

Review of how mechanical factors could influence the pathogenesis of emphysema.
Abstract
Transpulmonary pressure and the mechanical stresses of breathing modulate many essential cell functions in the lung via mechanotransduction. We review how mechanical factors could influence the pathogenesis of emphysema. Although the progression of emphysema has been linked to mechanical rupture, little is known about how these stresses alter lung remodeling. We present possible new directions and an integrated multiscale view that may prove useful in finding solutions for this disease.
Emphysema is a slowly progressing disease with no cure other than lung transplantation. The main risk factors of emphysema include cigarette smoking, environmental irritants, genetic factors, and indoor pollutants that lead to inflammation accompanied by gradual airspace enlargement (5). Consequently, lung elastic recoil and gas exchange become progressively compromised. The progression of the disease can be slowed by quitting smoking; however, the rate of decline of lung function in emphysematous patients remains higher than in healthy individuals even after smoking cessation (42).
After decades of studying emphysema, we still do not fully understand its pathogenesis or progression. The majority of emphysema research focuses on uncovering and blocking signaling pathways. As a result, new molecules, including various enzymes, cytokines, or intracellular and extracellular matrix (ECM) constituents that appear to play a role in the pathogenesis and/or progression, are constantly being discovered. Unfortunately, this reductionist approach has not led to a major breakthrough. The complexity of the disease thus highlights the need for cross-disciplinary and integrative approaches.
Cells and the ECM in the lung are under the influence of transpulmonary pressure (Ptp) and the incessant mechanical stresses (force normalized by cross-sectional area) of breathing. In the emphysematous lung, the ECM is damaged, and the stresses of breathing are capable of rupturing the ECM molecules and tissues (34). Perhaps more importantly, these mechanical stresses also influence the basic functioning of adherent cells (12). For example, the mRNA level of collagen type I is upregulated in the presence of cyclic stretching of lung fibroblast in a substrate-dependent manner (8). This phenomenon, called mechanotransduction (28), is well appreciated in many areas of medical science, such as vascular biology (53); however, little attention has been devoted to mechanical stress-induced lung remodeling in emphysema.
In this brief review, we first summarize how the composition and structure of the parenchyma determine organ level function in the normal and emphysematous lung. Based on limited literature, we next review how mechanical factors could influence the pathogenesis of emphysema. The progressive nature of emphysema has been shown to be influenced by mechanical stresses due to rupture of alveolar walls (59). However, little is known about how the heterogeneous distribution of mechanical stresses in the emphysematous lung alters lung remodeling. Finally, we conclude by considering several possible directions that might prove useful in finding solutions for this disease.
Structure of the Normal and Emphysematous Lungs
The lung parenchyma surrounds the airway tree and consists of a large number of gas-exchanging alveoli that form a fine network. The internal surface of the alveoli is lined by alveolar types I and II epithelial cells, which are covered by a thin liquid film. The surface tension at the air-liquid interface contributes to lung elastic recoil. The type II cells release surfactant that reduces the surface tension. The alveolar septa are composed of interstitial cells and the ECM. Cells can slightly alter the local tension on the ECM fibers by contraction. Alternatively, the mechanical stresses of Ptp and breathing are transmitted to the cells via the ECM fibers, mostly collagen and also elastin, both of which are embedded in a soft gel, the proteoglycan (PG) matrix (58). Within the ECM are the fibroblast cells that shape, maintain, and repair the ECM, and these processes are influenced by both biochemical and mechanical cues (36). FIGURE 1 summarizes the general organization of the parenchyma from the scale of an alveolar duct surrounded by alveoli to ECM structure and cell-ECM interactions within the wall.
FIGURE 1.

Structure and complexity of the parenchyma at three length scales
Top: a terminal bronchiole (TB) leading to an alveolar duct (AD). Bottom left: a zoom into a single air-filled alveolus (A) with type I (E1) and type II (E2) alveolar epithelial cells covered by a thin liquid layer. The surfactant (S) molecules at the air-liquid interface. Secretion of lamellar bodies (LB) by the E2 cell is also shown. Bottom right: a schematic representation of the extracellular matrix of the alveolar septal wall with various components, including amorphous elastin (El), wavy collagen (C), complex proteoglycans (PG), basement membrane (BM), and fibroblast cells (F). Image is adapted from Ref. 48 and is used here with permission from Am J Respir Crit Care Med.
During emphysema, the lung structure is gradually destroyed (FIGURE 2). At the largest scale, lung volume is increased. At intermediate scales, the network of alveolar septal walls becomes grossly heterogeneous, with many walls missing and some under high tensile stress. A recent study even suggests that small airways disappear before distal airspace enlargement occurs (40). At an even smaller scale, the ECM is significantly remodeled within the septal walls. This pathological remodeling is heterogeneous with local fibrotic-like collagen deposition as well as decreased elastin content (10). However, the remodeled ECM fibers are visibly disorganized, often showing evidence of rupture (34). In human emphysema, PG content also changes and often decreases (41, 67). Another hallmark of emphysema is the appearance of inflammatory cells, mainly macrophages, neutrophils, and T cells (15), that release various enzymes such as matrix metalloproteinases (MMPs) that can cleave ECM molecules and promote remodeling (52). Finally, fibroblast can undergo oxidative stress-induced apoptosis (4).
FIGURE 2.
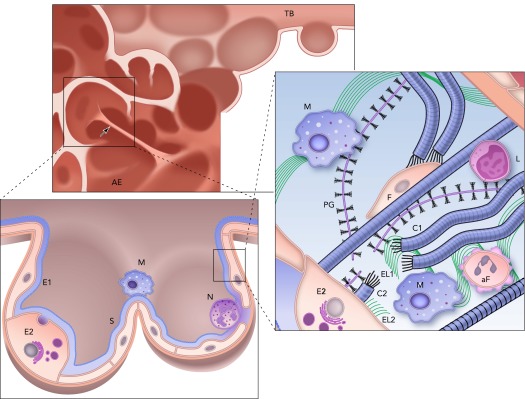
Structure and complexity of the parenchyma at three length scales in the emphysematous lung
Top: the remnants of a terminal bronchiole (TB) leading to airspace enlargement (AE). Note the single straight septal wall under larger than average mechanical stress (arrow). Bottom left: zoomed in image of an enlarged airspace region that used to be two alveoli lined with type I (E1) and type II (E2) epithelial cells covered by a thin liquid layer and surfactant (S). Inflammatory cells, including a macrophage (M) and a neutrophil (N), are also shown. Right: a schematic representation of the extracellular matrix of the septal wall with various components including amorphous elastin (El) and collagen (C), and reduced amount of proteoglycans (PG) and fibroblast (F). Compared with the normal ECM in FIGURE 1, here two macrophages (M) and an apoptotic fibroblast (aF) are also seen. Additionally, collagen C is straight because it is under tension, whereas the wavy fragments of collagen C1 and C2 show a rupture site, where elastin (El1 and El2) as well as PG fragments are found. Note also that a macrophage is recruited near this rupture site by the fragments. L denotes a lymphocyte.
Functional Mechanical Properties of the Normal and Emphysematous Lungs
We first attempt to provide a mechanistic link between structural components in FIGURE 1 and the functional mechanical properties of the lung (60). Since some of the links are unknown, there remains some speculation. Nevertheless, such structure-function relations are useful since lung function is easily measured, whereas structure and composition are less available noninvasively due to limited resolution (e.g., CT imaging) or extent (biopsy). The solid green curve in FIGURE 3 shows a conceptual pressure-volume (P-V) curve of the normal lung inflated from the collapsed state to total lung capacity (TLC). In region 1, Ptp increases with little change in volume due to substantial airway and alveolar collapse. In region 2, Ptp begins to exceed the critical airway opening pressures and airways start to open in avalanches that recruit alveolar airspaces with a steep volume increase (1, 57). As the open regions begin to expand, surface film expansion and possibly proteoglycan compression and shear contribute. In region 3, surface film and alveolar tissue elasticity drive volume changes. Local collagen and elastin fiber alignment and crimp unfolding can occur together with stretching of the elastin. Once most alveoli are recruited, collagen fiber recruitment dominates region 4 with a sharp increase in lung stiffness. During deflation, these processes are likely reversed, although the kinetics of surfactant adsorption-desorption result in lower closing than opening pressures. Consequently, the P-V curve shows a prominent hysteresis, and the amount of trapped air determines residual volume (RV).
FIGURE 3.

Schematic representation of the pressure-volume curve
Schematic representation of the pressure-volume (P-V) curve of a normal (green) and an emphysematous (red) lung during inflation from the collapsed state to total lung capacity (TLC) starting at zero pressure and volume, deflation to residual volume (RV), and during breathing with tidal volume from functional residual capacity (FRC). The regions labeled 1, 2, 3, and 4 correspond approximately to regions of different mechanisms contributing to the curve. The arrow shows the direction of inflation (see text for further explanation). The y-axis is given in percent of TLC of the normal lung.
During breathing, different processes determine lung mechanics (60), primarily because no airway or alveolar collapse occurs. Also, the extent of surface film stretch is much less than during inflation from RV to TLC. Since surface tension toward end-expiration is low, most of the hysteresis loop is thought to be generated by ECM viscoelasticity (49). Thus the low stiffness along the P-V curve during tidal breathing is mostly determined by the mechanics of the ECM. The ECM stiffness is dominated by fiber alignment and stretching of elastin and perhaps of collagen, with additional small contribution from shearing the proteoglycans. The contractile tone of fibroblasts and smooth muscle cells may also influence the slope of the loop.
In the emphysematous lung (FIGURE 3, red curves), the deflation P-V curve is shifted upward. In mice, the inflation P-V curve is also shifted left and up (21) as in the figure, but in rats it changes shape such that the elbow is shifted slightly to the right of the knee of the normal P-V curve (65). To our knowledge, the situation in humans is not known. A left (right) shift of the knee is consistent with a lower (higher) airway opening pressure. The higher opening pressure can be explained by reduced tethering in the emphysematous lung not being able to pull airways open. The reduced tethering is a consequence of fewer septal walls, with possibly reduced stiffness. A left shift could represent reduced airway wall stiffness as a result of airway wall remodeling and local fibrosis enhancing recruitment (21). As inflation proceeds, perhaps similar mechanisms including airway opening in region 2, surface stretching, PG compression, and fiber folding in region 3 can occur. However, absolute lung volume is now higher than in the normal lung because the recoil pressure of the parenchyma and perhaps the pleura are lower.
The small loop represents breathing with increased hysteresis as a consequence of septal wall remodeling (9). The reason is that surface tension appears to be normal in various animal models of emphysema (43) and, due to tissue loss, mean airspace diameters are larger and surface tension effects through Laplace's law should be even smaller than in the normal lung. It is the folding, sliding, and rearrangement of the disorganized fibers during cyclic stretching of the tissue that should lead to higher energy loss (9). The loop is also shifted left and up with a significantly higher slope or dynamic compliance. Little is known about what processes contribute to these dynamic changes. However, the missing septal walls and their remodeled state are likely main contributors to the increased compliance. Additionally, since PGs positively influence tissue stiffness (11), their loss in human emphysema (41, 67) could also contribute to increased lung compliance. Although in the normal lung the bronchial and vascular trees contribute little to lung elastic recoil (54), their contribution is not known in the emphysematous lung.
Enter the Mechanical Stresses
The next question is how the structure of the normal lung in FIGURE 1 transitions to a grossly different structure in emphysema as shown in FIGURE 2. Many different mechanisms have been proposed that involve, for example, complex interactions of proteases (14), inflammation (48), apoptosis (16), or perhaps some combinations of them. Molecular mechanisms have also been advanced based on the idea of homeostatic lung structure maintenance, which can be vulnerable to proteases and oxidants, leading to cell damage and airspace enlargement (63). However, these processes occur in the presence of Ptp and breathing. Is it possible that the mechanical stresses alter the ECM maintenance program and contribute to the pathogenesis and/or the progression of emphysema? We next briefly discuss this possibility.
Gas exchange requires that fresh air travels down the airways and enters the alveoli. Since the airways and alveoli are collapsible, maintaining an open access to the gas-exchange region requires a static distension of the lung provided by Ptp that generates a preexisting tensile stress, or prestress, in the tissue (60). A component of this static stretch is related to gravity due to the weight of the lung. The open access alone does not draw air into the alveoli; the pressure needs to be lower in the alveoli than at the mouth, which is provided by the rhythmic stretching of breathing superimposed on the static prestress. Both the prestress and the cyclic stretch have an enormous influence on how adherent cells work in the lung. Since the discovery of stretch-induced surfactant release in the lung by type II epithelial cells (71), many other cells have been shown to respond to stretch. For example, lung fibroblasts that play a role in ECM maintenance respond to static stretch by phosphorylation of mitogen-activated protein kinases (7), whereas they increase type I collagen expression following cyclic stretch (8). Although fibroblasts from patients with COPD show reduced capability for tissue repair (64), it is not known how the repair processes are influenced by the changes in static and dynamic stresses in emphysema illustrated in FIGURE 3.
Mechanical Stress and Pathogenesis
Often the progressive nature and the pathogenesis of emphysema are not discussed separately, even though separate mechanisms may govern the two processes. Virtually nothing is known about the role of mechanical stresses in the very early phase of pathogenesis. The inhaled cigarette smoke is delivered into the alveoli by regional inspiratory flows, and the agents in the smoke trigger local inflammation. Various signaling and cellular processes in this early inflammatory phase eventually lead to the release of MMPs and/or reactive oxygen species (ROS), and both MMPs (13, 18, 44, 51) and ROS (19, 23, 33) are capable of degrading ECM fibers and PGs. Since emphysema is known to preferentially attack the upper lobes, it was proposed that the higher prestress due to lung weight in the upper lobes plays a role in this process (68). Indeed, the lack of a difference in inflammation between the upper and lower lobes (72) supports the argument that somehow mechanical stresses are involved in upper lobe emphysema.
There are several possibilities to explain these findings. For instance, the higher static component of stress in the upper lobes can influence signaling through integrin-mediated mechanotransduction leading to an increased release of MMPs, specifically MMP-9 (29), or enhanced oxidative stress. Although this hypothesis needs to be tested, there is evidence that extracellular superoxide dismutase, which functions as a superoxide anion scavenger attenuating oxidative stress, protects mouse lungs against cigarette smoke- or elastase-induced airspace enlargement (73). Unfortunately, it is not known whether and how mechanical stresses influence oxidative stress in the emphysematous lung. It has also been reported that cyclic mechanical stretch of fetal lung fibroblasts increases MMP-2 activity and decreases its inhibitor, TIMP-2 (22). However, changes in lung volume during breathing is larger in the dependent regions of the lung, which makes it unlikely that the cyclic component of stress would be responsible for the enhanced emphysema in the upper lobes. If epithelial signaling is involved, then one should also note that changes in lung volume would first unfold the epithelium before stretching the cells (66). An alternative hypothesis is that cigarette smoke triggers inflammation nearly uniformly in the lung, but the higher mechanical stresses in the upper regions accelerate enzyme activity in the ECM.
Recently, we reported that, in normal lung tissue, increased static stress on elastin fibers accelerates the cleavage by elastase due to two mechanisms: 1) stretch on the entropic elastin fibers unfolds elastase binding sites, and 2) a larger stretch of the fiber increases the unbinding off rate of elastase, making the overall cleavage process faster (32). Furthermore, we have recently found that the off rate of elastase on elastin fibers from elastase-induced emphysematous mouse lungs is in fact higher and more stretch dependent than that in normal lungs (FIGURE 4). Although the molecular mechanism at the level of the binding site is unclear, this finding has important implications. The accelerated cleavage in the already damaged fibers can act as a feedback loop at least in two ways. First, following cleavage, exposed lysyl-derived cross-links are chemotactic for monocytes (26). This may serve to maintain local inflammation and further production of MMPs and/or ROS. Interestingly, in a murine model of elastase-induced emphysema, blocking elastin fragments reduced macrophage recruitment into the lung and eliminated airspace enlargement (25). A second possible mechanism is that the cleavage of elastin weakens the fibers, which in turn become more stretched by the prestress that can further accelerate cleavage. This positive feedback loop should eventually lead to rupture of the fibers. Mechanical failure of fibers within the wall weakens the wall itself, with increased likelihood of failure, which is likely central to the progressive nature of emphysema (59, 70).
FIGURE 4.

Unbinding rate measured in normal lung tissue and in emphysematous lung tissue
Unbinding rate (Koff) measured in normal lung tissue and in emphysematous lung tissue obtained at day 3 or 42 following porcine pancreatic elastase treatment of mice using fluorescent recovery after photobleaching (FRAP), as described in Ref. 32. Each bar represents the mean and SD of 50–100 individual FRAP curve measurements. The effect of static uniaxial strain on Koff is significant (P < 0.001). Additionally, independent of strain, the effects of treatment are also significant (P < 0.001), with an interaction that almost reached the significance level (P = 0.052) (Sato S, Bartolak-Suki E, Suki B, unpublished observations).
Mechanical Stress and Progression
Determining the influence of mechanical stresses on the progression of emphysema in a systematic way is difficult because one cannot use “inhibitors” to eliminate Ptp or breathing. However, a recent study used a clever approach to modulate mechanical stresses by graded levels of Ptp during the regrowth of the mouse lung following pneumonectomy (24). Specifically, two levels of plombage, partially filling the thoracic cavity with sterile dental wax, were employed to reduce or abolish mechanical stress, which is thought to be the driving force during regrowth. Plombage following pneumonectomy significantly reduced Ptp, regrowth, and cell proliferation. This has significant implications for the progression of emphysema, since it has been reported that an imbalance between lung cell apoptosis and proliferation contributes to the progression of emphysema (38). Following cigarette smoke extract treatment of rats, tissue destruction was seen by 4 wk that showed progression to 8 wk, and spontaneous repair began at 12 wk. Interestingly, a 4-wk treatment of rats with a peroxisome proliferator activated receptor agonist or granulocyte and macrophage-colony stimulating factor was able to prevent the progression and decreased cell apoptosis. Although the exact mechanism of how mechanical stress would influence the apoptotic process in the emphysematous lung is not known, in cultured human aortic smooth muscle cells, cyclic stretch was found to upregulate the microRNA miR-21 expression, which regulates both proliferation and apoptosis (55). These studies suggest that physiological levels of mechanical stress are needed for proper cellular turnover and ECM maintenance. When the mechanical stress is subphysiological as in emphysema (FIGURE 3), an imbalance of proliferation and apoptosis is created that contributes to the progression of lung destruction. However, apoptosis alone does not degrade the tissue. Therefore, MMP release and enzyme and/or ROS activity is necessary for any further ECM destruction, and this process is substantially influenced by mechanical forces.
The relentless progression of emphysema cannot be explained only in terms of the biology or biochemistry of the disease. The almost unnoticed residual inflammation has been suggested to keep the “cigarette burning” (48) even after cessation of smoking. However, just like apoptosis, inflammation alone does not eliminate alveolar septal walls. Previous experiments have demonstrated (34) and computational modeling (47, 59) supports the notion that, when enzymatic processes sufficiently weaken the septal walls, mechanical stresses of breathing can rupture the walls. When this happens, the stress in the wall before rupture is redistributed among the septal walls of the neighboring alveoli. Consequently, there will be neighbors carrying an increased prestress, which in turn unfolds more binding sites and increases the cleaving rate (32) or enhances mechanotransduction to release more MMPs. These mechanisms in turn increase the probability that the wall mechanically fails. A single wall failure can thus trigger a series of additional ruptures, serving as a positive feedback for further tissue destruction (59). This cascade of events can explain the persistent progression of emphysema with gradual and irreversible decline in function. Furthermore, following a single rupture, the neighbors will not rupture immediately. First, a new mechanical equilibrium is established in which the neighboring walls carry an increased prestress with altered enzyme activity and local mechanotransduction. This new equilibrium may be stable for a while, during which cells may try to compensate for the altered mechanical conditions by aberrant remodeling of the ECM.
The quiet periods can be interrupted by exacerbations. Although an exacerbation is triggered by viral or bacterial infections (50, 56), esophageal pressures during forceful coughing can exceed 200 cmH2O (37), which eventually lead to failure of septal walls with irreversible decline in function. Recently, we tested this hypothesis by delivering frequent deep inspirations to mice 2, 7, or 21 days after elastase treatment (61). At 21 days, deep inspirations changed the distribution of airspace diameters and increased septal wall thickness and the number septal ruptures. These observations suggest that, once a critical remodeling has been reached, acute and sufficiently high mechanical stresses will lead to irreversible changes in structure and function similar to COPD exacerbations. This has indeed been corroborated in a longitudinal study on COPD patients, because the rate of CT-based lung structure destruction was higher in patients with frequent exacerbations (62). Although much remains to be done since nothing is known about how exacerbations might alter mechanotransduction, it is feasible to conclude that the combination of fatigue of damaged fibers, aberrant mechanotransduction, and exacerbations significantly contribute to the slow yet irreversible structure-function deterioration shown in FIGURES 2 AND 3.
New Areas Requiring Research
Three more issues warrant discussion. First, little is known about how individual ECM constituents remodel and fail. The thin blood-gas barrier is supported by the basement membrane composed mostly of type IV collagen and laminin. It has been speculated that the basement membrane may be mechanically compromised in emphysema (69). Collagen fibers, including types I and III, have been directly shown to fail at stresses of normal breathing (34). Although cross-links within and among the ECM components contribute to the strength of the tissue, they likely fail during wall rupture. Furthermore, during ECM remodeling, the cross-linking enzyme lysyl oxidase should play a key role, but its normal function is likely impaired (45). Thus future research should examine the regulation of cross-linking enzymes by ECM composition and stiffness.
The second issue is related to the stiffness of the septal walls. It is now well established that substrate stiffness fundamentally alters cell signaling in a variety of cells (31) including stem cells (17). For example, although severe tissue stiffening has traditionally been regarded as a consequence of fibrosis, it has recently been shown that local ECM stiffness plays an important role in the development of the fibrotic response of fibroblasts (39). In a bleomycin-injured murine model of fibrosis, normal tissue stiffness increases up to sixfold. When cultured lung fibroblasts were exposed to such changes in substrate stiffness, they transitioned from a quiescent state to a state with increasing proliferation and matrix synthesis as well as decreases in matrix proteolytic gene expression. These results suggest a feedback mechanism between matrix stiffening and fibroblast activation that amplifies fibrosis. Surprisingly, little is known about how septal wall stiffness changes in emphysema, most likely due to the highly heterogeneous nature of the tissue structure. Nevertheless, this knowledge is much needed for the understanding of local mechanical stimuli-induced ECM remodeling. What is clear, however, is that the increase in lung compliance is in large part due to the loss of septal walls. Even if septal walls stiffen in emphysema due to local fibrosis, the substantial loss of walls can still result in increased lung compliance. Indeed, although total lung collagen increased in elastase-induced emphysema in mice, the failure stress decreased and lung compliance increased significantly (30). Furthermore, if local ECM stiffness influences mechanotransduction in emphysema, research should also be directed toward actin and focal adhesions that are essential in mechanosensing (4). This area requires much further research.
Finally, we briefly discuss the nature of local stretch pattern. Many studies have reported that both the frequency and the amplitude of stretch substantially influence cellular behavior (20, 36, 64). Furthermore, the pattern of stretch during breathing is not monotonous, but its amplitude and frequency change from cycle to cycle. Such variations have been found to influence surfactant metabolism in type II epithelial cells (2, 3) as well as lung fibroblast signaling in engineered tissue (29). It is thus likely that fibroblast signaling in vivo is also sensitive to stretch pattern. Indeed, preliminary studies using fibroblast from normal and emphysematous mice show difference in ATP synthase level that is a function of variability in stretch (6). Because variability of tidal volume significantly increases from 26% in normal to 43% in emphysema (35), the stretch pattern itself may have a broad effect on general cell signaling in the lung. An interesting possibility arises from the observation that MMP-1, interstitial collagenase, is released by type II epithelial cells in human end-stage emphysema (27). Since type II cells are sensitive to stretch pattern (2), the altered breathing variability in emphysematous patients (35) likely contributes to aberrant mechanotransduction-induced MMP-1 expression that can drive collagen remodeling to weaken its failure properties (34). This idea warrants further experimental investigation.
To summarize, we present a conceptual diagram in FIGURE 5 that illustrates how feedback mechanisms including mechanical stresses, both static and cyclic, local ECM stiffness, and stretch pattern may interact at various spatial and temporal scales with cellular signaling to produce MMPs and/or ROS that can degrade the tissue. The diagram also shows how mechanical stresses can propagate injury and maintain a slow but relentless progression of the disease. Specifically, the red pathways show a possible mechanism of self-sustained progression even after smoking cessation once a critical level of tissue destruction has been reached. Notice also that fiber failure and ultimately wall rupture play a key role since they directly generate airspace enlargement, by redistributing mechanical stresses they feed back to breathing pattern, and, through exposure of fragments, they also maintain inflammation to drive additional signaling. Indeed, fragments of elastin (25, 26), proteoglycans (20), as well as collagen (46) are chemotactic; that is, they attract inflammatory cells. Furthermore, the red dashed line represents the added effect of exacerbations on the rate of progression (61, 62). The proposed concepts attempt to organize the mechanobiology of emphysema to stimulate further experiments in this direction.
FIGURE 5.

Schematic diagram showing the complexity of multiscale signaling and mechanics with embedded feedback mechanisms influencing ECM composition and lung function in emphysema
The arrows represent known or possible links. Notice the feedback from alveolar wall and network mechanics to static stress distribution and dynamic stresses, which decrease transpulmonary pressure and alter breathing pattern. The blue at the bottom represents internal or external triggers. The red pathways show a possible mechanism of self-sustained progression. Fiber and wall failure (red) play a key role here since they directly generate airspace enlargement, feed back to breathing pattern, and, through exposure of fragments, maintain inflammation and drive signaling. The red dashed line represents the added effect of exacerbations on the steady progression.
Conclusion
In this review, we discussed the processes that contribute to the P-V curve of the lung and how these may change in emphysema. We presented arguments that mechanical stresses play an important but not well understood role in both the pathogenesis and progressive nature of emphysema through lung remodeling as well as tissue destruction. However, we did not discuss single molecular pathways in mechanotransduction because they are not yet known; instead, we presented an integrated multiscale view of how mechanical stresses might influence the pathogenesis and progression. Studying mechanotransduction and stress failure is nevertheless important for at least two reasons. Uncovering the cellular pathways of mechanotransduction may open new directions in emphysema research, with the possibility of discovering novel therapeutic targets. Additionally, understanding mechanical stress-induced failure can have implications for quality of life such as exercise tolerance, the type of exercise without eliciting an exacerbation, as well as proper mechanical ventilation of patients that require ventilator support.
Footnotes
This study was supported by National Heart, Lung, and Blood Institute Grant HL-098976.
No conflicts of interest, financial or otherwise, are declared by the author(s).
Author contributions: B.S. and E.B.-S. conception and design of research; B.S. and E.B.-S. prepared figures; B.S. drafted manuscript; B.S. approved final version of manuscript; S.S. and E.B.-S. performed experiments; S.S., M.V.S., and A.T. analyzed data; S.S., H.P., M.V.S., A.T., and E.B.-S. edited and revised manuscript; H.P., M.V.S., and A.T. interpreted results of experiments.
References
- 1.Alencar AM, Arold SP, Buldyrev SV, Majumdar A, Stamenovic D, Stanley HE, Suki B. Physiology: dynamic instabilities in the inflating lung. Nature 417: 809–811, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Arold SP, Bartolak-Suki E, Suki B. Variable stretch pattern enhances surfactant secretion in alveolar type II cells in culture. Am J Physiol Lung Cell Mol Physiol 296: L574–L581, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arold SP, Suki B, Alencar AM, Lutchen KR, Ingenito EP. Variable ventilation induces endogenous surfactant release in normal guinea pigs. Am J Physiol Lung Cell Mol Physiol 285: L370–L375, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Baglole CJ, Bushinsky SM, Garcia TM, Kode A, Rahman I, Sime PJ, Phipps RP. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 291: L19–L29, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 343: 269–280, 2000 [DOI] [PubMed] [Google Scholar]
- 6.Bartolák-Suki E, Suki B. Effects of glucose level and variability in stretch on fibroblast mitochondrial ATP-synthase from normal and emphysematous mouse lungs. Am J Respir Crit Care Med 187: A1887, 2013 [Google Scholar]
- 7.Boudreault F, Tschumperlin DJ. Stretch-induced mitogen-activated protein kinase activation in lung fibroblasts is independent of receptor tyrosine kinases. Am J Respir Cell Mol Biol 43: 64–73, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breen EC. Mechanical strain increases type I collagen expression in pulmonary fibroblasts in vitro. J Appl Physiol 88: 203–209, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Brewer KK, Sakai H, Alencar AM, Majumdar A, Arold SP, Lutchen KR, Ingenito EP, Suki B. Lung and alveolar wall elastic and hysteretic behavior in rats: effects of in vivo elastase treatment. J Appl Physiol 95: 1926–1936, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Cardoso WV, Sekhon HS, Hyde DM, Thurlbeck WM. Collagen and elastin in human pulmonary emphysema. Am Rev Respir Dis 147: 975–981, 1993 [DOI] [PubMed] [Google Scholar]
- 11.Cavalcante FS, Ito S, Brewer KK, Sakai H, Alencar AM, Almeida MP, Andrade JS, Jr, Majumdar A, Ingenito EP, Suki B. Mechanical interactions between collagen and proteoglycans: implications for the stability of lung tissue. J Appl Physiol 98: 672–679, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Chiquet M, Gelman L, Lutz R, Maier S. From mechanotransduction to extracellular matrix gene expression in fibroblasts. Biochim Biophys Acta 1793: 911–920, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Churg A, Wang RD, Tai H, Wang X, Xie C, Dai J, Shapiro SD, Wright JL. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. Am J Respir Crit Care Med 167: 1083–1089, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Churg A, Wright JL. Proteases and emphysema. Curr Opin Pulm Med 11: 153–159, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Cosio MG, Majo J, Cosio MG. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest 121: 160S–165S, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 7: 53, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell 126: 677–689, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Foronjy RF, Okada Y, Cole R, D'Armiento J. Progressive adult-onset emphysema in transgenic mice expressing human MMP-1 in the lung. Am J Physiol Lung Cell Mol Physiol 284: L727–L737, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Gao F, Koenitzer JR, Tobolewski JM, Jiang D, Liang J, Noble PW, Oury TD. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J Biol Chem 283: 6058–6066, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakansson L, Venge P. The molecular basis of the hyaluronic acid-mediated stimulation of granulocyte function. J Immunol 138: 4347–4352, 1987 [PubMed] [Google Scholar]
- 21.Hantos Z, Adamicza A, Janosi TZ, Szabari MV, Tolnai J, Suki B. Lung volumes and respiratory mechanics in elastase-induced emphysema in mice. J Appl Physiol 105: 1864–1872, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawwa RL, Hokenson MA, Wang Y, Huang Z, Sharma S, Sanchez-Esteban J. Differential expression of MMP-2 and -9 and their inhibitors in fetal lung cells exposed to mechanical stretch: regulation by IL-10. Lung 189: 341–349, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi A, Ryu A, Suzuki T, Kawada A, Tajima S. In vitro degradation of tropoelastin by reactive oxygen species. Arch Dermatol Res 290: 497–500, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Hoffman AM, Shifren A, Mazan MR, Gruntman AM, Lascola KM, Nolen-Walston RD, Kim CF, Tsai L, Pierce RA, Mecham RP, Ingenito EP. Matrix modulation of compensatory lung regrowth and progenitor cell proliferation in mice. Am J Physiol Lung Cell Mol Physiol 298: L158–L168, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest 116: 753–759, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hunninghake GW, Davidson JM, Rennard S, Szapiel S, Gadek JE, Crystal RG. Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 212: 925–927, 1981 [DOI] [PubMed] [Google Scholar]
- 27.Imai K, Dalal SS, Chen ES, Downey R, Schulman LL, Ginsburg M, D'Armiento J. Human collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysema. Am J Respir Crit Care Med 163: 786–791, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J 20: 811–827, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Ito I, Nagai S, Handa T, Muro S, Hirai T, Tsukino M, Mishima M. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. Am J Respir Crit Care Med 172: 1378–1382, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Ito S, Ingenito EP, Brewer KK, Black LD, Parameswaran H, Lutchen KR, Suki B. Mechanics, nonlinearity, and failure strength of lung tissue in a mouse model of emphysema: possible role of collagen remodeling. J Appl Physiol 98: 503–511, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Janmey PA, Winer JP, Murray ME, Wen Q. The hard life of soft cells. Cell Motil Cytoskeleton 66: 597–605, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jesudason R, Sato S, Parameswaran H, Araujo AD, Majumdar A, Allen PG, Bartolak-Suki E, Suki B. Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys J 99: 3076–3083, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD. Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxidants Redox Signal 10: 261–268, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kononov S, Brewer K, Sakai H, Cavalcante FS, Sabayanagam CR, Ingenito EP, Suki B. Roles of mechanical forces and collagen failure in the development of elastase-induced emphysema. Am J Respir Crit Care Med 164: 1920–1926, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Kuratomi Y, Okazaki N, Ishihara T, Arai T, Kira S. Variability of breath-by-breath tidal volume and its characteristics in normal and diseased subjects. Ventilatory monitoring with electrical impedance pneumography. Japan J Med 24: 141–149, 1985 [DOI] [PubMed] [Google Scholar]
- 36.Laurent GJ, Chambers RC, Hill MR, McAnulty RJ. Regulation of matrix turnover: fibroblasts, forces, factors and fibrosis. Biochem Soc Trans 35: 647–651, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Lavietes MH, Smeltzer SC, Cook SD, Modak RM, Smaldone GC. Airway dynamics, oesophageal pressure and cough. Eur Respir J 11: 156–161, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Lee JH, Hanaoka M, Kitaguchi Y, Kraskauskas D, Shapiro L, Voelkel NF, Taraseviciene-Stewart L. Imbalance of apoptosis and cell proliferation contributes to the development and persistence of emphysema. Lung 190: 69–82, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol 190: 693–706, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Pare PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 365: 1567–1575, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merrilees MJ, Ching PS, Beaumont B, Hinek A, Wight TN, Black PN. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease. Respir Res 9: 41, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohamed Hoesein FA, Zanen P, de Jong PA, van Ginneken B, Boezen HM, Groen HJ, Oudkerk M, de Koning HJ, Postma DS, Lammers JW. Rate of progression of CT-quantified emphysema in male current and ex-smokers: a follow-up study. Respir Res 14: 55, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouded M, Egea EE, Brown MJ, Hanlon SM, Houghton AM, Tsai LW, Ingenito EP, Shapiro SD. Epithelial cell apoptosis causes acute lung injury masquerading as emphysema. Am J Respir Cell Mol Biol 41: 407–414, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen YT. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 78: 1077–1087, 1998 [PubMed] [Google Scholar]
- 45.Osman M, Keller S, Hosannah Y, Cantor JO, Turino GM, Mandl I. Impairment of elastin resynthesis in the lungs of hamsters with experimental emphysema induced by sequential administration of elastase and trypsin. J Lab Clin Med 105: 254–258, 1985 [PubMed] [Google Scholar]
- 46.Overbeek SA, Braber S, Koelink PJ, Henricks PA, Mortaz E, LoTam Loi AT, Jackson PL, Garssen J, Wagenaar GT, Timens W, Koenderman L, Blalock JE, Kraneveld AD, Folkerts G. Cigarette smoke-induced collagen destruction; key to chronic neutrophilic airway inflammation? PLos One 8: e55612, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parameswaran H, Majumdar A, Suki B. Linking microscopic spatial patterns of tissue destruction in emphysema to macroscopic decline in stiffness using a 3D computational model. PLos Comput Biol 7: e1001125, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Retamales I, Elliott WM, Meshi B, Coxson HO, Pare PD, Sciurba FC, Rogers RM, Hayashi S, Hogg JC. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 164: 469–473, 2001 [DOI] [PubMed] [Google Scholar]
- 49.Sakai H, Ingenito EP, Mora R, Abbay S, Cavalcante FS, Lutchen KR, Suki B. Hysteresivity of the lung and tissue strip in the normal rat: effects of heterogeneities. J Appl Physiol 91: 737–747, 2001 [DOI] [PubMed] [Google Scholar]
- 50.Seemungal TA, Harper-Owen R, Bhowmik A, Jeffries DJ, Wedzicha JA. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur Respir J 16: 677–683, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shapiro SD. Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. Am J Respir Crit Care Med 150: S160–S164, 1994 [DOI] [PubMed] [Google Scholar]
- 52.Shapiro SD, Senior RM. Matrix metalloproteinases. Matrix degradation and more. Am J Respir Cell Mol Biol 20: 1100–1102, 1999 [DOI] [PubMed] [Google Scholar]
- 53.Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res 91: 769–775, 2002 [DOI] [PubMed] [Google Scholar]
- 54.Smith JC, Butler JP, Hoppin FG., Jr Contribution of tree structures in the lung to lung elastic recoil. J Appl Physiol 57: 1422–1429, 1984 [DOI] [PubMed] [Google Scholar]
- 55.Song J, Hu B, Qu H, Bi C, Huang X, Zhang M. Mechanical stretch modulates microRNA 21 expression, participating in proliferation and apoptosis in cultured human aortic smooth muscle cells. PLos One 7: e47657, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stockley RA, O'Brien C, Pye A, Hill SL. Relationship of sputum color to nature and outpatient management of acute exacerbations of COPD. Chest 117: 1638–1645, 2000 [DOI] [PubMed] [Google Scholar]
- 57.Suki B, Barabasi AL, Hantos Z, Petak F, Stanley HE. Avalanches and power-law behaviour in lung inflation. Nature 368: 615–618, 1994 [DOI] [PubMed] [Google Scholar]
- 58.Suki B, Ito S, Stamenovic D, Lutchen KR, Ingenito EP. Biomechanics of the lung parenchyma: critical roles of collagen and mechanical forces. J Appl Physiol 98: 1892–1899, 2005 [DOI] [PubMed] [Google Scholar]
- 59.Suki B, Lutchen KR, Ingenito EP. On the progressive nature of emphysema: roles of proteases, inflammation, and mechanical forces. Am J Respir Crit Care Med 168: 516–521, 2003 [DOI] [PubMed] [Google Scholar]
- 60.Suki B, Stamenovic D, Hubmayr RD. Lung parenchymal mechanics. In: Comprehensive Physiology, The Respiratory System, Respiration Mechanics: Organ, Cell, Molecule, edited by Fredberg JJ, Sieck GC, Gerthoffer WT. Hoboken, NJ: Wiley-Blackwell, 2011, p. 1317–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szabari MV, Parameswaran H, Sato S, Hantos Z, Bartolak-Suki E, Suki B. Acute mechanical forces cause deterioration in lung structure and function in elastase-induced emphysema. Am J Physiol Lung Cell Mol Physiol 303: L567–L574, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanabe N, Muro S, Hirai T, Oguma T, Terada K, Marumo S, Kinose D, Ogawa E, Hoshino Y, Mishima M. Impact of exacerbations on emphysema progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 183: 1653–1659, 2011 [DOI] [PubMed] [Google Scholar]
- 63.Taraseviciene-Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest 118: 394–402, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, Kawasaki S, Ahn Y, Fredriksson K, Skold CM, Mueller KC, Branscheid D, Welker L, Watz H, Magnussen H, Rennard SI. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med 178: 248–260, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tolnai J, Szabari MV, Albu G, Maar BA, Parameswaran H, Bartolak-Suki E, Suki B, Hantos Z. Functional and morphological assessment of early impairment of airway function in a rat model of emphysema. J Appl Physiol 112: 1932–1939, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tschumperlin DJ, Margulies SS. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J Appl Physiol 86: 2026–2033, 1999 [DOI] [PubMed] [Google Scholar]
- 67.van Straaten JF, Coers W, Noordhoek JA, Huitema S, Flipsen JT, Kauffman HF, Timens W, Postma DS. Proteoglycan changes in the extracellular matrix of lung tissue from patients with pulmonary emphysema. Mod Pathol 12: 697–705, 1999 [PubMed] [Google Scholar]
- 68.West JB. Distribution of mechanical stress in the lung, a possible factor in localisation of pulmonary disease. Lancet 1: 839–841, 1971 [DOI] [PubMed] [Google Scholar]
- 69.West JB, Mathieu-Costello O. Strength of the pulmonary blood-gas barrier. Respir Physiol 88: 141–148, 1992 [DOI] [PubMed] [Google Scholar]
- 70.Winkler T, Suki B. Emergent structure-function relations in emphysema and asthma. Crit Rev Biomed Eng 39: 263–280, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science 250: 1266–1269, 1990 [DOI] [PubMed] [Google Scholar]
- 72.Wright JL. Airway inflammatory cells in upper and lower lobes in lungs of patients with and without emphysema. Pathol Res Practice 183: 297–300, 1988 [DOI] [PubMed] [Google Scholar]
- 73.Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, Crapo JD, Rahman I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci USA 107: 15571–15576, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]