

Abstract
ADAPT-NMR (Assignment-directed Data collection Algorithm utilizing a Probabilistic Toolkit in NMR) supports automated NMR data collection and backbone and side chain assignment for [U-13C, U-15N]-labeled proteins. Given the sequence of the protein and data for the orthogonal 2D 1H-15N and 1H-13C planes, the algorithm automatically directs the collection of tilted plane data from a variety of triple-resonance experiments so as to follow an efficient pathway toward the probabilistic assignment of 1H, 13C, and 15N signals to specific atoms in the covalent structure of the protein. Data collection and assignment calculations continue until the addition of new data no longer improves the assignment score. ADAPT-NMR was first implemented on Varian (Agilent) spectrometers [Bahrami, A., Tonelli, M., Sahu, S.C., Singarapu, K.K., Eghbalnia, H.R., Markley, J.L., 2012. PLoS ONE 7, e33173.]. Because of broader interest in the approach, we present here a version of ADAPT-NMR for Bruker spectrometers. We have developed two AU console programs (ADAPT_ORTHO_run and ADAPT_NMR_run) that run under TOPSPIN Versions 3.0 and higher. To illustrate the performance of the algorithm on a Bruker spectrometer, we tested one protein, chlorella ubiquitin (76 amino acid residues), that had been used with the Varian version: the Bruker and Varian versions achieved the same level of assignment completeness (98% in 20 hours). As a more rigorous evaluation of the Bruker version, we tested a larger protein, BRPF1 bromodomain (114 amino acid residues), which yielded an automated assignment completeness of 86% in 55 hours. Both experiments were carried out on a 500 MHz Bruker AVANCE III spectrometer equipped with a z-gradient 5 mm TCI probe. ADAPT-NMR is available at http://pine.nmrfam.wisc.edu/ADAPT-NMR in the form of pulse programs, the two AU programs, and instructions for installation and use.
Keywords: ADAPT-NMR, Bruker, Reduced Dimensionality, Fast NMR data collection, Computational Biology, Structural Biology
INTRODUCTION
NMR spectroscopy offers unparalleled approaches to understanding protein structure, dynamics, and function in a solution environment. The important first step in such studies is the assignment of NMR signals to specific atoms in the covalent structure of the protein. This task has been made more reliable and robust by the application of triple-resonance NMR methods to proteins labeled uniformly with 13C and 15N. Because peaks within 3D spectra of labeled proteins are sparse, reduced dimensionality methods that combine the 13C and 15N dimensions in a variety of tilted planes have proved highly successful in speeding up the times required to collect data for NMR experiments with 1H, 13C, and 15N chemical shifts in the three orthogonal axes [1–3]. However, not all data from a series of 3D experiments is required for successful peak assignments. ADAPT-NMR pioneered an approach that achieves rapid data collection combined with assignment [4]. A reworked and improved version of PINE-NMR [5] served as the probabilistic assignment engine used by ADAPT-NMR. Once an initial body of data has been collected, the ADAPT-NMR assignment engine chooses the next experiment and tilted plane within that experiment to collect on the basis its ability to best improve the current level of probabilistic assignments. ADAPT-NMR estimates the probability of each subnet of the global network of states by calculating a pseudo-energy term that evaluates peak picking and assignment quality according to iterative updates of the generation of spin systems and peak assignments. The new data are then incorporated into the assignment set, and the next experiment and tilted plane within that experiment are chosen. This process proceeds until further data collection no longer improves the extent and quality of assignments.
ADAPT-NMR was implemented initially on Varian (Agilent) spectrometers. In adapting the approach to Bruker BioSpin spectrometers, we were able to reuse the MATLAB part of the Varian version that carries out 2D peak picking, 3D peak generation, and experiment type and tilt angle prediction, because it utilizes frequency-domain data generated by NMRPipe. However, many other parts of ADAPT-NMR had to be modified to account for differences in pulse programming software: VNMRJ scripts (Varian) and AU programs (Bruker). We describe below the implementation of the ADAPT-NMR algorithm on Bruker spectrometers and tests of its performance with [U-13C, U-15N]-labeled proteins.
MATERIALS AND METHODS
Development of Pulse Sequences
The Bruker pulse sequences for the series of 3D experiments used by ADAPT-NMR [HNCO, HN(CA)CB, HNCA, HN(CO)CA, HN(CA)CO, CBCA(CO)NH and C(CO)NH] were modified for reduced dimensionality (2D) through co-evolution of the two indirect dimensions (15N and 13C). These pulse sequences, along with the appropriate acquisition parameter settings, are available from (http://pine.nmrfam.wisc.edu/ADAPT-NMR/). As an example, we show how the conventional HNCO (parameter setting HNCOGPWG3D) was modified to an ADAPT-NMR version:
# ifdef HIFI F1PH(calph(ph4, +90), caldel(d0, +in0) & caldel(d10, +in10) & caldel(d29, +in29) & caldel(d30, -in30) & caldel(d31, +in31)) F2PH(calph(ph5, +90), caldel(d60, +in60)) # else F1PH(calph(ph4, +90), caldel(d0, +in0)) F2PH(calph(ph5, +90), caldel(d10, +in10) & caldel(d29, +in29) & caldel(d30, -in30) & caldel(d31, +in31)) # endif/*HIFI*/
aqseq is set to 321 in the pulse sequence with 15N set to be at the 2nd dimension (inner loop). When the ZGOPTNS flag HIFI is turned on, the real and imaginary parts of N are acquired without independent time evolution by using a dummy delay (d60) and by setting TD2 to 2, and the 15N chemical shift co-evolves with that for 13C. To ensure high resolution along the 13C dimension in the HIFI coevolution version, semi-constant time evolution is used in the 15N dimension:
# ifdef HIFI
“FACTOR2=d30*10000000*2/td1”
“in30=FACTOR2/10000000”
# else “FACTOR2=d30*10000000*2/td2”
“in30=FACTOR2/10000000”
# endif/*HIFI*/
“if (in30 > in10) {in31 = 0;}else {in31=in10-in30;}”
“if (in30 > in10) {in30 = in10;}”
With “in10=inf2/4” and “in29=in10”, where inf2 is defined by the spectral width of the 15N dimension, the semi-constant time period is defined by setting “d30=d23/2+p14/2+d31”, in which “d23=16m” assuming 1JNC ≈ 15 Hz. The pulse sequence is shown in Fig. 1, with the main modification highlighted by the dashed rectangle. To achieve fast and fully-automated NMR data acquisition, processing, and NMR signal assignment, all original 3D NMR experiments were adapted to reduced dimensionality by synchronizing the chemical shift evolution (see the F1PH line above with the definition of HIFI) of 13N and 15C through correlating inf2 (N) = 1/SWN * COS(ϑ) and inf1 (C) = 1/SWc * SIN(ϑ), in which ϑ is the angle between the 1H-15N and 1H-13C planes.
Figure 1.
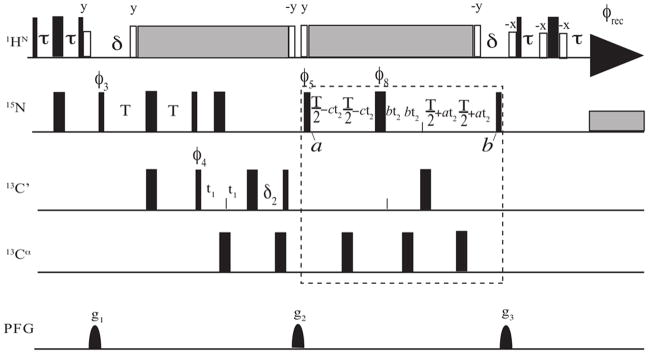
Pulse sequence for the ADAPT-NMR version of HNCO. The time constants were; t (d26) = 2.3 ms, T (d23) = 16 ms, d (d21) = 5.5 ms. The 15N chemical shift evolution was modified to a semi-constant time version, as shown in the box, in order to match the possible requirement for high resolution along the 13C dimension. The 15N and 13C chemical shift evolutions are synchronized by correlating the time incremental interval inf2 (N) and inf1 (C) to a common tilted angle between the two orthogonal planes. On the 500 MHz spectrometer, the 13C′ and 13Cα 90° and 180° selective pulses were achieved by using 384 μs Q5 and 307 μs Q3 pulses, respectively.
TOPSPIN Parameter Files
TOPSPIN parameter sets for 3D experiments were used for the collection of orthogonal plane data. The universal carrier positions for various nuclei were: 1H (4.76 ppm, H2O frequency); 15N (118 ppm); 13Cα shaped pulse (56 ppm); 13Caliphatic, 13Cα or 13Cβ, shaped pulse (45 ppm); 13C′ shaped pulse (176 ppm). The 1H, 15N, 13Cα, 13Caliphatic and 13C′ dimensions were covered, respectively, by 1024, 32, 64, 64, and 64 complex data points (respective spectral widths of 16 ppm, 36 ppm, 32 ppm, 70 ppm, and 22 ppm). As described below, these parameters can be optimized during data collection to yield faster data acquisition and improved assignments.
Acquisition parameters were adjusted manually to improve water suppression so as to obtain optimal signal-to-noise ratio; the last INEPT delay used in all NMR experiments (d26 was set to 2.3 ms with the soft water selective pulse p11 (power level, sp1, is manually optimized) to be 1ms.
These settings ensure a universal phase correction for the direct-detected 1H dimension among all the experiments. In addition, the receiver phase (ph31) was adjusted (flipped) to achieve phasing agreement in data processing among all experiments.
The ADAPT_ORTHO_run AU Program
ADAPT_ORTHO_run is the AU program designed to carry out automated data collection and Fourier transformation to generate 2D orthogonal planes from each of the experiment types in the experiment list file (Fig. 2A). The AU program runs on TopSpin (version 3.0 and patch level 4 or higher are required). ADAPT_ORTHO_run requires three input files: parameters.txt (ADAPT_NMR parameter file), ORTHO_list.txt (experiment list file), and nmrpipe.par (NMRpipe parameter file). The parameters.txt file specifies preset data collection parameters, which are updated to match the experiments in the experiment list file. ORTHO_list.txt supplies default or modified parameters specifying the number of scans, number of increments, carrier positions, and spectral widths.
Figure 2.
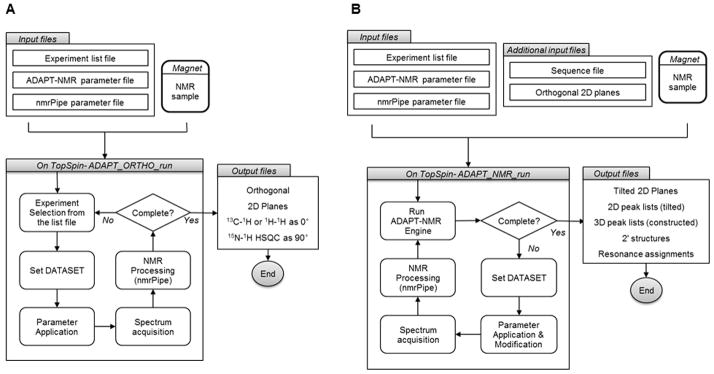
Overall flowchart for the two ADAPT-NMR modules. (A) ADAPT_ORTHO_run, (B) ADAPT_NMR_run.
Pre-installation of NMRpipe is required for transformation of time-domain data to frequency-domain data. NMRpipe parameters such as phasing, extracting, zero filling, and solvent filters must be specified in the NMRpipe parameter file (nmrpipe.par). Time-domain orthogonal planes collected by ADAPT_ORTHO_run are Fourier transformed by NMRpipe automatically and simultaneously. The AU program runs through the experiments specified by the experiment list file (Fig. 2A). ADAPT_ORTHO_run generates transformation script files for each data directory and executes them to produce the orthogonal planes.
The ADAPT_NMR_run AU program
A second AU program running under TopSpin, ADAPT_NMR_run, collects 2D tilted plane data by integrating with the ADAPT-NMR and magnet operation modules (Fig. 2B). Four input files are required to run this program: parameters.txt (ADAPT_NMR parameter file), ADAPT_list.txt (experiment list file), nmrpipe.par (NMRpipe parameter file), and a protein sequence file. Unlike ADAPT_ORTHO_run, all the information from the parameters.txt is read by the program for the refined ADAPT-NMR settings such as peak picking, assignment level, and digital resolution. The format of the experiment list differs from ORTHO_list.txt; ADAPT_list.txt does not contain carrier positions and spectral widths for direct and indirect dimensions because ADAPT_NMR_run acquires them from the orthogonal planes produced by ADAPT_ORTHO_run. However, the number of increments (ni) and scans (nt) are adjustable to achieve better resolution. The most important feature of ADAPT_NMR_run is that it always runs the ADAPT-NMR engine before collecting data for discovering the best tilt angle and experiment type. For achieve this, ADAPT-NMR picks 2D peaks from both the orthogonal planes and tilted planes, and constructs peaks from them in 3D space. If the number of 3D peaks constructed from the tilted planes of a certain experiment is less than that predicted from the orthogonal planes, ADAPT-NMR determines the best tilt angle to be collected for filling the gap. Once this criterion is satisfied for all experiment types in ADAPT_list.txt, ADAPT-NMR starts probabilistic assignment. Additional details about ADAPT-NMR have been published [3]. Currently supported experiments are, 1H,15N HSQC, HNCO, HN(CA)CO, HNCA, HN(CO)CA, HN(CA)CB, CBCA(CO)NH, HBHA(CO)NH, C(CO)NH, and H(CCO)NH. However, the use of ADAPT-NMR for side chain experiments such as HBHA(CO)NH, C(CO)NH, and H(CCO)NH is only recommended for small proteins (less than 5 kD). If the completeness of the assignment does not exceed the assignment_level parameter defined in parameters.txt, the software will acquire data from the experiment types and tilt angles needed fill gaps in the assignment. When the specified assignment level is reached or if the collection of additional data fails to improve the result, ADAPT_NMR_run stops.
Installation of ADAPT-NMR on a Bruker Spectrometer
Execution of the script file install.py installs the MATLAB libraries, ADAPT-NMR executables, pulse sequence, the TopSpin parameters for the experiments, and the two AU programs (ADAPT_ORTHO_run and ADAPT_NMR_run).
Test Results
Two proteins were selected as tests of ADAPT-NMR on Bruker spectrometers: chlorella ubiquitin (76 residues) and human BRPF1 bromodomain (117 residues). Chlorella ubiquitin served as a control, because it had been tested with the Varian (Agilent) version of ADAPT-NMR. The sample contained 1.1 mM [U-13C, 15N]-chlorella ubiquitin in 10 mM phosphate buffer at pH 6.6 containing 0.04% NaN3, 90% H2O, and 10% D2O. We selected the BRPF1 bromodomain as representative of a larger and more challenging protein. The sample contained 1.0 mM [U-13C, U-15N]-BRPF1 bromodomain in 20 mM Tris-HCl buffer at pH 6.8 containing 150 mM NaCl, 10 mM DTT, 90% H2O, and 10% D2O.
Data were collected at 25 °C on a 500 MHz Bruker AVANCE III spectrometer equipped with a z-gradient 5 mm TCI probe. We used TopSpin 3.0 with patch level 4 on a CentOS 5.5 workstation linked to the NMR spectrometer.
RESULTS
The 1H carrier in all NMR experiments was set at the position of the water signal as determined from a 1D 1H experiment by applying a very short (0.5 ms) excitation pulse. The 1H pulse width was adjusted manually by using the command “getprosol”.
The orthogonal 1H-15N and 1H-13C planes were checked by turning off the ZGOPTNS flag “HIFI” and by setting TD1 or TD2 to be 1. Water suppression was optimized manually by adjusting the power level (sp1) of the soft water selective pulse (p11). Then, parameters were set for running ADAPT-NMR in the reduced dimensionality mode by turning on the ZGOPTNS flag “HIFI”, by setting TD2 to 2 (for the real and imaginary part of N), and by changing TD1 to achieve the desired resolution.
The input tables we used for the automatic collection of orthogonal and tilted 2D planes for the two proteins are shown in Table 1. The observed nucleus was 1H, and the indirect dimension in the reduced-dimensionality 3D experiments was a linear combination of the 15N and 13C frequencies corresponding to the angle of the tilted plane.
Table 1.
Input table for orthogonal plane collection. The same values for the number of scans, increment, carrier position, and spectral width were used for tilted plane collection (see Table 2).
| (A) Chlorella ubiquitin
| ||||||
|---|---|---|---|---|---|---|
| Experiment | Keyword | # of scans | # of increments | Name of the plane | Carrier position (ppm) | Spectral width (ppm) |
| 1H,15N-HSQC | ubiq | 2 | 128 | ubiq_NHSQC | 118.0 | 36.0 |
| HNCO | ubiq | 4 | 64 | ubiq_HNCO_0 | 176.0 | 20.0 |
| HN(CO)CA | ubiq | 4 | 64 | ubiq_HNCOCA_0 | 56.0 | 32.0 |
| HNCA | ubiq | 8 | 64 | ubiq_HNCA_0 | 56.0 | 32.0 |
| CBCA(CO)NH | ubiq | 8 | 50 | ubiq_CBCACONH_0 | 45.0 | 70.0 |
| HN(CA)CB | ubiq | 8 | 64 | ubiq_HNCB_0 | 45.0 | 70.0 |
| HN(CA)CO | ubiq | 8 | 128 | ubiq_HNCACO_0 | 176.0 | 20.0 |
| (B) BRPF1 bromodomain
| ||||||
|---|---|---|---|---|---|---|
| Experiment | Keyword | # of scans | # of increments | Name of the plane | Carrier position (ppm) | Spectral width (ppm) |
| 1H, 15N-HSQC | Hbromo | 2 | 128 | Hbromo_NHSQC | 118.0 | 36.0 |
| HNCO | Hbromo | 4 | 64 | Hbromo_HNCO_0 | 176.0 | 22.0 |
| HN(CO)CA | Hbromo | 4 | 64 | Hbromo_HNCOCA_0 | 56.0 | 32.0 |
| HNCA | Hbromo | 8 | 64 | Hbromo_HNCA_0 | 56.0 | 32.0 |
| CBCA(CO)NH | Hbromo | 8 | 58 | Hbromo_CBCACONH_0 | 45.0 | 70.0 |
| HN(CA)CB | Hbromo | 32 | 80 | Hbromo_HNCB_0 | 45.0 | 70.0 |
| HN(CA)CO | Hbromo | 16 | 64 | Hbromo_HNCACO_0 | 176.0 | 22.0 |
Because ubiquitin is known to provide sharp and well-dispersed peaks, we used the default numbers of scans for each experiment. To achieve better resolution, we changed the spectral widths for the HNCO and HN(CA)CO experiments from the default value of 22.0 ppm to 20.0 ppm. For BRPF1 bromodomain, we increased the numbers of default number of scans to those shown in Table 1B to account for the weak peak intensities in the HN(CA)CB and HN(CA)CO experiments. The number of increments for both proteins was optimized to satisfy both speed and resolution.
ADAPT-NMR took about 20 h to collect and assign signals from chlorella ubiquitin, and 55 h for BRPF1 bromodomain (Table 2). Of this time, 2–4 h was used with each protein for repetitive runs of ADAPT_ORTHO_run to determine the optimal parameters for the orthogonal 2D planes. In the first stage, ADAPT-NMR collected a certain number of tilt angles from each experiment in turn; 2D tilted planes recorded from each experiment are shown in the table without parentheses. This process took very little time because the 3D construction was made without deep analysis of the quality of the 3D peaks and the agreement between experiment types. This number of 2D tilted planes was sufficient to identify enough peaks to begin the assignment. In the second stage, ADAPT-NMR ran the torsion angle prediction module and resonance assignment module. By executing these modules, ADAPT-NMR determined what experiment needed to be further collected and what angle should be collected for the experiment. The planes collected by this procedure are shown in Table 3 within parentheses. Whereas ADAPT-NMR spent approximately one minute to suggest a new angle when it did not run the torsion angle prediction resonance assignment modules, it took about one hour for the final stage with the torsion angle prediction and resonance assignment modules.
Table 2.
Orthogonal planes and tilted planes recorded by ADAPT-NMR on Bruker DRX 500 MHz spectrometer.
| (A) Chlorella ubiquitin
| |||
|---|---|---|---|
| Experiment | Orthogonal planes | Angles of tilted planes | Time |
| 1H,15N-HSQC | 90° | - | 40 min |
| HNCO | 0° | 73°, 81°, | 53 min |
| HN(CO)CA | 0° | 76°, 50°, (30°)1 | 2 h 30 min |
| HNCA | 0° | 35°, 58°, 122°, 45°, (17°)1 | 1 h 56 min |
| CBCA(CO)NH | 0° | 46°, 39°, 32°, (54°, 28°, 62)1 | 3 h 38 min |
| HN(CA)CB | 0° | 37°, 50°, 71°, 22° | 3 h 16 min |
| HN(CA)CO | 0° | 49°, 72°, 37° | 5 h 7 min |
| ADAPT-NMR running between data collections | 2 h (approx) | ||
|
| |||
| Total time | 20 h (approx) | ||
| (B) BRPF1 bromodomain
| |||
|---|---|---|---|
| Experiment | Orthogonal planes | Angles of tilted planes | Time (h) |
| 1H,15N-HSQC | 90° | - | 39 min |
| HNCO | 0° | 16°, 54°, | 51 min |
| HN(CO)CA | 0° | 29°, 20°, 56° | 1 h 20 min |
| HNCA | 0° | 24°, 52°, 42°, 34° | 3 h 9 min |
| CBCA(CO)NH | 0° | 29°, 70°, 47°, 40°, (18°, 53°, 43°, 34)1 | 5 h 32 min |
| HN(CA)CB | 0° | 18°, 63°, 54°, 43°, 30°, (23°, 70)1 | 26 h 52 min |
| HN(CA)CO | 0° | 16°, 58°, 48°, 32°, 38°, (40°, 66°, 20) 1 | 12 h 11 min |
| ADAPT-NMR running between data collections | 4 h (approx) | ||
|
| |||
| Total time | 55 h (approx) | ||
Tilt angles in parentheses were recorded after the first continuous recording of tilted planes in the experiment list queue. The ADAPT-NMR engine specified these experiment types and angles on-the-fly to the ADAPT_NMR_run AU program.
DISCUSSION
The quality and completeness of the chemical shift assignments are illustrated in Fig. 3. The color scheme represented follows that of the PINE-NMR webserver [5]. Colors indicate assignment probabilities: ≥99% (green), >85%–99% (cyan), >50%–85%, 50% (red), no candidate spin system identified (gray). As expected, the result is very good for chlorella ubiquitin, (Fig. 3A). The resonance assignments were nearly complete (98%), disregarding prolines and the first residue which could not be assigned with confidence. The result from the Bruker version of ADAPT-NMR was equivalent to that achieved with the Varian (Agilent) version carried out on a 600 MHz Avance spectrometer. ADAPT-NMR required more time and achieved a lower level of assignment completeness with the BRPF1 bromodomain (Fig. 3B) owing to the low sensitivity and resolution of the CBCA(CO)NH, HN(CA)CB and HN(CA)CO experiments. Nevertheless, 86% of the resonances were picked and assigned automatically in two days. The remaining 14% were easily assigned by using the ADAPT-NMR Enhancer program [6], which enables manual picking and editing of peaks and assignments. ADAPT-NMR Enhancer is available for download from <http://pine.nmrfam.wisc.edu.edu/adapt-nmr-enhancer>.
Figure 3.

Assignment results from tests of ADAPT-NMR for Bruker with two proteins: (A) chlorella ubiquitin (76 residues), and (B) human BRPF1 bromodomain (117 residues). The color scheme represented follows that of the PINE-NMR webserver [5]. Colors indicate assignment probabilities: ≥99% (green), >85%–99% (cyan), >50%–85%, 50% (red), no candidate spin system identified (gray).
Many factors limit structure-function investigations of proteins, including the quality and stability of protein samples, availability and cost of NMR spectrometer time, the lack of automation, and the need for NMR spectral expertise. The current version of ADAPT-NMR can help to overcome some of these limitations by combining rapid data collection and automated resonance assignment. We are developing a future version of ADAPT-NMR that will be capable of detecting the low sensitivity of certain experiments (e.g., HN(CA)CB, CBCA(CO)NH, HN(CA)CO) and then do one of three things: (1) collect a regular 3D data set, (2) collect a nonuniform sampling 3D data set, or (3) use the tilted plane data to reconstruct the 3D spectrum in manner of radially-sampled non-uniform sampling data. It is also clear that the addition of NOESY data will improve the extent and quality of assignments made by ADAPT-NMR.
HIGHLIGHTS.
ADAPT-NMR supports combined protein NMR data collection and assignment
A variety of 3D triple-resonance experiments are collected as 2D tilted-planes
We describe the development and testing of ADAPT-NMR for Bruker spectrometers
Acknowledgments
This project was supported by the National Institutes of Health Institute of General Medical Sciences through grant 8P41 GM103399 to JLM. Preparation of the bromodomain was funded by an award from the American Heart Association (10BGIA3420014), and from the National Institutes of Health Institute of General Medical Sciences (R15GM104865) to KCG.
Abbreviations used are
- AU
TopSpin macro programming language
- BMRB
Biological Magnetic Resonance Data Bank
- HIFI-NMR
High-resolution Iterative Frequency Identification for NMR
- PINE-NMR
Probabilistic Interaction Network of Evidence
- TD
parameter in TopSpin representing the number of sampled Time Domain data points
- TopSpin
Bruker’s software package for NMR data acquisition
- VNMRJ
Varian (Agilent)’s software package for NMR data acquisition
- ZGOPTNS
ZG options, parameter in TopSpin used for conditional pulse program execution
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kupce E, Freeman R. Reconstruction of the three-dimensional NMR spectrum of a protein from a set of plane projections. J Biomol NMR. 2003;27(4):383–7. doi: 10.1023/a:1025819517642. [DOI] [PubMed] [Google Scholar]
- 2.Atreya HS, Szyperski T. G-matrix Fourier transform NMR spectroscopy for complete protein resonance assignment. Proc Natl Acad Sci U S A. 2004;101(26):9642–7. doi: 10.1073/pnas.0403529101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eghbalnia HR, Bahrami A, Tonelli M, Hallenga K, Markley JL. High-resolution iterative frequency identification for NMR as a general strategy for multidimensional data collection. J Am Chem Soc. 2005;127(36):12528–36. doi: 10.1021/ja052120i. NIHMSID: NIHMS6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahrami A, Tonelli M, Sahu SC, Singarapu KK, Eghbalnia HR, Markley JL. Integrated protein NMR data collection and assignment by the ADAPT-NMR approach. Plos One. 2012;7(3):e33173. doi: 10.1371/journal.pone.0033173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol. 2009;5(3):e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee W, Bahrami A, Markley JL. ADAPT-NMR Enhancer: complete package for reduced dimensionality in protein NMR spectroscopy. Bioinformatics. 2013;29(4):515–7. doi: 10.1093/bioinformatics/bts692. [DOI] [PMC free article] [PubMed] [Google Scholar]