

Abstract
StyA2B represents a new class of styrene monooxygenases that integrates flavin-reductase and styrene-epoxidase activities into a single polypeptide. This naturally-occurring fusion protein offers new avenues for studying and engineering biotechnologically relevant enantioselective biochemical epoxidation reactions. Stopped-flow kinetic studies of StyA2B reported here identify reaction intermediates similar to those reported for the separate reductase and epoxidase components of related two-component systems. Our studies identify substrate epoxidation and elimination of water from the FAD C(4a)-hydroxide as rate-limiting steps in the styrene epoxidation reaction. Efforts directed at accelerating these reaction steps are expected to greatly increase catalytic efficiency and the value of StyA2B as biocatalyst.
Keywords: catalytic mechanism, flavoprotein, monooxygenase, Rhodococcus opacus 1CP, styrene epoxidation
Introduction
Styrene monooxygenases (StyAB; EC 1.14.14.11) are two-component enzymes performing regio- and enantioselective oxidations (Scheme 1).1,2 The smaller NADH-dependent flavin reductase component StyB produces reduced FAD (FADred) which is taken up by the epoxidase component StyA.2–6 Then oxygen gets also incorporated in StyA and thus activated to a FAD C(4a)-hydroperoxide (FADHOOH) intermediate allowing StyA to perform a variety of biotechnologically relevant epoxidation and sulfoxidation reactions.1–7
Scheme 1.
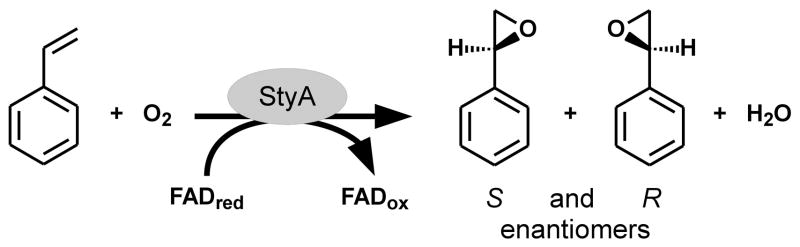
The enantioselective epoxidation of styrene by means of StyA, molecular oxygen, and FADred yields the almost pure S-enantiomer of styrene oxide.
Recently, a novel self-sufficient styrene monooxygenase (StyA2B) from Rhodococcus opacus 1CP was identified that comprises the reductase and epoxidase components in a single polypeptide chain.4 The fused StyA2B protein may have several advantages over conventional two-component StyAB systems.2,4 One major advantage would be the more efficient translocation of FADred from the reductase to the epoxidase active site, so that more epoxide per NADH can be gained.
Steady-state kinetic characterization revealed that the reductase (3.7 U mg−1) as well as epoxidase (0.02 U mg−1) activity of StyA2B are far behind that of two-component StyAB enzymes of pseudomonads (reductase: 200 U mg−1 and epoxidase 2.1 U mg−1).3,4,6 One reason might be that this fused type evolved more recently.8 To gain more insight into the catalytic features of StyA2B, we set out to investigate the kinetics of the reductive and oxidative half-reaction of StyA2B using stopped-flow spectroscopy. The results provide an understanding of the catalytic mechanism of StyA2B and reveal different rate-limiting steps between one-and two-component styrene monooxygenases.
Materials and Methods
His-tagged StyA2B was provided via gene expression (pSRoA2B_P1 in the host E. coli BL21 pLysS) and purification via Ni-NTA affinity chromatography as described previously.4 The protein concentration was either determined by BCA-assay or estimated from the 280 nm absorbance applying the molar extinction coefficient of 71.550 mM−1 cm−1 (StyA2B apo-protein). If FADox was still bound to the protein for the latter protein determination procedure the absorbance of FAD (ε280 20.5 mM−1 cm−1, ε450 11.3 mM−1 cm−1) was considered. Total amount of free oxidized FAD (FADox) in samples was determined after heat denaturation and separation of the protein pellet via centrifugation. Approximately 360 mg pure StyA2B protein out of 6-L fermentation broth was obtained after Ni-NTA purification and subsequent ammonium sulfate precipitation. Protein was stored at −20°C in a storage buffer (100 mM Tris-HCl, pH 7.25, containing 50% v/v glycerol and 100 mM ammonium sulfate) as described elsewhere.4 In order to equilibrate the protein in reaction buffer and to remove unbound flavin, protein samples were passed through a desalting column (Bio-Gel P6, 10 ml; Biorad) prior to experiments.
In general, low-salt Tris-HCl buffers (25 to 100 mM, pH about 7.25) were applied to study the enzyme. Anaerobic conditions were established for redox-titration experiments or kinetic studies, respectively. Therefore a tonometer equipped with a titration port (fixed Hamilton syringe) and a quartz cuvette was used to make samples anaerobic by sequential evacuating and backfilling with purified nitrogen gas via a Schlenk line as reported earlier.5 Kinetic studies were performed by stopped-flow experiments and absorbance or fluorescence data were recorded according Kantz and coworkers.5,9 When studying the oxidative half reaction reduced enzyme was loaded in one drive syringe and mixed with aerobic buffer containing styrene from the other syringe. Reductive half reaction was investigated as follows. Aerobic enzyme was reacted with aerobic NADH. The kinetics of the reduction reaction were found to be independent of NADH concentration suggesting that NADH binds in rapid equilibrium within the 3 ms dead-time of the stopped-flow instrument. The observed kinetics thus represent the first-order kinetic of hydride-transfer from NADH to FAD. All experiments were initiated under pseudo-first order reaction conditions. Observed rate constants were computed by exponential fitting corresponding to the time-dependent evolution of intermediates in the reaction sequence.
In studies of the oxidative half reaction, FADox bound to StyA2B was reduced by titration with sodium dithionite. Data obtained were fitted and plotted using KaleidaGraph version 4.1. Rates were calculated from at least three independently measured traces and the standard error observed was about 15% or less. Numerical modeling was performed with KINSIM11 in order to estimate extinction coefficients of the observed flavin-oxygen adducts.
Results and Discussion
The StyA2B preparation obtained contained about 0.7 mol of FADox per mole of protein. The remaining 30% of StyA2B were supposed to be apo-protein under these aerobic conditions. The absorbance spectrum of protein-bound FADox showed maxima at 357 nm and 456 nm with distinct shoulders at higher wavelengths (Figure 1; under oxidized conditions). Addition of free FADox to StyA2B samples with sub-stoichiometric FADox yielded similar spectra implying that binding of FADox is reversible. The molar extinction coefficients of FADox bound to StyA2B were calculated to ε357 9.8 mM−1 cm−1 and ε456 10.7 mM−1 cm−1.
Figure 1.

Electronic absorbtion spectra representative of StyA2B(FADox), and the StyA2B(FADHOOH), and StyA2B(FADHOH) reaction intermediates. About 25 μM StyA2B and FADox were mixed in 25 mM Tris-HCl buffer (pH 7.25) and spectrophotometrically analysed under oxidizing conditions and after anaerobically titrating with dihionite in the presence or absence of styrene and then exposing to an aerobic atomosphere to genereate stable oxygen intermediates.
Spectra representative of intermediates, which accumulate in the reaction of the StyA2B(FADred) with oxygen and styrene, are compared with the spectrum of StyA2B(FADox) in Figure 1. Spectra were recorded after first preparing StyA2B(FADred) by titration of StyA2B(FADox) with dithionite (stock solution: 2.5 mM) in the presence or absence of 200 μM styrene and then opening the cuvette to an aerobic atmosphere and mixing. In the absence of styrene a spectrum with an absorbance maximum at 378 nm was observed, while in the presence of styrene the absorbance maximum occurred at 370 nm. These observations are in congruence with earlier studies on StyA of Pseudomonas putida S125,9 where during catalysis a FAD (C4a)-hydroperoxide (FADHOOH; maximum absorbance at 382 nm) and FAD C(4a)-hydroxide (FADHOH; maximum absorbance at 368 nm) intermediate were formed.
In previous studies of StyA Pseudomonas putida S129 a minimum of four chemical steps were needed to fully describe the styrene epoxidation reaction: 1) reaction of StyA(FADred) with molecular oxygen to form StyA(FADHOOH), 2) epoxidation of styrene yielding styrene oxide and StyA(FADHOH), 3) elimination of water from StyA(FADHOH) to yield StyA(FADox), and 4) release of FADox into the medium.2a However, for the reaction of StyA2B(FADred) with oxygen and styrene a minimum of five exponentials was needed to satisfactorily describe the absorbance and fluorescence stopped-flow data (Figure 2). This suggests that four reaction intermediates are required to describe the oxidative half reaction of StyA2B as illustrated in Scheme 2. Interestingly, in case of 50 μM styrene in the second phase of the reaction an increase in absorbance at 450 nm was observed (Figure 2). This absorbance change, amounting to 5% of the projected maximum of FADox, has been modeled as uncoupled elimination of hydrogen peroxide occurring in parallel with productive epoxidation of styrene (Scheme 2). It was not found in presence of excess styrene (550 μM) indicating that reactive FADHOOH is stabilized by the substrate. Extinction coefficients computed for the observed reaction intermediates are in a range expected for those compounds and similar to those previously reported in the literature.5,9
Figure 2.

Oxidative half-reaction of 16 μM StyA2B with chemically reduced FAD (only protein bound FAD was used) was investigated by rapid mixing experiments (25 mM Tris-HCl, pH 7.25, 15°C, various styrene: 50 or 550 μM). Reactions were followed with absorbance (375 nm, 390 nm, and 450 nm) and fluorescence (520 nm, excitation at 375 nm). Results were combined and analysed by exponential fitting. Best fits derived with 5 rate constants are shown here as solid lines passing through the experimental data points. Values of the best fitting rate constants are listed in the legend of Scheme 2.
Scheme 2.

Proposed steps of the oxidative half-reaction of StyA2B are shown. Styrene epoxidation (S to SO) yields FADHOH. In parallel uncoupled formation of hydrogen peroxide occurs. Extinction coefficients of FAD-species were calculated via numeric modelling approaches and used to assign the FAD species most likely present.5 Rate constants from exponential fitting were k1 = 85 s−1, k2 = 1.65 s−1, k3 = 0.165 s−1, k4 = 0.065 s−1, k5 = 0.009 s−1 in reactions with 50 μM styrene and k1 = 85 s−1, k2 = 52 s−1, k3 = 0.11 s−1, k4 = 0.06 s−1, k5 = 0.006 s−1 in reactions with 550 μM styrene. Upon rapidly mixing the reduced enzyme with buffer containing styrene in air-saturated buffer, the first observed rate kinetic phase occurs as a single-exponential increase in absorbance corresponding to formation of a FADHOOH intermediate. The proceeding second-order kinetic steps of oxygen and styrene binding to the enzyme are relatively rapid and are thought for this reason not to significantly contribute to the observed kinetics of peroxide formation.
The oxidative half-reaction of StyA2B resembles that of StyA from Pseudomonas putida S12.5 However, there are some striking differences. First, the StyA2B rates are much slower (e.g. StyA epoxidation rate is about 104 s−1).2a Second, the uncoupling at sub-stoichiometric amounts of FAD is rather uncommon for styrene monooxygenases. Third, during catalysis we observed a tremendous stabilization of StyA2B(FADHOH). This intermediate is highly fluorescent as shown earlier.9,12,13 With low amounts of styrene less FADHOH was formed, but when supplying excess substrate it built up and decomposed to FADox extremely slowly as indicated by the very late increase in absorbance at 450 nm (Figure 2; after 10 s). For phenol hydroxylase from Trichosporon cutaneum, stabilization of FADHOH was strongly dependent on the type of substrate and increased in the presence of monovalent anions.13–15 In case of StyA from Pseudomonas putida S12 an ionic strength and pH dependent stabilizing effect was observed as well.9 For p-hydroxyphenylacetate 3-hydroxylase (HPAH) from Acinetobacter baumannii, stabilization of the respective FMN C(4a)-hydroxide was attributed to interaction with a conserved serine.16 However, in case of StyA2B the FADHOH intermediate was formed under normal catalytic conditions and remained very stable as StyA2B(FADHOH) which significantly distinguishes StyA2B from those other flavoprotein monooxygenases.
Transient kinetic data recorded at 450 nm monitoring the reductive half-reaction of StyA2B are presented in Figure 3. In these studies, StyA2B was mixed rapidly in the stopped-flow instrument with NADH and various concentrations of FADox under anaerobic conditions. An initial exponential drop in absorbance (kobs ~ 32 s−1) corresponding to the reduction of StyA2B(FADox) by NADH was followed by steady state turnover of the pool of free FADox (not shown).
Figure 3.

Reaction of 6.9 μM StyA2B with 250 μM NADH and oxygen in the presence and absence of 250 μM styrene as investigated by rapid mixing experiments (25 mM Tris-HCl, pH 7.25, 15°C) in the presence or absence of 15.8 μM added FADox. Absorbance at 340 nm and fluorescence emission at 520 nm were monitored and combined results were analysed by exponential fitting (see Table 1).
To study the rate of NADH and FADox binding, StyA2B was prepared with sub-stoichiometric or excess FADox and reacted in the stopped-flow instrument with excess NADH, either in the absence or presence of styrene under aerobic conditions. Depending on the samples mixed, different numbers of exponentials were needed to satisfactorily fit the data (Figure 3, Table 1). Addition of NADH or FADox first to StyA2B and rapid mixing of the other compound did not differentiate the results. This indicates that NADH and FADox bind rapidly and with high affinity to StyA2B such that the kinetics of the FAD-reduction reaction is not influenced by the preceding FADox- and NADH-binding steps.
Table 1.
Rates observed after mixing StyA2B with NADH in presence of protein-bound or surplus FADox (see Figure 3).
| Signal | FADox | Observed rate constants (k in s−1) from best fits | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Without styrene | With styrene | ||||||||
| Abs. | Bound | 32 | 1.6 | 0.01 | - | 32 | 1.6 | 0.01 | - |
| Surplus | 32 | 0.13 | - | - | 32 | 0.13 | - | - | |
|
| |||||||||
| Rel. Flu. | Bound | 32 | 1.6 | 0.01 | - | 32 | 1.6 | 0.15 | - |
| Surplus | 32 | 0.38 | 0.25 | 0.03 | 32 | 2.2 | 0.13 | 0.03 | |
The kinetics of hydride transfer were monitored by absorbance at 340 nm and fluorescence emission at 520 nm corresponding to NADH oxidation and FAD reduction, respectively (Figure 3). The observed rate constant of this reaction (kred = 32 s−1) was similar when the reaction was run anaerobically. The next phase of the reaction (1.6 s−1) is characterized by slight decreases in 340 nm absorbance and fluorescence signals. We postulate therefore the formation of StyA2B(FADHOOH) from StyA2B(FADred) reacting with oxygen. After a few seconds, a slower decrease in absorbance together with an increase in fluorescence was observed indicating that StyA2B(FADred) and/or StyA2B(FADHOOH) slowly re-oxidized and the formed FADox became again reduced. The picture changed somewhat with FADox present in excess. Again fast reduction of protein-bound FAD was observed followed now by a steady-state reduction of surplus FADox (kred steady-state = 0.13 s−1). StyA2B(FADred) subsequently reacted with oxygen to StyA2B(FADHOOH) and re-oxidized again as indicated by the regain of fluorescence and different rate constants observed (Table 1).
In both cases (stoichiometric and surplus FADox) with presence of styrene the fusion protein still performs an initial fast reduction followed by a steady-state turnover of FADox. FADHOH was formed and stabilized by StyA2B and the rates observed for formation of the highly fluorescent intermediate (Table 1: 0.15 or 0.13 s−1) were comparable to those determined with chemically reduced FAD (k3 = 0.165 or 0.11 s−1). Therefore, styrene epoxidation and corresponding FADHOH formation seem independent of the FADred source. Interestingly, on-going activity of the reductase moiety and so continuous production of reduced FADred did not serve to competitively displace FADHOH from the active site of the epoxidase. This suggests that fusion of the StyA2 and StyB protein components does not significantly affect the behaviour of individual active sites, and is indicative for a diffusive transfer of FAD between both protein components.17,18
Conclusions
The herein investigated self-sufficient and naturally fused styrene monooxygenase StyA2B presents an interesting candidate enzyme for enantioselective biocatalytic applications (Scheme 1). Initial studies revealed that the epoxidase component of StyA2B is rather slow.4 Here we find that the reductive half-reaction of StyA2B proceeds rapidly on a time scale similar to that of the reductase component, StyB of the two-component SMO. The following steps including the transport of FADred to the epoxidase active site of StyA2B and subsequent reaction of FADred with oxygen to form FADHOOH are also rapid and not rate limiting in catalysis. We find the ultimate bottleneck of the oxidative half-reaction of StyA2B occurs at the stages of the styrene epoxidation and FADHOH dehydration, which occur much more slowly than observed in the single component SMO.5 Interestingly, monovalent anions, specific substrates, or low temperatures are not needed to stabilize StyA2B(FADHOH) as with other monooxygenases.5,12–15 Thus, StyA2B provides a valuable system to study FADHOH formation and its interaction with an enzyme.
The rather slow styrene turnover reaction of StyA2B appears to be due to the rate-limiting elimination of water from the FADHOH intermediate. Work focused on accelerating this reaction step including site-directed mutagenesis of residues directly involved in the stabilization of FADHOH16 and directed evolution to further enhance the epoxidase activity2,19 has the potential to better tune StyA2B as an efficient biocatalyst. Naturally fused styrene monooxygenases are rarely found in nature,8 thus comparisons of the mechanism of StyA2B with homologous systems is expected to further provide significant mechanistic insight.
Highlights.
First mechanistic insights into a naturally occurring styrene monooxygenase fusion protein.
Similar flavin intermediates observed as for conventional SMOs.
H2O-elimination of FAD C(4a)-hydroxide found to be severely rate limiting.
Substrate styrene stabilizes FAD C(4a)-hydroxide
Reductase activity does not influence the epoxidase.
Acknowledgments
The authors acknowledge the financial support by NIH SC1 GM081140 to George Gassner and by NWO EIB.10.004 to Willem van Berkel. Dirk Tischler was granted by a pre-doctoral Fulbright scholarship.
Abbreviations
- SMO
styrene monooxygenase
- StyA and StyB
epoxidase and reductase subunit of conventional two-component SMOs
- StyA2B
naturally fused self-sufficient SMO comprising reductase (B) and epoxidase (A2)
- FADox and FADred
oxidized and reduced FAD
- FADHOOH and FADHOH
FAD (C4a)-hydroperoxide and FAD C(4a)-hydroxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dirk Tischler, Email: dirk-tischler@email.de.
Willem J.H. van Berkel, Email: willem.vanberkel@wur.nl.
George T. Gassner, Email: gassner@sfsu.edu.
References
- 1.Hartmans S, van der Werf MJ, de Bont JAM. Bacterial degradation of styrene involving a novel flavin adenine dinucleotide-dependent styrene monooxygenase. Appl Environ Microbiol. 1990;56:1347–1351. doi: 10.1128/aem.56.5.1347-1351.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Montersino S, Tischler D, Gassner GT, van Berkel WJH. Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv Synth Catal. 2011;353:2301–2319. [Google Scholar]; b) van Berkel WJH, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 3.Otto K, Hofstetter K, Röthlisberger M, Witholt B, Schmid A. Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J Bacteriol. 2004;186:5292–5302. doi: 10.1128/JB.186.16.5292-5302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tischler D, Eulberg D, Lakner S, Kaschabek SR, van Berkel WJH, Schlömann M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J Bacteriol. 2009;191:4996–5009. doi: 10.1128/JB.00307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantz A, Gassner GT. Nature of the reaction intermediates in the flavin adenine dinucleotide-dependent epoxidation mechanism of styrene monooxygenase. Biochem. 2011;50:523–532. doi: 10.1021/bi101328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tischler D, Kermer R, Gröning JAD, Kaschabek SR, van Berkel WJH, Schlömann M. StyA1 and StyA2B from Rhodococcus opacus 1CP: a multifunctional styrene monooxygenase system. J Bacteriol. 2010;192:5220–5227. doi: 10.1128/JB.00723-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikodinovic-Runic J, Coulombel L, Francuski D, Sharma ND, Boyd DR, Moore O, Ferrall R, O’Connor KE. The oxidation of alkylaryl sulfides and benzo[b]thiophenesby Escherichia coli cells expressing wild-type and engineered styrene monooxygenase from Pseudomonas putida CA-3. Appl Microbiol Biotechnol. 2013;97:4849–4858. doi: 10.1007/s00253-012-4332-5. [DOI] [PubMed] [Google Scholar]
- 8.Tischler D, Gröning JAD, Kaschabek SR, Schlömann M. One-component styrene monooxygenases: an evolutionary view on a rare class of flavoproteins. Appl Biochem Biotechnol. 2012;167:931–944. doi: 10.1007/s12010-012-9659-y. [DOI] [PubMed] [Google Scholar]
- 9.Kantz A, Chin F, Nallamothu N, Nguyen T, Gassner GT. Mechanism of avin transfer and oxygen activation by the two-component avoenzyme styrene monooxygenase. Arch Biochem Biophys. 2005;442:102–116. doi: 10.1016/j.abb.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 10.Ukaegbu UE, Kantz A, Beaton M, Gassner GT, Rosenzweig AC. Structure and ligand binding properties of the epoxidase component of styrene monooxygenase. Biochem. 2010;49:1678–1688. doi: 10.1021/bi901693u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waschsstock DH, Pollard TD. Transient state kinetics tutorial using KINSIM. Biophs J. 1994;67:1260–1273. doi: 10.1016/S0006-3495(94)80598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghisla S, Entsch B, Massey V, Husain M. On the structure of flavin-oxygen intermediates involved in enzymatic reactions. Eur J Biochem. 1977;76:139–148. doi: 10.1111/j.1432-1033.1977.tb11579.x. [DOI] [PubMed] [Google Scholar]
- 13.Maeda-Yorita K, Massey V. On the reaction mechanism of phenol hydroxylase. New information obtained by correlation of fluorescence and absorbance stopped flow studies. J Biol Chem. 1993;268:4134–4144. [PubMed] [Google Scholar]
- 14.Detmer K, Massey V. Effect of monovalent anions on the mechanism of phenol hydroxylase. J Biol Chem. 1984;259:11265–11272. [PubMed] [Google Scholar]
- 15.Taylor MG, Massey V. Decay of the 4a-hydroxy-FAD intermediate of phenolydroxylase. J Biol Chem. 1990;265:13687–13694. [PubMed] [Google Scholar]
- 16.Thotsaporn K, Chenprakhon P, Sucharitakul J, Mattevi A, Chaiyen P. Stabilization of C4a-hydroperoxyflavin in a two-component flavin-dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J Biol Chem. 2011;286:28170–28180. doi: 10.1074/jbc.M111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison E, Kantz A, Gassner GT, Sazinsky MH. Structure and mechanism of styrene monooxygenase reductase: new insight into the FAD–transfer. Biochem. 2013;52:6063–6075. doi: 10.1021/bi400763h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Webb BN, Ballinger JW, Kim E, Belchik SM, Lam KS, Youn B, Nissen MS, Xun L, Kang CH. Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J Biol Chem. 2010;285:2014–2027. doi: 10.1074/jbc.M109.056135. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tinikul R, Pitsawong W, Sucharitakul J, Nijvipakul S, Ballou DP, Chaiyen P. The transfer of reduced FMN from LuxG oxidoreductase to luciferase occurs via free diffusion. Biochem. 2013 doi: 10.1021/bi4006545. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Jung ST, Lauchli R, Arnold FH. Cytochrome P450: Taming a Wild Type Enzyme. Curr Opin Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]