.1, 118.5, 105.0 (2C), 60.8, 55.9 (2C), 49.9, 32.7, 26.1, 24.7; ESIMS: m/z 320 [M + H]+. Anal. HPLC [tR (min) (content)]: 4.23 (95.6%) from solvent a or 5.82 (96.6%) from solvent b.
4.9.8. (E)-2-(3-(3,4,5-Trimethoxyphenyl)acryloyl)isoindolin-1-one (40)
Yield 55%; brown solid, mp; 104 °C; 1H NMR (300 MHz, CDCl3): δ 8.23 (1H, d, J = 7.0 Hz), 7.83–7.68 (2H, m), 7.33–7.11 (3H, m), 6.81 (2H, s), 3.90 (6H, s), 3.83 (3H, s), 3.72 (3H, s). 13C NMR (75 MHz, CDCl3): δ 168.7, 165.4, 153.5 (2C), 146.2, 130.3, 128.3 (2C), 127.9, 124.8, 124.4, 123.8, 123.4, 111.9, 105.9 (2C), 60.8, 56.1 (2C), 72.7; ESIMS m/z [M + Na]+ 376. Anal. HPLC [tR (min) (content)]: 4.65 (95.9%) from solvent a or 7.93 (95.3%) from solvent b.
4.9.9. (E)-1-(4-Benzylpiperazin-1-yl)-3-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (41)
Yield 75%; pale yellow solid, mp: 135 °C; 1H NMR (300 MHz, CDCl3): δ 7.55–7.48(1H, d, J = 15.2 Hz), 7.30–7.20 (5H, m), 6.69 (1H, d, J = 15.2 Hz), 6.68 (2H, s), 3.86 (6H, s), 3.84 (3H, s), 3.70 (4H, br s), 3.52 (2H, s), 2.45 (4H, m). 13C NMR (75 MHz, CDCl3): δ 164.8, 153.4 (2C), 142.9, 137.4, 130.7, 129.0 (2C), 128.3 (2C), 128.9, 127.3, 116.1, 105.2 (2C), 63.0, 60.6, 56.0 (2C), 53.2, 53.02, 45.7, 42.1; ESIMS: m/z 397 [M + H]+, HRESIMS m/z [M + H]+ 397.2151 (calcd for C23H28N2O4, 397.2151). Anal. HPLC [tR(min) (content)]: 17.2 (96.4%) from solvent a or 9.47 (98.0%) from solvent b.
4.9.10. (E)-1-(4-Methylpiperazin-1-yl)-3-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (42)
Yield 80%; pale yellow solid, mp: 130 °C; 1H NMR (300 MHz, CDCl3): δ 7.58–7.48 (1H, d, J = 15.8 Hz), 6.73–6.65 (1H, d, J = 15.8 Hz), 6.69 (2H, s), 3.89 (6H, s), 3.84 (3H, s), 3.57 (8H, brm), 2.30 (3H, s). 13C NMR (75 MHz, CDCl3): δ 164.7, 153.4 (2C), 143.0, 140.0, 130.7, 115.9, 105.3 (2C), 60.5, 56.0 (2C), 55.1 (2C), 46.0 (2C), 41.9; ESIMS: m/z 321 [M + H]+, HRESIMS m/z [M + H]+ 321.1821 (calcd for C17H24N2O4, 321.1821). Anal. HPLC [tR (min) (content)]: 5.20 (95.8%) from solvent a.
4.9.11. (E)-1-(4-Phenylpiperazin-1-yl)-3-(3,4,5-trimethoxyphenyl) prop-2-en-1-one (43)
Yield 83%; pale yellow oil; 1H NMR (300 MHz, CDCl3):δ 7.60–7.53 (1H, d, J = 15.2 Hz), 7.23 (2H, t, J = 8.6 Hz), 6.91 (3H, m), 6.74 (1H, d, J = 15.2 Hz), 6.70 (2H, s), 3.90 (6H, s), 3.83 (3H, s), 3.90–3.79 (4H, brm), 3.20 (4H, m). 13C NMR (75 MHz, CDCl3): δ 164.8, 153.5 (2C), 150.9, 143.3, 140.1, 130.6, 129.2 (2C), 120.6, 116.7 (2C), 115.8, 105.4 (2C), 60.5, 56.1 (2C), 49.8 (2C), 46.8, 43.4; HRESIMS m/z [M + H]+ 383.1965 (calcd for C22H26N2O4, 383.1965). Anal. HPLC [tR (min) (content)]: 7.08 (96.0%) from solvent a or 6.82 (95.6%) from solvent b.
4.10. Aldose reductase inhibition studies
4.10.1. Expression and purification of human recombinant ALR2
Recombinant human ALR2 was overexpressed in Escherichia coli and purified from bacterial cultures essentially as described previously [24] with a minor modification. Chromatography over AffiGel Blue (Bio-Rad) affinity matrix was used as a final purification step [25].
4.10.2. ALR2 assay
The activity of ALR2 was measured as described previously [24]. The change in the absorbance at 340 nm due to NADPH oxidation was followed in a Lamda35 spectrophotometer (Perkin-Elmer, Shelton, USA).
4.11. ALR2 inhibition and determination of IC50 values
For inhibition studies concentrated stocks of compounds prepared in dimethyl sulfoxide (DMSO) were used and the final concentration of DMSO was not more than 1%of the assay volume. Various concentrations of the above mentioned analogues were added to assay mixture of ALR2 and incubated for 5 min before initiating the reaction by NADPH as described above. The percentage inhibition was calculated considering the activity in the absence of compound as 100%. The IC50 values were determined by nonlinear regression analysis of the plot of percent inhibition versus log compound concentration.
4.12. Inhibition of sorbitol formation under high glucose conditions by 3c, 3e and 2j in ex vivo system
4.12.1. In vitro incubation of RBC
5 mL blood was collected from healthy male volunteers after overnight fasting in heparinized tubes. Red blood cells were separated by centrifugation and washed three times with isotonic saline at 4 °C. Washed RBC were suspended in Kreb’s-ringer bicarbonate buffer, pH 7.4 (pre-equilibrated with 5% CO2). Duplicate samples were incubated at 37 °C in presence of 5% CO2 for 3 h under normal (5.5 mM) and high glucose (55 mM) conditions [24]. The effect of 3c, 3e and 2j on sorbitol accumulation was evaluated by incubating the RBC with different concentrations of 3c, 3e and 2j.
4.12.2. Estimation of sorbitol in RBC
At the end of the incubation period, RBC were homogenized in 9 volumes of 0.8 M perchloric acid. The homogenate was centrifuged at 5000× g at 4 °C for 10 min and the pH of the supernatant was adjusted to 3.5 with 0.5 M potassium carbonate. The sorbitol content of the supernatant was measured by a fluorometric method as described previously using a spectrofluorometer (Jasco-FP-6500) [26].
4.13. Molecular docking studies
Molecular docking was done by discovery (discover 2.7) package from (Biosystems Technologies, San Diego, CA, USA), on an O2 (R12000) workstation (Silicon Graphics, Mountain View, CA, USA). All ligands were minimized to least energy conformations and taken for docking studies. Crystal structure of human ALR2 was downloaded from Brookhaven data bank (PDB: 1PWM) and protein structure was minimized by using charmM force field. All water molecules were removed. Docking was done by discovery ligand fit module with created sphere about 12 Å around the active site in the protein. After docking, poses were viewed by DS viewer and calculation of binding energy was done using the discovery programmer. GOLD was also used for docking of all compounds into ALR2 active site. In the docking procedure all parameters were kept in default. ALR2 active site was defined by from single residue Tyr 48 with 12 Å cavity detection. Fitness of each ligand was generated by based on the GOLD fitness score. The docked poses were viewed by discovery viewer, silver screen 1.1.1 and ligand scout software. Structure-based pharmacophore was generated by MOE and catalyst software’s.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge the keen interest of Dr. J. S. Yadav, Director, IICT, Hyderabad, India in the present work. We also acknowledge V. Raju Nayak at Biomedical Informatics Centre at National Institute of Nutrition (ICMR) for his help in molecular docking studies. The work in GBR’s lab was supported by grants from Life Sciences Research Board of Defense Research and Development Organization, Government of India. VRSR, GS, RSC, and PM acknowledge CSIR and University Grants Commission, Govt. of India, respectively for financial support.
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2012.09.014.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Diabetes Care. 2004;27:1047. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M. Nature. 2001;414:813. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 3.Kinoshita JH. Exp. Eye Res. 1990;50:567–573. doi: 10.1016/0014-4835(90)90096-d. [DOI] [PubMed] [Google Scholar]
- 4.Bhatnagar A, Srivastava SK. Biochem. Med. Metab. Biol. 1992;48:91–121. doi: 10.1016/0885-4505(92)90055-4. [DOI] [PubMed] [Google Scholar]
- 5.Kador PF, Robison WG, Kinoshita JH. Annu. Rev. Pharmacol. Toxicol. 1985;25:691–714. doi: 10.1146/annurev.pa.25.040185.003355. [DOI] [PubMed] [Google Scholar]
- 6.Pfeifer MA, Schumer MP, Gelber DA. Diabetes. 1997;46:S82. doi: 10.2337/diab.46.2.s82. [DOI] [PubMed] [Google Scholar]
- 7.Raskin P, Rosenstock J. Am. J. Med. 1987;83:298–306. doi: 10.1016/0002-9343(87)90702-9. [DOI] [PubMed] [Google Scholar]
- 8.(a) Newman DJ, Cragg GM, Snader KM. J. Nat. Prod. 2003;66:1022. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]; (b) Lee K-H. J. Nat. Prod. 2010;73:500–516. doi: 10.1021/np900821e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Sumathykutty MA, Rao JM. Phytochemistry. 1991;30:2075–2076. [Google Scholar]; (b) Reddy SV, Srinivas PV, Praveen B, Hara KK, Raju BC, Murthy USN, Rao JM. Phytomedicine. 2004;11:697–700. doi: 10.1016/j.phymed.2003.04.004. [DOI] [PubMed] [Google Scholar]; (c) Srinivas PV, Rao JM. Phytochemistry. 1999;52:957. [Google Scholar]; (d) Srinivas PV, Anuradha V, Tiwari AK, Rao JM. J. Piper. Ind. Focus Pepper. 2005;2:21. [Google Scholar]; (e) Srinivas PV, Tiwari AK, Sarma VUM, Anuradha V, Haribabu T, Rao DK, Khan IA, Rao JM. J. Ethnopharmacol. 2006;108:445. [Google Scholar]; (f) Rao VRS, Kumar GS, Sarma VUM, Raju SS, Babu KH, Babu KS, Babu TH, Rekha K, Rao JM. Tetrahedron Lett. 2009;50:2774. [Google Scholar]; (g) Rao VRS, Suresh G, Babu KS, Raju SSN, Vishnu Vardhan MVPS, Ramakrishna S, Rao JM. Tetrahedron. 2011;67:1885–1892. [Google Scholar]
- 10.(a) Kirtikar KR, Basu BD. Indian Medicinal Plants. vol. III. New Delhi, India: Periodical Experts Book Agency; 1933. p. 2128. [Google Scholar]; (b) The Wealth of India. Raw Materials. vol. VIII. CSIR, New Delhi: 1969. p. 83. [Google Scholar]; (c) Parmar VS, Jain SC, Bisht KS, Jain R, Taneja P, Jha A, Tyagi OD, Prasad AK, Wengel J, Olsen CE, Boll PM. Phytochemistry. 1997;46:597. [Google Scholar]
- 11.Greco O, Rossiter S, Kanthou C, Folkes LK, Wardman P, Tozer GM, Dachs GU. Mol. Cancer Ther. 2001;1:151. [PubMed] [Google Scholar]
- 12.Folkes LK, Wardman P. Biochem. Pharmacol. 2001;61:129. doi: 10.1016/s0006-2952(00)00498-6. [DOI] [PubMed] [Google Scholar]
- 13.Bandini M, Melchiorre P, Melloni A, Umani-Ronchi A. Synthesis. 2002:1110. [Google Scholar]
- 14.(a) Yadav JS, Abraham S, Reddy BVS, Sabitha G. Synthesis. 2001:2165. [Google Scholar]; (b) Komoto I, Kobayashi S. J. Org. Chem. 2004;69:680. doi: 10.1021/jo0353177. [DOI] [PubMed] [Google Scholar]; (c) Harrington PE, Kerr MA. Synlett. 1996:1047. [Google Scholar]; (d) Zhan Z-P, Yang R-F, Lang K. Tetrahedron Lett. 2005;46:3859. [Google Scholar]; (e) Bartoli G, Bosco M, Giuli S, Giuliani A, Lucarelli L, Marcantoni E, Sambri L, Torregiani E. J. Org. Chem. 2005;70:1941. doi: 10.1021/jo048776w. [DOI] [PubMed] [Google Scholar]; (f) Firouzabadi H, Iranpoor N, Nowrouzi F. Chem. Commun. 2005;6:789. doi: 10.1039/b412653j. [DOI] [PubMed] [Google Scholar]; (g) Jia Y-X, Zhu S-F, Yang Y, Zhou Q-L. J. Org. Chem. 2006;71:75. doi: 10.1021/jo0516537. [DOI] [PubMed] [Google Scholar]
- 15.(a) Jung LS, June LJ, Hyeak K-C, Moo JY, Min LB, Hyo KB. Tetrahedron Lett. 2009;50:484–487. [Google Scholar]; (b) Chunchi L, Jianming H, Sastry MNV, Hulin F, Zhijay T, Tsung L-J, Yao C-F. Tetrahedron. 2005;61:11751–11757. [Google Scholar]; (c) Ming C-C, Shijay G, Sastry MNV, C-Fa Y. Tetrahedron Lett. 2005;46:4971–4974. [Google Scholar]; (d) Bimal B-K, Miguel F, Clarissa A. Tetrahedron Lett. 2005;46:2479. [Google Scholar]; (e) Shun W-Y, Shun J-J, Teck L-P. Synlett. 2003;15:2377–2379. [Google Scholar]
- 16.(a) Yi H, Zou G, Li Q, Chen Q, Tang J, He M-Y. Tetrahedron Lett. 2005;46:5665–5668. [Google Scholar]; (b) Raghavan S, Subramanian GS. Tetrahedron. 2011;67:7529–7539. [Google Scholar]
- 17.(a) Ishiwata H, Sone H, Kigoshi H, Yamada K. Tetrahedron. 1994;50:12853–12854. [Google Scholar]; (b) Crimmins MT, King BW, Tabet EA, Chaudhary K. J. Org. Chem. 2001;66:894–902. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]; (c) Crimmins TM, Shamszad M, Mattson AE. Org. Lett. 2010;12:2614. doi: 10.1021/ol1008203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Martin SF, Williamson SA, Gist RP, Smith KM. J. Org. Chem. 1983;48:5170–5180. [Google Scholar]; (b) Schmidt B, Kunz O, Biernat A. J. Org. Chem. 2010;75:2389–2394. doi: 10.1021/jo1002642. [DOI] [PubMed] [Google Scholar]
- 19.Basavaiah D, Rao KV, Reddy RJ. Chem. Soc. Rev. 2007;26:1581–1588. doi: 10.1039/b613741p. [DOI] [PubMed] [Google Scholar]
- 20.Park B-S, Son D-J, Choi S-S, Takeoka GR, Han SO, kim T-W, Lee S-E. Phytother. Res. 2008;22:1195. doi: 10.1002/ptr.2432. [DOI] [PubMed] [Google Scholar]
- 21.Saraswat M, Muthenna P, Suryanarayana P, Petrash JM, Reddy GB. Asia Pac. J. Clin. Nutr. 2008;17:558–565. [PubMed] [Google Scholar]
- 22.Morrison AD, Clements RS, Winegord AI. J. Clin. Investig. 1972;51:3114–3123. doi: 10.1172/JCI107138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Griffiths DG, Partis MD, Sharp RN, Beechey RB. FEBS Lett. 1981;134:261–263. doi: 10.1016/0014-5793(81)80615-1. [DOI] [PubMed] [Google Scholar]; (b) Partis MD, Griffiths DG, Roberts GC, Beechey RB. J. Protein Chem. 1983;2:263–277. [Google Scholar]
- 24.Muthenna P, Suryanarayana P, Gunda SK, Petrash JM, Reddy GB. FEBS Lett. 2009;583:3637–3642. doi: 10.1016/j.febslet.2009.10.042. [DOI] [PubMed] [Google Scholar]
- 25.Petrash JM, Harter TM, Devine CS, Olins PO, Bhatnagar A, Liu S, Srivastava SK. J. Biol. Chem. 1992;267:24833–24840. [PubMed] [Google Scholar]
- 26.Reddy GB, Satyanarayana A, Balakrishna N, Ayyagari R, Padma M, Viswanath K, Petrash JM. Mol. Vis. 2008;14:593–691. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.