

Abstract
The survival of patients with oral squamous cell carcinoma (OSCC) has not changed significantly in several decades, leading clinicians and investigators to search for promising molecular targets. To this end, we performed comprehensive genomic analysis of gene expression, copy number, methylation and point mutations in OSCC. Integrated analysis revealed more somatic events than previously reported, identifying four major driver pathways (mitogenic signaling, Notch, cell cycle, TP53) and two additional key genes (FAT1, CASP8). The Notch pathway was defective in 66% of patients, and in follow-up studies of mechanism, functional NOTCH1 signaling inhibited proliferation of OSCC cell lines. Frequent mutation of CASP8 defines a new molecular subtype of OSCC with few copy number changes. Although genomic alterations are dominated by loss of tumor suppressor genes, 80% of patients harbored at least one genomic alteration in a targetable gene, suggesting that novel approaches to treatment may be possible for this debilitating disease.
Keywords: Integrated genomics, head and neck/oral cancers, NOTCH1, CASP8
Introduction
Head and neck squamous cell carcinoma (HNSCC) includes a diverse group of tumors from the upper aerodigestive tract (1). Oral squamous cell carcinoma (OSCC), a subset of the disease, has a five-year survival rate of only about 50% (1). Limited genomic analysis to date has revealed few molecular alterations that might be exploited clinically as biomarkers for treatment selection or druggable targets. Until recently, TP53 was the only known recurrent mutation, with frequencies ranging from 60 to 80%. We and others recently showed NOTCH1 mutations in 15% of patients by whole exome sequencing (2, 3), thereby opening new avenues of investigation. The Notch pathway in cancer was first recognized in hematopoietic malignancies as oncogenic (4), but in OSCC, NOTCH1 mutations are believed to be tumor-suppressive (2, 3, 5).
Many copy number alterations have been observed in OSCC (1, 6–8). The Retinoblastoma pathway is frequently altered through deletion of CDKN2A (p16) or amplification of Cyclin D1 (CCND1) (1), promoting proliferation. Amplification of the epidermal growth factor receptor (EGFR), also known to promote proliferation, has been reported in 10–30% of patients (1). An EGFR-targeting antibody (cetuximab) shows clinical benefit in combination with radiotherapy for HNSCC patients (9) and is approved for clinical use alone or in combination with radiotherapy or chemotherapy, but no biomarkers are known to predict response to the treatment (10).
Despite the genomic alterations previously observed in OSCC, no therapeutically targetable genomic subtypes have been identified, and therefore, surgery, radiation and cisplatin still form the backbone of standard therapy for the disease. Here we report a multi-platform, integrated genomic analysis that further elucidates signaling pathways exploited by the cancer and identifies several therapeutically targetable genomic subtypes. These findings offer new opportunities for pre-clinical investigation of OSCC with new molecularly-targeted agents.
Results
Copy number alterations
Genome-wide copy number alterations (CNA) as well as tumor purity and tumor ploidy were computed from high-quality SNP arrays from 38 OSCC patients (Supplementary Figure S1A and Tables S1–S2). Thirty-two percent (12/38) of the tumors were diploid, 32% (12/38) were triploid, and 37% (14/38) were tetraploid or had higher ploidy (Supplementary Table S2). We identified 11 regions of focal gain and 33 regions of focal loss (Supplementary Fig. S1B and Table S3), as well as eight recurrent chromosomal arm gains and 10 arm losses (Supplementary Fig. S2A). We found an average of 16.3 focal and 7.3 arm-level CNAs per sample, and 74% (28/38) of tumors had at least 20 CNAs (Supplementary Table S4). The numbers of focal and arm-level CNAs were correlated (R=0.772), suggesting a common mechanism for their development. Together with the high level of polyploidy, those CNA findings suggest that genomic instability is a dominant feature of OSCC.
Detailed analysis of the recurrent CNA events identifies genes and pathways important to the disease. We found five arm-level CNAs occurred in >50% of the tumors (Supplementary Fig. S2A). Included were gains of 8q (63%, 24/38) and 3q (58%, 22/38) and losses of 3p (76%, 29/38), 8p (53%, 20/38) and 18q (58%, 22/38). Forty-seven percent (18/38) of the tumors showed both 3q gain and 3p loss, and 39% (15/38) showed both 8q gain and 8p loss. Focal deletions of FHIT and CSMD1 suggest them as candidate tumor suppressors (on 3p and 8p, respectively), as reported previously in HNSCC (7, 8). Loss of 8p correlated with reduced disease-free survival (p=0.047) and increased extracapsular spread (p=0.021), and it showed a trend toward reduced overall survival (p=0.067). Candidate cancer drivers on 3q include TP63, PIK3CA, TERC and PRKCI; 8q contains MYC. There were focal gains in regions containing CCND1, EGFR, MYC and TP63 and focal deletions in regions containing the tumor suppressor CDKN2A. Interestingly, focal 9p deletions involving the CDKN2A locus sometimes occurred against a background of overall 9p copy number gain. Twenty-six percent of tumors showed 9p gains, nearly as common as the 32% (12/38) of tumors with losses of 9p. That observation strongly suggests that there is an oncogenic driver on 9p. One possible candidate is NFIB, since 3 of the tumors showed high-level amplification (>4 copy gains) of a region on 9p that includes the gene. High-level amplifications were strongly associated with high gene expression (Fig. 1A). Only CCND1 (22%, 8/38) and EGFR (16%, 6/38) demonstrated high level amplifications in >10% of samples. Other high level amplifications that were seen in at least 5% of samples include TP63, MYC and regions on 2q11.2, 5p, 9p23, 14q24.3, and 20p12.1.
Fig. 1.
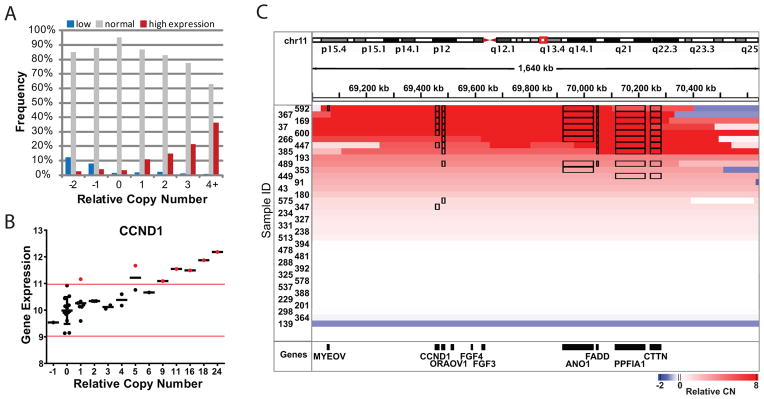
Integrated analysis of gene expression and copy number alterations. A) Copy number and outlier gene expression plot. Frequency indicates how often a gene with the indicated relative CN shows low, normal or high transcript expression. Frequencies were calculated from all genes and all samples with the indicated relative CN. High or low expression identifies samples outside of the 95% confidence interval for expression as described in B below. B) Gene expression vs. copy number for CCND1. Each dot represents one sample. Black horizontal bars designate gene expression mean. Red bars indicate the 95% confidence interval of gene expression for samples with relative CN=0. C) Modified IGV plot for the region around CCND1. Chromosome location is shown at the top. Each row is a sample, and the relative CN is indicated by color. Black boxes around a gene indicate it as an outlier with respect to expression.
Copy number alterations and gene expression
To identify genes under selection within amplified and deleted regions, we compared CNA with expression data. We tested the correlation between gene expression and relative copy number, which normalized copy number to the tumor ploidy, and absolute copy number, which normalized copy number to a normal ploidy of 2. A strong correlation between relative copy number and gene expression (FDR <0.01) was evident for 1721 genes (Supplementary Table S5). Similar analysis for absolute copy number identified only 224 genes at the same significance threshold (Supplementary Table S5). Overall, relative copy number was more strongly correlated with gene expression (i.e., had lower p-values) than was absolute copy number (Supplementary Fig. S2B), which may suggest that polyploid tumors adjust their overall gene expression to compensate for ploidy. Protein expression was also found to be associated with relative copy number, even for low level copy number gains (Supplemental Fig. S2C).
The distribution of relative copy number and gene expression for cyclin D1 (CCND1) is shown in Fig. 1B. Many genes in the focal amplicon on chromosome 11, which includes CCND1, were among the most strongly correlated (CCND1, p=5.66e-9). Although CCND1 is usually described as the oncogenic driver in this amplicon (7, 8) many other genes were also highly expressed, and it is possible that other genes contribute to tumor development (Fig. 1C). Overall, 35 genes, including ORAOV1, IKBKB, FADD and BIRC2, showed stronger correlations than did CCND1. Some of these may even be therapeutic targets, for example knock-down of ORAOV1 (oral cancer over-expressed 1) was shown to reduce tumor growth in OSCC (11).
CNA and microRNA expression data were compared in a similar manner, but the correlations were much weaker. No microRNAs were significant at an FDR <0.01, and only 18 were significant at FDR <0.1 (Supplementary Table S6). Included among the 18 were miR-30b and miR-30d on chromosome 8q, miR-31 on 9p and miR-15b on 3q. Those observations suggested that microRNAs were not frequently regulated by copy number in OSCC.
mRNA and microRNA expression
We evaluated global mRNA and microRNA expression patterns by hierarchical clustering. The most variably expressed mRNAs and microRNAs revealed two major clusters that correlated with the differentiation state of the tumor (p<0.002, Fig. 2A). Expression cluster 1 contained seven out of eight poorly differentiated tumors, whereas expression cluster 2 contained six of six well-differentiated tumors. The genes and microRNAs most differentially expressed between the two groups include squamous differentiation genes, as well as stromal and immune genes (Supplementary Table S7). Expression cluster 1 shows some similarity to the “mesenchymal” subtype defined by Walter et.al. (12), and expression cluster 2 shows similarity to their “basal” subtype. Interestingly, expression cluster 1 tumors contained fewer point mutations than did expression cluster 2 tumors (median 53 vs. 81, p=0.0073) and exhibited higher average ploidy (3.33 vs. 2.62, p=0.033) (Fig. 2B). All non-users of tobacco were in expression cluster 1 (p<0.007).
Fig. 2.

Integrated mRNA and microRNA expression and DNA methylation clusters. A) Clustered heatmap of highly variable mRNAs and microRNAs. Sample IDs are shown at the bottom. B) Plots showing total number of mutations (top) and average ploidy (bottom) per sample for expression clusters 1 and 2. Median or mean values are shown by bars. P-values are indicated in the upper left and calculated by Mann Whitney test or Student’s t-test. C) Clustered heatmap of bimodally methylated probes. Methylated probes are shown in red. D) Plot showing the total number of CNAs per sample for methylation clusters 1 and 2.
DNA methylation
We identified 3694 differentially methylated probes that yielded two primary clusters by unsupervised hierarchical clustering (Fig. 2C and Supplementary Table S8). Methylation cluster 1 showed a higher frequency of DNA methylation, and six samples in methylation cluster 1 exhibited an even higher frequency of methylation, perhaps indicating a CpG island methylator phenotype (CIMP) similar to that observed in colorectal carcinoma (CRC) (13). As in CRC CIMP patients, the samples in methylation cluster 1 exhibited fewer copy number alterations (Fig. 2D); however, other CIMP markers, such as MLH1 methylation and increased numbers of mutations, were not observed in methylation cluster 1. Tobacco use was associated with the methylation clusters (p<0.009); eight out of nine tobacco non-users were in methylation cluster 2 (Fig. 2C).
Notch pathway
NOTCH1 was mutated at 9% in our cohort (Supplementary Tables S9–S11). To explore the mutation rate of NOTCH1 further, we sequenced it in a panel of 44 HNSCC cell lines that had previously been characterized for TP53 mutations and tumorigenicity (14, 15). Sixteen candidate alterations of NOTCH1 identified in those cell lines (Fig. 3A and Supplementary Table S12) included three homozygous frame-shift mutations and 2 homozygous nonsense mutations. As expected, NOTCH1 protein was essentially absent in all three of the truncating mutant cell lines examined (Fig. 3B). The missense mutations found in HN31 (C478F) also occurred in a primary tumor of skin (5), suggesting that it is functional mutation.
Fig. 3.

NOTCH1 is detrimental to HNSCC cell lines. A) NOTCH1 genomic alterations identified in HNSCC cell lines. Mutations of unknown significance are indicated with an *. B) Western blot for total NOTCH1 protein. WT=wild-type, MS=missense, NS=non-sense. FL indicates full length NOTCH1, and TM indicates the transmembrane form. Actin is shown as a loading control. C) Flow cytometry for GFP in HN31 cells infected with retrovirus to express activated NOTCH1 (ICN1). The frequency of events is shown inside each gate. D) Relative cell fractions for the “horse-race” experiment. GFP-positive fraction was normalized to the frequency at day 4. Vector controls are shown with solid lines and ICN1-expressing cells are shown with dashed lines. E) Xenograft tumor growth in an orthotopic mouse model of tongue cancer. HN31 or UM-SCC-47 cell lines were infected with virus to express the full-length NOTCH1 (NFL), the activated form of NOTCH1 (ICN1), or the empty vector. F) Cell cycle analysis of HN31 cells expressing ICN1 or empty vector. Samples were analyzed at day 3 post infection. G) Western blot for p21 (CDKN1A) protein in cells expressing ICN1 or vector. Samples were collected that the indicated time post-infection. Actin is shown as a loading control. H) Senescence-associated beta-galactosidase staining in cells expressing ICN1 or vector. Cells with positive staining appear blue.
To study the role of Notch signaling in HNSCC, we used a retroviral construct to express the cleaved/activated form of NOTCH1 (ICN1) in five cell lines that harbor missense or truncating NOTCH1 mutations. The infections were performed at low multiplicity of infection so that only a fraction of the population (20–50%) expressed the activated NOTCH1 (Fig. 3C). Under those conditions, the relative fraction of ICN1-expressing cells decreased to less than 40% of its original value in 16 days (Fig. 3D). In sharp contrast, the percentage of GFP-positive cells following infection with control virus expressing only GFP remained essentially unchanged for the duration of the experiment. To test it in a more natural context we expressed full-length wild-type NOTCH1 (NFL) in HNSCC cell lines. This was also detrimental to cells, and the effect was further enhanced by activation of NOTCH1 through growth on recombinant ligand (JAG1) (Supplementary Fig. S3). To examine the effect of NOTCH1 in vivo, mouse xenografts were performed in an orthotopic tongue cancer model. HN31 cells expressing the empty vector, ICN1 or NFL were injected into mouse tongues. Both ICN1 and NFL caused a dramatic reduction in tumor size compared with the vector control (Fig. 3E). Similar results were obtained with the cell line UMSCC47 expressing NFL (Fig. 3E). These results lend functional support to the hypothesis, gleaned from mutation data, that the Notch pathway has a tumor-suppressive effect in OSCC.
To begin to understand the mechanisms behind this tumor suppressive effect cell cycle analysis was performed on cells expressing ICN1 (Fig. 3F). Persistent activation of NOTCH1 led to a significant arrest in the G1 phase (i.e, 67% at 3 days post-infection, Fig. 3F), accompanied by induction of p21 (CDKN1A) (Fig. 3G). Cells expressing ICN1 had a rounded morphology and stopped proliferating (Fig. S4A). Tumor cells infected with ICN1 also showed induction of β-galactosidase by day 5 post-infection (Fig. 3H and S4B) suggesting they had become senescent, despite the absence of a flattened morphology typically associated with senescence. Although cleavage of caspase-3 (CASP3) and PARP could not be detected on western blots in lysates from ICN1 infected cells at these early time points (data not shown), we cannot rule out the possibility that at least a fraction of the cells underwent apoptotic death as approximately 15% of the ICN1 infected population had sub-G1 DNA content by cell cycle analysis (Fig. 3F).
To further expand the role of Notch signaling in HNSCC alterations to Notch pathway members were found in 66% (23/35) of the tumors (Fig. 4A and Supplementary Table S13). We identified gains in JAG1, JAG2 and NUMB and losses in MAML1, all of which have been shown to modulate Notch signaling (16). JAG1 and JAG2 have the potential either to inhibit Notch signaling through cis-inhibition or to promote Notch signaling through activation of the receptor (16). Since all other alterations to the Notch pathway indicate a tumor suppressor role for Notch in OSCC, amplifications were likely to inhibit the pathway. TP63 alterations (34%, 12/35) were based on expression specific to the deltaN isoform (ΔN-p63) because that isoform was shown to have oncogenic functions in vitro (17), and its expression was higher than that of the TA isoform in tumors (18). ΔN-p63 directly inhibits NOTCH1 expression and Notch signaling during squamous epithelial differentiation (19–21). TP63 was mutated in 9% (3/35) of the tumors (Fig. 4A) in ways likely to disrupt the tumor-suppressive functions of the TA-p63 isoform.
Fig. 4.

Driver pathways and key genes in OSCC. A) Pathway alteration in each sample is indicated by a black box. Sample IDs are shown at the top and pathway names at the left. Selected clinical variables and clusters from Fig. 3 are indicated with colored boxes. B) Notch pathway. C) Mitogenic signaling pathway. D) Cell cycle pathway. E) TP53 pathway. Pathway diagrams indicate the genes included in each pathway. Copy gains are indicated with red, copy losses with blue, mutations with black and methylation with purple. Percentages shown indicate the frequency of that event.
FAT1
FAT1 was mutated in 30% (12/40) of patients, and two thirds of those were inactivating nonsense, frameshift, or splice site. That is the highest frequency of FAT1 mutation yet observed in HNSCC (2, 3). FAT1 was also focally deleted, yielding a combined inactivation rate of 46% (16/35) (Fig. 4A). FAT1 is a member of the cadherin family of proteins that play important roles in differentiation and control of cell growth. It is very a large transmembrane cadherin protein that regulates developmental programs in humans, mice and flies (22–24) and may also regulate invasion and migration in squamous epithelium (23, 25).
Mitogenic signaling pathway
Alterations in genes important for mitogenic signaling were found to be altered in 63% (22/35) of the tumors (Fig. 4A). Included were activating mutations in HRAS, PIK3CA and BRAF and copy number gains in EGFR, PIK3CA, AKT1, RPS6KB1 and MYC (Fig. 4B). Elevated EGFR expression was previously demonstrated in the majority of HNSCC (1), but the abundance of additional genomic alterations downstream of EGFR may explain the limited clinical responsiveness of HNSCCs to treatment with EGFR inhibitors alone (9, 26).
Cell cycle pathway
Two of the most common genomic alterations in OSCC, amplification of CCND1 and loss of CDKN2A, should facilitate cell cycle progression (Fig. 4D). Together those events are found in 94% (33/35) of tumors. Cell cycle alterations were a nearly universal feature of OSCC.
TP53 pathway
TP53 mutations were observed in 60% (24/40) of the tumors. The majority of mutations were missense, but 13% (12/40) of the tumors harbored a splice site mutation. Splice site mutations, which generally result in no protein expression (14), have been reported as only 2% of all TP53 mutations across tumor types (27). Overall, TP53 mutations were associated with more CNAs (p=0.0051) but no difference in number of mutations. TP53 mutation is a common feature of OSCC that has shown potential for clinical utility as a prognostic/predictive biomarker or target for therapy (28).
Caspase 8
CASP8 was mutated in 10% (4/40) of the tumors (Fig. 4A). CASP8 mutated patients harbored significantly fewer total copy number alterations (p=0.0136) (Fig. 5A). We validated this finding in The Cancer Genome Atlas (TCGA) HNSCC data. Indeed, CASP8 mutations were also strongly associated with fewer focal CNAs in the TCGA cohort (p<0.0001) (Fig. 5B). The only other TCGA tumor site with frequent CASP8 mutations thus far is CRC (13), and remarkably we observed a similar association in these tumors (p<0.0001) (Fig. 5C).
Fig. 5.

CASP8 mutations are associated with fewer CNAs and RAS-family mutations. A) Number of CNAs by CASP8 mutation status in OSCC. B) Focal CNAs by CASP8 status in TCGA HNSCC samples. C) Focal CNAs by CASP8 status in TCGA CRC samples. Median values are shown by the bar. P-values were calculated by Mann Whitney test. D) Number of samples with various CASP8 and Ras-family genotypes. P-values were calculated with Fisher’s exact test. E) CASP8 genomic alterations identified in HNSCC cell lines. Mutations of unknown significance are indicated with an *. F) Frequency of tumor formation based on CASP8 mutation status. Mean values are shown by the bar. P-value was calculated by Mann Whitney test. G) Difference in final tumor volume by CASP8 mutation status. P-value was calculated by t-test. Sample numbers are the same as in H. H) Overall mouse survival by CASP8 mutation status. P-value was calculated by log-rank test. Mouse tumor data are from Sano et.al.(14)
Additionally, CASP8 mutations were strongly correlated with HRAS mutations in our cohort (p=0.0016) (Fig. 5D). Three of the four tumors with CASP8 mutations had an HRAS mutation, and three of the four HRAS mutant tumors had a CASP8 mutation. That association was also validated in the TCGA HNSCC cohort (Fig. 5D), in which seven of eight HRAS mutant tumors also had a CASP8 mutation. In CRC neither HRAS, which was almost never mutated, nor KRAS or NRAS correlated with CASP8; however BRAF mutations did correlate (13). Five of 21 BRAF mutant CRC tumors also had a CASP8 mutation (p=0.0009) (Fig. 5D). The data suggest a functional relationship between some types of RAS-RAF pathway mutations and CASP8 mutations in certain tumors.
In order to begin to understand the functional consequences and relevance of CASP8 mutations, this gene was sequenced in a panel of HNSCC cell lines. Seven candidate mutations were identified. All mutations were heterozygous. CASP8 mutant cell lines were found to be more aggressive in the orthotopic tongue cancer model. They were more tumorigenic (Fig. 5F) and generated larger (Fig. 5G) and more lethal tumors (Fig. 5H). The spectrum of CASP8 mutations in tumors and cell lines (nonsense, splice site, and frame shift) suggests that they likely to be loss-of-function. Expression of a CASP8 shRNA in the HN4 cell line, which reduced CASP8 protein level (Fig. S5A), resulted in larger tumors in the orthotopic tongue cancer model (Fig. S5B). These data are consistent with CASP8 mutations causing a loss-of-function and more aggressive tumors.
Targetable events
We next sought to identify targetable genomic alterations. We found 76 targetable genes with at least one sample containing a copy gain with increased expression or activating mutation (Supplementary Table S14). Forty-five of those genes are therapeutic targets of an agent already approved for use in cancer (Figure 6). In total, 80% (28/35) of the tumors exhibited one targetable genomic alteration, and 54% (19/35) exhibited two or more. Some of the most interesting candidates were TNK2 (17%, 6/35), PTK2/FAK (14%, 5/35) and SRC-family kinases (SRC, LYN, YES1, 29%, 10/35). Candidates with therapeutics already in clinical trials included aurora-family kinases (AURKA/C, 14%, 5/35) and PI3 kinase signaling members (PIK3CA, AKT1/2/3, 34%, 12/35). Those genes may represent novel avenues for therapy of OSCC.
Fig. 6.

Targetable genomic alterations. Genes and frequencies are shown on the left. Approved therapeutics targeting each listed gene are shown on the right. Sample names are along the top. Only copy number events that also demonstrated high expression are shown.
Discussion
We have performed the first comprehensive integrated genomic analysis of OSCC, with a principal goal of identifying cancer drivers, developing molecular classifications for improved prognosis and treatment, and defining new subtypes.
For the first time, we find evidence for oncogenic alterations in the majority of OSCC tumors and targetable alterations in 80% of tumors. That finding is important with respect to the development of novel approaches to therapy because it suggests that therapy directed toward genomic alterations may not depend on synthetic lethal approaches directed toward tumor suppressor genes, as previously suggested (2). Additionally, the majority of tumors showed alterations in multiple targetable genes that are candidates for combination therapy, which is likely to be more successful than single-agent therapy. Although integrated genomic analysis should identify the most likely candidate oncogenic events, they are still candidate events until validated in functional studies. It is hoped that some of these events will prove useful in the treatment of OSCC patients, but it is likely that many of these events will not.
Many of the targetable alterations identified in this study demonstrated high gene expression in combination with small increases in copy number. These alterations may be clinically relevant as the downstream signaling changes could impact response to conventional or targeted therapies. Recent reports indicate that multiple genes from large regions of copy number change can have a cumulative impact on the growth of tumors (29, 30). Our analysis clarifies the genomic landscape of OSCC and paves the way for subsequent studies to address the significance of these recurring alterations.
A mitogenic signaling pathway that includes EGFR, HRAS, and PI3K was altered in >60% of the tumors. EGFR inhibitors have shown modest success in HNSCC (9), but the identification of numerous genomic alterations downstream of EGFR suggests that inhibition of this pathway through other targets should be pursued. PI3K signaling is the most obvious oncogenic target in the pathway that has not yet been robustly pursued for clinical treatment of HNSCC. Inhibitors of this pathway are currently in various stages of clinical development.
The genomic landscape of OSCC is dominated by loss of tumor suppressor genes, most frequently CDKN2A and TP53, which are each lost in the majority of tumors. A surprising new candidate tumor suppressor is FAT1, which was the third most frequently inactivated gene. Like NOTCH1, FAT1 is a large transmembrane protein involved in cell-cell signaling that is cleaved at the membrane by a furin-like convertase and by γ-secretase (23, 31–33). FAT1 has been shown to control migration and invasion in OSCC (25), and recent work has linked FAT1 to wnt signaling in glioblastoma and possibly other tumor types (24). In this study inactivation of FAT1 was shown to promote nuclear localization of β-catenin, activation of wnt pathway genes and proliferation. While the wnt pathway is important in many epithelial tumor types, disruption of this pathway has not generally been associated with OSCC (1, 34). Additional alterations in the wnt pathway were not frequently identified in our analysis, so it is still unclear if FAT1 functions as a tumor suppressor in OSCC by regulating the wnt pathway. The wnt and Notch pathways have been shown to coordinately regulate differentiation in flies and control proliferation mouse intestines. However, in the intestines NOTCH1 acts as an oncogene, and wnt signaling was shown to interact with activation of NOTCH1 to promote proliferation. It is interesting to speculate that perhaps FAT1 is involved in developmental signaling with NOTCH1 and TP63 in OSCC, but it is likely that because NOTCH1 is a tumor suppressor in OSCC the signaling networks may be different from those in other tissues.
Notch signaling is an even more frequently disrupted tumor suppressor than previously observed. The Notch pathway is disrupted in 66% of cases, the key genes being NOTCH1 and TP63. We do not observe any correlation or mutual exclusivity between Notch and TP63 defects, even though the two genes are known to interact functionally. We have identified alterations in four additional genes that regulate Notch signaling, highlighting the importance of Notch in OSCC carcinogenesis. In functional experiments, growth inhibition was seen with activated NOTCH1 in all cell lines tested, including some with wild-type NOTCH1 sequence (data not shown)(35). These data emphasize the key role of Notch signaling in OSCC. Although Notch regulates differentiation, Notch pathway alterations were not associated with the differentiation state of the tumor, and Notch disruption did not completely prevent squamous differentiation (data not shown). It is likely that Notch signaling regulates many aspects of tumor physiology and response to treatment. It will be important to identify robust biomarkers for Notch pathway function that can be analyzed in future clinical trials.
There was a strong correlation between CASP8 mutations and HRAS mutations. HRAS-mutant tumors were generally a subset of the CASP8 mutant tumors, suggesting that CASP8 mutation is permissive for HRAS mutation. Consistent with that hypothesis, it has been shown that oncogenic HRAS sensitizes fibroblasts to TRAIL-induced apoptosis and CASP8 cleavage (36). For reasons not known, CASP8 mutant tumors also showed fewer CNAs. These data suggest that CASP8 mutations may prevent global CNAs in OSCC and other tumor types. However, this finding is contrary to the increased chromosomal instability found in a mouse model of CASP8 loss in B-cell lymphoma (37), suggesting that tissue or species-specific factors may influence the role of CASP8 in genomic stability. Interestingly, alterations to other members of the extrinsic apoptosis pathway (FADD, BIRC2, BIRC3) were not associated with fewer CNAs and may suggest that this phenotype is related to a caspase independent function of CASP8. If inactivation of CASP8 is shown to prevent genomic instability in HNSCC then it may indicate a novel avenue for therapeutic intervention; caspase inhibition might sensitize tumors with wild-type CASP8 to chemotherapeutics that induce genomic alterations. Additionally, as the majority of HNSCC tumors have frequent CNAs, the inverse correlation with CASP8 mutation may provide an important clue for unraveling the mechanism(s) that give rise to genomic instability in OSCC.
Overall, the genome of OSCC contains many alterations, including frequent oncogenic drivers and novel candidate therapeutic targets. It will be important to incorporate genomic analysis into prospective clinical trials, as these findings will only become useful by extending them to well annotated and clinically relevant cohorts. Although the sample size described here is not large (N=40), only the most robust findings were reported and we expect that the majority of these results will be validated in larger cohorts. It is exciting to have identified alterations that can be used clinically as biomarkers for treatment selection or targets for new molecularly based treatments. It may soon be possible to make individualized biologically-based treatment decisions that can improve the survival and/or quality of life for patients suffering from OSCC.
Methods
Tissue processing
Fresh-frozen surgically resected non-recurrent tumor and matched non-malignant adjacent tissue were obtained from consented patients treated for HNSCC at The University of Texas M.D. Anderson Cancer Center, under an Institutional Review Board approved protocol. Frozen tissue was embedded in optimal cutting temperature compound and cryosections from the top and middle of specimens were stained with hematoxylin and eosin prior to being evaluated by a pathologist for the presence of > 60% tumor nuclei content or absence of tumor (i.e., normal). Samples that passed this criterion were completely sectioned and washed once in PBS prior to isolating genomic DNA using an ArchivePure DNA purification kit (5Prime).
Genomic analysis summary
Exome DNA was captured with Nimblegen reagents (Nimblegen) and sequenced on SOLiD or Illumina machines. SNP analysis was performed on SNP6.0 arrays (Affymetrix) and the data was analyzed with Partek (v6.6, Partek Inc), ASCAT (v1.0, (38)), GISTIC (v2.0.12, (39)), Circos (v0.53, (40)) and R software. DNA methylation was determined by using the HumanMethylation450 arrays (Illumina) and analyzed in R. Total RNA was isolated with TRI reagent (Sigma-Aldrich) and hybridized to Human Exon 1.0ST arrays (Affymetrix) and Human microRNA microarrays rel12.0 (Agilent). RNA data analysis was performed in R. Additional details of the genomic analysis and integrated analysis are described in the supplemental methods.
Sequencing NOTCH1 and CASP8 in cell lines
PCR primers were designed for amplification of each exon of NOTCH1. The PCR primers were designed to include sequencing primers, and the amplified exons were sequenced by Sanger chemistry on an ABI3730 instrument as previously described (41). DNA sequencing reads were processed using SNPdetector (42).
HNSCC cell lines
All HNSCC cell lines used in this study were collected from the original source of the cell line or ATCC and were STR profiled for authentication. The assembly, characterization, and STR profiling of these cell lines is described in detail in Zhao et.al.(15).
Western blot analysis
Protein lysates from the indicated cells were collected in RIPA buffer and quantified with the DC Protein Assay Kit (Bio-Rad Laboratories). 30ug of protein was separated on 7.5% Mini-Protean TGX pre-cast gels (Bio-Rad Laboratories) or 10% SDS-PAGE gels and transferred to PDVF membranes. Membranes were blocked in 5% milk and probed with primary antibodies to NOTCH1 (C-20, sc6014, Santa Cruz Biotechnology), p21WAF1 (OP64, Calbiochem), Caspase-8 (551243, BD Pharmingen) or beta-actin (sc81178, Santa Cruz Biotechnology), and secondary antibodies to rabbit or mouse IgG linked to HRP (#7074 & #7076, respectively, Cell Signaling Technologies) in 2.5% milk. HRP was detected with ECL Detection reagents (Amersham, GE Healthcare Life Sciences).
Horse race & FACS
The intracellular domain of human NOTCH1 (ICN1, a1760-2555) was cloned into the MigR1 (murine stem cell virus-based) retroviral vector that coexpresses GFP as described previously. Retroviral supernatants were prepared as described previously (43). HNSCC cell lines were infected with retroviral supernatant in the presence of polybrene. GFP fractions were measured by flow cytometry (LSRFortessa, BD Biosciences) and analyzed with FlowJo Software (TreeStar). Relative cell fraction was generated by normalizing the % GFP positive on each day to the % GFP positive on day 4.
Senescence assay
Cells were infected with ICN-GFP or MigR1 GFP for 3 days, re-plated at 25,000 cells/well into 6-well plates for an additional 3 days before fixation in 2% formaldehyde/0.2% glutaraldehyde and staining with a Senescence β-galactosidase Kit from Cell Signaling.
Cell cycle analysis
HN31 cells were infected with either ICN1-GFP or MigR1-GFP to achieve over 90% infection and cells were analyzed for cell cycle distribution by propidium iodide staining 3 days post-infection. Cells were trypsizined, washed in PBS, fixed at room temperature by addition of cold 70% ethanol and stored at 4 C before adding propidium iodide/RNase and analyzing on an iCyt Eclipse Flow Cytometer.
Orthotopic nude mouse model of oral cavity cancer
All animal experimentation was approved by the Animal Care and Use Committee (ACUC) of the University of Texas MD Anderson Cancer Center. Our orthotopic nude mouse model of oral cavity cancer has been previously validated and described in the literature (44). Male athymic nude mice were obtained from the NCI. UM-SCC-47 and HN31 parental cells or cells stably expressing Notch intracellular domain (ICN), full length Notch (NFL), or MIGR1 vector control (vector) were harvested from subconfluent culture by trypsinization and washed with PBS. For CASP8 experiments HN4 parental cells or cells stably expressing CASP8shp30 (clone V2LHS_112730), CASP8shp31 (clone V2LHS_112731), or GIPZ vector control (vector) were harvested from subconfluent culture by trypsinization and washed with PBS. Groups of 8–10 mice were injected into the tongues with (5×104) cells suspended in 30 μL of PBS as described previously (44). Mice were examined twice a week for 10 weeks where tumor size and weight loss were assessed and recorded. Tongue tumors were measured with microcalipers, and tumor volume calculated as (A)(B2)π/6, where A is the longest dimension of the tumor and B is the dimension of the tumor perpendicular to A. In order to compare the tumor growth between cell lines the mean tumor volume of each cell line was plotted for each day calculated. Mice were euthanized by CO2 asphyxiation when they lost more than 20% of their preinjection body weight. Primary tumors were resected, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Overall survival was determined by Kaplan-Meier plots and compared using log-rank statistics.
Supplementary Material
Statement of significance.
This is the first integrated genomic analysis of oral squamous cell carcinoma. Only through integrated multi-platform analysis was it possible to identify 4 key pathways. We also discovered a new disease subtype associated with CASP8 and HRAS mutation. Lastly, many candidate targetable events were found and provide hope for future genomically driven therapeutic strategies.
Acknowledgments
Financial support: This work was supported by the Cancer Prevention Research Institute of Texas grant RP100233; National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research grant RC2DE020958; NIH Specialized Program of Research Excellence grants P50CA097007; Cancer Center Support Grant P30CA0CA16672 and the Pantheon Program. C.R.P is a TRIUMPH Fellow and supported by the GSK Translational Research Fellowship.
We thank our patients for their courage and generosity. We thank the members of the Myers lab for moral support, technical support, and helpful discussions. This work was supported by the Cancer Prevention Research Institute of Texas grant RP100233 (to J.N. Myers); National Institutes of Health (NIH)/National Institute of Dental and Craniofacial Research grant RC2DE020958 (to J.N. Myers); NIH Specialized Program of Research Excellence grants P50CA097007; Cancer Center Support Grant P30CA0CA16672; an Institutional Research Grant from the University of Texas M.D. Anderson Cancer Center (to M.J. Frederick) and the Pantheon Program. C.R.P is a TRIUMPH Fellow and supported by the GSK Translational Research Fellowship. Expression and methylation data have been deposited in the Gene Expression Omnibus database (accession no. GSE41117).
Abbreviations
- OSCC
oral squamous cell carcinoma
- HNSCC
head and neck squamous cell carcinoma
- CNA
copy number alterations
- CIMP
CpG island methylator phenotype
- CRC
colorectal carcinoma
- ICN1
cleaved/activate NOTCH1
- NFL
full-length wild-type NOTCH1
- TCGA
The Cancer Genome Atlas
Footnotes
Conflicts: The authors disclose no potential conflicts of interest.
References
- 1.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 5.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:17761–6. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bockmuhl U, Petersen I. DNA ploidy and chromosomal alterations in head and neck squamous cell carcinoma. Virchows Arch. 2002;441:541–50. doi: 10.1007/s00428-002-0729-3. [DOI] [PubMed] [Google Scholar]
- 7.Gollin SM. Chromosomal alterations in squamous cell carcinomas of the head and neck: window to the biology of disease. Head Neck. 2001;23:238–53. doi: 10.1002/1097-0347(200103)23:3<238::aid-hed1025>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 8.Wreesmann VB, Singh B. Chromosomal aberrations in squamous cell carcinomas of the upper aerodigestive tract: biologic insights and clinical opportunities. J Oral Pathol Med. 2005;34:449–59. doi: 10.1111/j.1600-0714.2005.00343.x. [DOI] [PubMed] [Google Scholar]
- 9.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 10.Le Tourneau C, Siu LL. Molecular-targeted therapies in the treatment of squamous cell carcinomas of the head and neck. Curr Opin Oncol. 2008;20:256–63. doi: 10.1097/CCO.0b013e3282f9b575. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, Zeng X, Yang H, Wang Z, Shen J, Bai J, et al. Oral cancer overexpressed 1 (ORAOV1): a regulator for the cell growth and tumor angiogenesis in oral squamous cell carcinoma. Int J Cancer. 2008;123:1779–86. doi: 10.1002/ijc.23734. [DOI] [PubMed] [Google Scholar]
- 12.Walter V, Yin X, Wilkerson MD, Cabanski CR, Zhao N, Du Y, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8:e56823. doi: 10.1371/journal.pone.0056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Network TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sano D, Xie TX, Ow TJ, Zhao M, Pickering CR, Zhou G, et al. Disruptive TP53 mutation is associated with aggressive disease characteristics in an orthotopic murine model of oral tongue cancer. Clin Cancer Res. 2011;17:6658–70. doi: 10.1158/1078-0432.CCR-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao M, Sano D, Pickering CR, Jasser SA, Henderson YC, Clayman GL, et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin Cancer Res. 2011;17:7248–64. doi: 10.1158/1078-0432.CCR-11-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene. 2008;27:5148–67. doi: 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Lu H, Yan B, Romano RA, Bian Y, Friedman J, et al. DeltaNp63 versatilely regulates a Broad NF-kappaB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res. 2011;71:3688–700. doi: 10.1158/0008-5472.CAN-10-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Dotto GP. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat Rev Cancer. 2009;9:587–95. doi: 10.1038/nrc2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koster MI, Dai D, Roop DR. Conflicting roles for p63 in skin development and carcinogenesis. Cell Cycle. 2007;6:269–73. doi: 10.4161/cc.6.3.3792. [DOI] [PubMed] [Google Scholar]
- 21.Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. Embo J. 2001;20:3427–36. doi: 10.1093/emboj/20.13.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ciani L, Patel A, Allen ND, ffrench-Constant C. Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol Cell Biol. 2003;23:3575–82. doi: 10.1128/MCB.23.10.3575-3582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sopko R, McNeill H. The skinny on Fat: an enormous cadherin that regulates cell adhesion, tissue growth, and planar cell polarity. Curr Opin Cell Biol. 2009;21:717–23. doi: 10.1016/j.ceb.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. 2013;45:253–61. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishikawa Y, Miyazaki T, Nakashiro K, Yamagata H, Isokane M, Goda H, et al. Human FAT1 cadherin controls cell migration and invasion of oral squamous cell carcinoma through the localization of beta-catenin. Oncol Rep. 2011;26:587–92. doi: 10.3892/or.2011.1324. [DOI] [PubMed] [Google Scholar]
- 26.Harari PM, Wheeler DL, Grandis JR. Molecular target approaches in head and neck cancer: epidermal growth factor receptor and beyond. Semin Radiat Oncol. 2009;19:63–8. doi: 10.1016/j.semradonc.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–9. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 28.Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–61. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–9. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc Natl Acad Sci U S A. 2012;109:8212–7. doi: 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magg T, Schreiner D, Solis GP, Bade EG, Hofer HW. Processing of the human protocadherin Fat1 and translocation of its cytoplasmic domain to the nucleus. Exp Cell Res. 2005;307:100–8. doi: 10.1016/j.yexcr.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Tanoue T, Takeichi M. New insights into Fat cadherins. J Cell Sci. 2005;118:2347–53. doi: 10.1242/jcs.02398. [DOI] [PubMed] [Google Scholar]
- 33.Sadeqzadeh E, de Bock CE, Zhang XD, Shipman KL, Scott NM, Song C, et al. Dual processing of FAT1 cadherin protein by human melanoma cells generates distinct protein products. J Biol Chem. 2011;286:28181–91. doi: 10.1074/jbc.M111.234419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–34. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duan L, Yao J, Wu X, Fan M. Growth suppression induced by Notch1 activation involves Wnt-beta-catenin down-regulation in human tongue carcinoma cells. Biol Cell. 2006;98:479–90. doi: 10.1042/BC20060020. [DOI] [PubMed] [Google Scholar]
- 36.Nesterov A, Nikrad M, Johnson T, Kraft AS. Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2004;64:3922–7. doi: 10.1158/0008-5472.CAN-03-2219. [DOI] [PubMed] [Google Scholar]
- 37.Hakem A, El Ghamrasni S, Maire G, Lemmers B, Karaskova J, Jurisicova A, et al. Caspase-8 is essential for maintaining chromosomal stability and suppressing B-cell lymphomagenesis. Blood. 2012;119:3495–502. doi: 10.1182/blood-2011-07-367532. [DOI] [PubMed] [Google Scholar]
- 38.Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010;107:16910–5. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Wheeler DA, Yakub I, Wei S, Sood R, Rowe W, et al. SNPdetector: a software tool for sensitive and accurate SNP detection. PLoS Comput Biol. 2005;1:e53. doi: 10.1371/journal.pcbi.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zweidler-McKay PA, He Y, Xu L, Rodriguez CG, Karnell FG, Carpenter AC, et al. Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood. 2005;106:3898–906. doi: 10.1182/blood-2005-01-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sano D, Myers JN. Xenograft models of head and neck cancers. Head Neck Oncol. 2009;1:32. doi: 10.1186/1758-3284-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.