

Abstract
The central dogma of gene expression is that DNA is transcribed into messenger RNAs, which in turn serves as the template for protein synthesis. The discovery of extensive transcription of large RNA transcripts that do not code for proteins, termed long noncoding RNAs (lncRNAs) provide an important new perspective on the centrality of RNA in gene regulation. Here we discuss genome-scale strategies to discover and characterize lncRNAs. An emerging theme from multiple model systems is that lncRNAs form extensive networks of ribonucleoprotein (RNP) complexes with numerous chromatin regulators, and target these enzymatic activities to appropriate locations in the genome. Consistent with this notion, long noncoding RNAs can function as modular scaffolds to specify higher order organization in RNP complexes and in chromatin states. The importance of these modes of regulation is underscored by the newly recognized roles of long RNAs for proper gene control across all kingdoms of life.
Keywords: functional genomics, chromatin, histone modifications, epigenetics
INTRODUCTION
The centrality of RNA in the flow of genetic information came to light in Jacob and Monod’s 1961 paper “Genetic Regulatory Mechanisms”, establishing the concept of messenger RNAs (1). In the 50 years since this landmark paper, numerous regulatory RNAs of all shapes and sizes have been discovered (2, 3). Long noncoding RNA (lncRNAs) biochemically resemble mRNAs posited by Jacob and Monod, yet do not template protein synthesis. Rather, lncRNAs functioning as RNA genes to orchestrate genetic regulatory outputs. Today, lncRNA transcripts have emerged as a cryptic, but critical layer in the genetic regulatory code (Fig. 1, 2).
Figure 1.
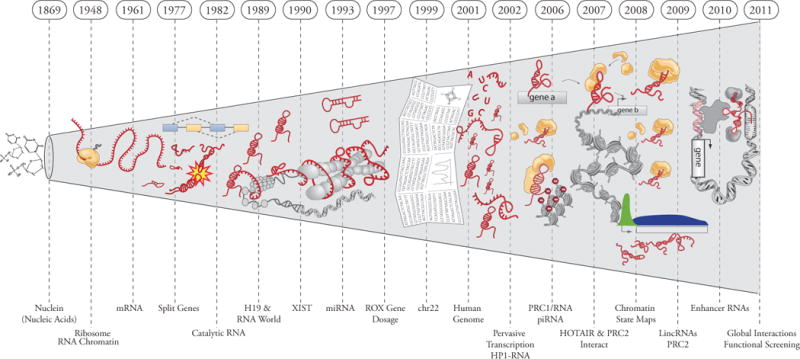
Timeline of discoveries of RNAs in biological regulation.
Figure 2. Anatomy of lncRNA loci.
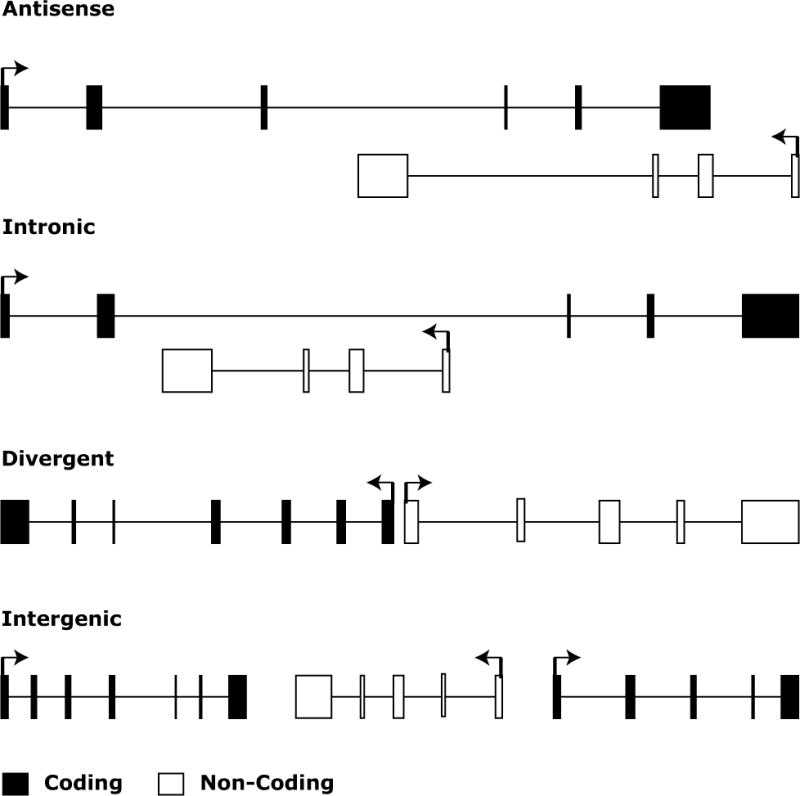
lncRNAs are often defined by their location relative to nearby protein coding genes. Antisense lncRNAs are lncRNAs that initiate inside of a protein coding gene and transcribe in the opposite direction that overlaps coding exons. Intronic lncRNAs are lncRNAs that initiate inside of an intron of a protein coding gene in either direction and terminates without overlapping exons. Bidirectional lncRNAs are transcripts that initiate in a divergent fashion from a promoter of a protein coding gene; the precise distance cut-off that constitutes bidirectionality is not defined but is generally within a few hundred base pairs. Finally, intergenic lncRNAs (also termed large intervening noncoding RNAs or lincRNAs) are lncRNAs with separate transcriptional units from protein coding genes. One definition required lincRNAs to be 5 kb away from protein coding genes (44).
Studies over the last several decades have pointed to the presence of large amounts of RNA that was transcribed but did not encode proteins (4–8). Some of this RNA was later explained by mRNA splicing and RNA genes comprising translation machinery and its regulation (i.e. ribosomal RNA, tRNA, RNase P, SRP-7S), yet, the vast majority was still unaccounted for. Biochemical experiments were able to characterize many abundant structural and regulatory RNAs by cellular localization and sequence similarity (5, 8–12), and genetic studies identified a few lncRNA genes involved in imprinting and other cellular processes (Xist, H19, AIR) (13, 14). Additional genetic studies also pointed to an emerging class of small regulatory RNAs such as microRNAs (12, 15–18) that regulate the translation of mRNAs to fine tune key genetic pathways. Collectively, these classical studies identified a diverse repertoire of RNAs, but may have only scratched the surface of functional RNAs in the cell.
The advent of full genome sequences enabled an unprecedented survey of the genomic landscape for new genes. Surprisingly, this prospecting for “genes” led to the discovery of numerous lncRNA genes, but not many more protein genes. At the same time, DNA microarray technology revealed that the genome encodes at least as many lncRNAs as known protein coding genes (19–22). In fact, further advancements in RNA sequencing and microarray technology allowed a consortium wide effort to define all the transcribed bases in the genome. At present, lncRNAs are operationally defined as RNA genes larger than 200 base-pairs that do not “appear” to have coding potential. Although this working definition is somewhat arbitrary, the size cut-off clearly distinguishes lncRNAs from small regulatory RNAs such as miRNAs or piRNAs. Some classically defined small nuclear RNAs are in fact greater than 200 nucleotides, but the lncRNA designation is only prospectively applied to newly recognized transcripts. The conclusion was that a vast majority of the genome was transcribed (23).
In contrast to the substantial progress in mapping lncRNAs, the functional roles for lncRNAs remained mostly elusive. In fact, the notion of such a wide-spread abundance of transcription was becoming controversial (23–25). More recently dozens of functional examples have emerged implicating lncRNAs in numerous cellular processes ranging from embryonic stem cell pluripotency, cell-cycle regulation and diseases such as cancer. Although lncRNAs exert a diverse spectrum of regulatory mechanisms across a variety of cellular pathways, a common theme is emerging. LncRNA drive the formation of ibonucleic-protein complexes, which in turn influence the regulation of gene expression.
Here we discuss multiple lines of evidence that point to lncRNAs as a key regulatory layer in global gene regulation. We review the the technological approaches for genome-wide discovery and characterization of lncRNAs, and highlight emerging mechanistic themes based on well-studied examples from diverse model systems. We further evaluate several emerging studies indicating important roles for lncRNAs in the etiology of a wide spectrum of diseases.
Genomic Discovery of lncRNAs
At the turn of the 21st century the scientific community was abuzz with great anticipation of the human genome project (26, 27). Perhaps at center stage was the burning question of “how many genes are there in the human genome?” Can the complexity of different organisms be explained by the sheer number of classic protein coding genes, their splicing diversity, or perhaps new types of regulation? This simple yet profound question drove the progress of many technologies, such as microarray and DNA sequencing, at an unprecedented rate. One of the first applications of automated Sanger sequencing in the mid 1990’s was the mapping of expressed sequence tags (ESTs) (28, 29) that identify fragments of genomic regions that were being actively transcribed. This first glimpse into the “transcriptome” in 1996 revealed an intriguing new notion that many genes would be lurking in yet undefined regions of the human genome (29). Yet limited by short sequence reads, lower coverage, and an incomplete reference human genome to align ESTs, it was remained elusive what these new genes may encode.
Tiling Microarrays
In addition to sequencing advances, new technologies were emerging to understand the regulation of gene-expression and de novo identification of new genes. In particular, the advent of DNA microarray technology allowed the ability to survey on the order of 20,000 gene or genomic loci. In parallel, the first complete human chromosome 22 sequence was released in 1999 (30). The combined power of microarrays and draft genome sequences provided the first glimpse into pervasive transcription of non-coding RNAs. Specifically, two independent studies reported initial estimates that there may be as many lncRNA genes as protein coding genes (21, 22). These studies used DNA microarrays with tiled or nested target sequences comprising the entirety of a chromosomal DNA sequence allowed for an unbiased survey of transcribed regions. Some limitations of tiling array studies included the potential for cross hybridization, the lack of strand-specific information if cDNAs were hybridized to the array, and the connectivity between transcribed regions was not known. Nonetheless, both studies were able to confirm expression from numerous non-coding loci by RT-PCR, RNA-blot analysis and evolutionary conservation studies, yet these findings were met with healthy skepticism that they may simply represent transcriptional noise. In a potentially interesting historical parallel, the discovery of “DNA-like RNA” upon phage infection in 1956 was a critical clue leading to “mRNA hypothesis” (31). Yet the significance of this finding was not recognized for at least five years since the “DNA like RNA” was less than 1% of total RNA (mostly rRNA) and thus assumed to be irrelevant noise.
Thus, one of the fruits of the Human Genome Project was the discovery of numerous new RNA genes, but not new protein genes. For example, the number of human miRNAs quickly rose from a handful to nearly 1,000 (32–34). In fact, further advancements in RNA sequencing, cDNA cloning and microarray technology of the next decade allowed a consortium-wide effort to define all the transcribed bases in the human genome. The conclusion was that a vast majority of the genome was transcribed (23). Despite the observed pervasive transcription throughout the genome, pinpointing functional RNA molecules was tantamount to finding needles in a haystack. In fact the notion of such a wide-spread abundance of transcription was becoming increasingly controversial (24, 25, 35–38).
Chromatin marks
A critical clue for hunting RNA genes came from chromatin, the DNA-protein complex where all eukaryotic genes reside (Fig. 3A). With full genome sequence in hand, chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) generated genomic maps of the chromatin architecture that has been termed the “epigenome” (39–41). Massively parallel sequencing of DNA sites occupied by histones and their modifications revealed numerous interesting domains of genomic architecture (39) (42). This included a clear signature of Polymerase II transcribed genes occupied by histone H3 lysine 4 trimethylation (H3K4me3) at the promoters of genes followed by histone H3 lysine 36 trimethylation (H3K36me3) along the transcribed unit (K4–K36 domains) (42) (43, 44).
Figure 3. Functional discovery pipeline of lncRNAs.
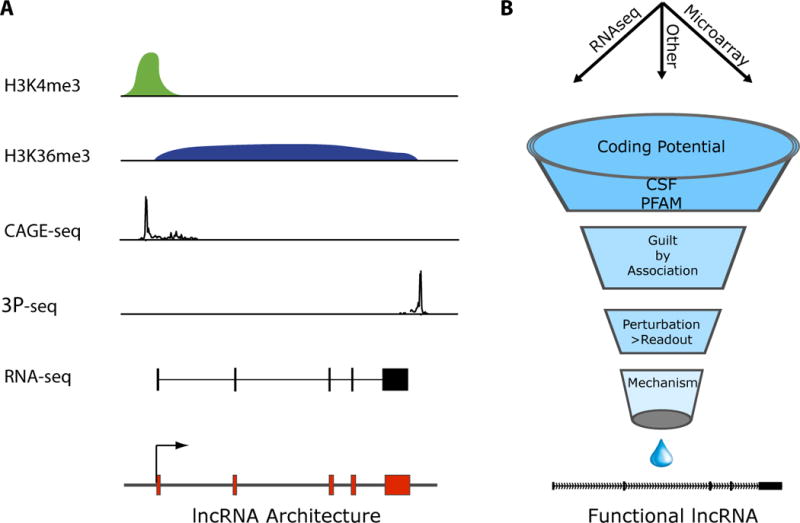
A, Genome-wide discovery of lncRNAs. Chromatin marks of transcription initiation (H3K4me3) and elongation (H3K36me3) define transcribed regions of the genome, while sequencing of capped RNA fragments (CAGE-tag) or poly-adenylation ends (3P-seq) defined the precise beginning and ends of transcripts. RNA-seq can directly define the primary structure of lncRNAs. B, Multiple bioinformatic tools and functional studies refine the set of lncRNAs associated with specific biological functions.
Surprisingly, surveying the entire mouse and human genomes by chromatin marks in several cell types revealed that approximately 5,000 K4–K36 domains represented lncRNAs (44). These lncRNAs had discrete gene loci that reside in previously unannotated intergenic regions between protein coding genes, and hence these RNAs were named large intervening noncoding RNAs (lincRNAs). Further analysis of these loci revealed highly conserved promoter regions that recruit the binding and direct regulation of key transcription factors (24, 44, 45). LincRNAs show sequence conservation throughout evolution over introns or untranscribed intergenic sequences, further suggesting their functionality (24, 44, 45). Moreover, expression patterns of lncRNAs are associated with numerous key cellular processes such as pluripotency, immune-response, and regulation of the cell cycle (44, 46–48). More recently, approximately a third of lincRNAs were found to associate with chromatin modifying complexes (49), and modulate key cellular pathways (48, 50–52).
RNA-seq
The advent of deep sequencing technology led to the ability to sequence cDNA at an unprecedented scale and throughput, termed RNA-Seq (53–57). These approaches have been coupled to computational methods allowing the reconstruction of transcripts and their isoforms at single nucleotide resolution (55–57). These studies have provided an unbiased identification of non-coding transcripts across many cell types and tissues (56, 58).
In addition to full-length reconstruction algorithms several applications have emerged to perform RNA-Seq. For example, a method termed “3-seq” targets the poly adenylated tail of cDNA to quantitatively measure the abundance of transcripts using more affordable short sequence reads (59). Moreover, a variant of this method can be employed to precisely map 3′ ends of transcripts (60). More recently, metabolically labeled mRNAs have been utilized to measure nascent transcription thereby providing insights into the pausing of polymerase and transcriptional dynamics (61). These and many other emerging technologies are providing ever deeper insights into the dynamic transcriptome (Fig. 3A).
Recently studies have utilized RNA sequencing and transcript abundance estimations to identify specific properties of distinct classes of large RNA genes. For example a recent study identified 8,000 large intergenic non-coding RNAs (lincRNAs) in the human genome by integrating numerous annotation sources in combination with RNA sequencing. This study revealed several global properties of lncRNAs, including a tendency for location next to developmental regulators, enrichment for tissue-specific expression patterns, identification of thousands of orthologous lincRNAs between human and mouse, and localization of hundreds of lincRNAs in gene deserts associated with genetic traits (58). By leveraging ever increasing depth of sequencing and read lengths has allowed some of the first steps towards characterizing lncRNAs on a global scale.
Genomic Characterization of lncRNAs
We next turn to discuss experimental and computational approaches to identify, map and derive hypothesis for lncRNA function (Fig. 3). By combining the above technical approaches, it now possible to identify all transcribed loci (K4–K36 chromatin domains) as well as precisely map the primary structure of the RNA products (RNA-seq). (44, 45, 56). These combined layers of information are synergistic where chromatin modifications identify stably transcribed gene loci and RNA sequencing allows detection of even very low abundance transcripts that alone could be argued as transcriptional noise. The additional chromatin information also indicates the promoter region of a given locus (H3K4me3) and the transcribed unit (H3K36me3) thereby assisting in the mapping of RNA transcript 5′ and 3′ ends. Through the successive addition of additional layers of information (such as conservation, coding potential patterns and anatomical properties) progress is being made towards identifying lncRNA gene families (43–45).
lncRNAs have been further defined based on anatomical properties of their gene loci. For example, antisense lncRNAs that overlap known protein coding genes, intronic lncRNAs that are encoded with in introns of protein coding genes, lncRNA that overlap protein coding genes termed overlapping transcripts and lincRNAs that are encoded completely within intergenic genomic space between protein coding loci (Fig. 2). Although, this anatomic characterization has been used initially it is likely that down the road many of these lncRNAs will share similar mechansistic and functional roles.
Excluding protein coding potential
Whether a RNA transcript codes for protein is fundamental to the definition of lncRNA but is in fact a challenging task. Many studies have assessed lncRNA coding potential by translating each lncRNA in all 3′ frames and performing homology queries (i.e. BLASTX) across large protein family and domain databases (i.e. Swissprot and PFAM). These informatic analyses are good initial indications of protein coding capacity, but may miss newly evolved protein sequences or very small open reading frames (< 50 amino acids). To address the former issue, Codon Substitution Frequency (CSF) analysis have been used to determine if codons for amino-acids are preferentially conserved through evolution, indicating preservation of protein coding potential (62, 63). CSF has been employed in multiple studies as an additional information layer of determining coding potential (44, 45, 56, 58, 64). Yet even these two methods combined could still miss small open reading frames buried in these long transcripts. Experimental methods such as ribosomal profiling that identifies putative RNAs that are bound and scanned by the ribosome has provided additional insights into those RNAs that may encode small peptides (65). Moreover, this method identifies the region of ribosomal occupancy thereby further honing on potential translated regions that can be used as refined input into informatics predictions such as CSF and BLASTX. Although some portion lncRNAs may encode small peptides, we note this does not rule out the potential dual nature of lncRNAs acting through RNA and their protein products. This has been exemplified by numerous mRNAs that contain regulatory noncoding RNA elements (p53, Sgrs, Oskar, VegT and others) (66–71).
Inference of lncRNAs functions by co-expression: Guilt by Association
With the mapping of thousands of lncRNA loci, the next challenge is to determine what lncRNAs do. A first step in hypothesis generation is to use the expression patterns of lncRNAs to identify specific cell types or biological processes associated with each candidate lncRNA. Some of the first expression studies of lncRNAs identified lncRNAs that are highly expressed in certain brain regions. In situ hybridization studies further confirmed these expression patters revealing exquisite patterns of expression in specific substructures of the mouse brain (72). A similar study by this group identified numerous lncRNAs that were tightly correlated with pluripotency transcription factors suggesting that many lncRNAs may function in stem cell pluripotency transcriptional networks (73).
More recently, an informatic method termed “Guilt by Association” allowed a global understanding of lncRNAs and protein coding genes that are tightly co-expressed and thus presumably co-regulated (44). This method identifies protein coding genes and pathways significantly correlated with a given lncRNA using gene-expression analyses. Thus, based on known functions of the co-expressed protein coding genes, hypotheses are generated for the functions and potential regulators of the candidate lncRNA. Moreover, this analysis revealed “families” of lncRNAs based on the pathways with which they do and do not associate. This approach has predicted diverse roles for lncRNAs, ranging from stem cell pluripotency to cancer (44). For example, numerous lncRNAs that were tightly correlated with p53 were induced in a p53 dependent manner, many more than would be expected by chance (44, 45, 48). These lncRNAs also were enriched for the p53-binding motif in their promoters. Moreover, one of these lncRNAs, termed lincRNA-p21 predicted to be associated with the p53 was found to be directly regulated by p53 and subsequently forms a lncRNA-RNP with a nuclear factor to serve as role as a global transcriptional repressor facilitating p53- mediated apoptosis (48). Similarly, several lncRNAs predicted to be associated with adipogenesis and pluripotency have recently been identified to be required for maintaining these cellular states [(46), Guttman, Sun et al. personal communication].
Other expression correlation analyses have revealed additional functional roles of lncRNAs. For example, a recent study profiling lncRNAs across over 130 breast cancers comprised on varying grades of the tumor and clinical information (74). This study identified numerous lncRNAs that are specifically up or down regulated in tumor subtypes. For example, it was identified that a lncRNA termed HOTAIR encoded in the HOXC cluster that was a strong predictor of breast cancer metastasis. In fact, enforced expression of HOTAIR was sufficient to drive breast cancer metastasis. More global expression studies of lncRNAs overlapping promoter regions of protein coding genes identified numerous lncRNAs associated with cell-cycle regulation (64). This lead to the functional characterization of a lncRNA termed PANDA that plays a critical role inhibiting p53-mediated apoptosis. The Guilt by Association methods are universally applicable to any biological system. For example, a family of telomere encoded lncRNAs in the malaria parasite (P. falciprum) was identified by their stage specific co-expression with PfsiP2 a key virulence transcription factor (75).
These and other correlative studies have started to identify specific roles of lncRNAs in global transcriptional regulation. Honing in on the pathways that lncRNAs are associated with nominate hypothesis-driven experiments that identified functional lncRNAs. Yet, the full scope of lncRNA transcriptional regulation and function is far from understood. To understand the more global regulatory roles of lncRNAs, comprehensive gain or loss of functional experiments need to be performed.
High throughput loss of function by RNA inference
Indeed, a very recent study performed a loss-of-function study across most (237) of the long intergenic non-coding RNAs (lincRNAs) expressed in mouse embryonic stem cells (ESC) and characterized the effects on gene expression. The authors demonstrated that knockdown of lincRNAs have major consequences on gene expression patterns, comparable to knockdown of well-known ESC regulators. Intriguingly, this global screen determined that lincRNAs primarily affect gene expression in trans. Perhaps more importantly dozens of lincRNAs were found to be functionally required in the maintenance of the pluripotent state. Further investigation into the molecular circuitry of ESCs showed that lincRNA genes are regulated by key transcription factors and that lincRNA transcripts physically bind to multiple chromatin regulatory proteins to affect shared gene expression programs. This study provided the first glimpse of global lincRNA functional properties, mechanisms and highlights their key role in the circuitry controlling ESC state.
LNCRNAS IN GENE REGULATION
lncRNAs bind to and target chromatin regulators
The intimate connection between RNA and chromatin–the DNA-protein complex where all eukaryotic genes reside–was recognized over 40 years ago (76). In 1975, Paul and Duerksen made the surprising finding that biochemically purified chromatin contained twice as much RNA as DNA, raising the idea that RNA may influence chromatin structure and gene regulation (5). Through the years, it has been demonstrated that RNA is required for proper chromatin structure and recruitment of the chromatin modifying complexes to DNA (14). Yet the specific RNA species associated remained elusive. Genetic studies in the ensuing decades revealed a few lncRNAs that were associated with heterochromatin formation and imprinting (i.e. Xist (77), Air (78), H19 (79)). Breakthroughs over the last few years have revealed numerous examples of lncRNAs in controlling the access or dismissal of regulatory proteins from chromatin (Table 1). Here, we will first focus on the protein binding partners of lncRNAs, next review the targeting mechanism of lncRNAs, and lastly discuss the emerging mechanistic themes of lncRNAs in gene regulation.
TABLE 1. Protein partners of lncRNAs.
Many of these lncRNA-protein complexes function at chromatin.
| Protein | LncRNA | LncRNA Function | Refs |
|---|---|---|---|
| CTCF | SRA | Enhances insulator function of CTCF. | (92) |
| DNMT3b | pRNA | Targets DNMT3b in cis to rRNA locus via RNA:DNA:DNA triplex for cytosine methylation and gene silencing. | (90) |
| G9a | Kcnq1ot1, Air | Targets the H3K9 methylase G9a in cis for imprinting. | (87, 88) |
| Glucocorticoi d receptor | Gas5 | Binds to GR as a decoy and titrates GR away from target genes | (102) |
| hnRNP-K | Linc-p21 | Targets hnRNP-K in trans to mediate p53-dependent gene repression. | (48) |
| LSD1-coREST | HOTAIR, many others | Targets LSD1 complex to demethylate H3K4me2 to enforce gene silencing. | (49, 84) |
| MLL-WDR5 | HOTTIP, some eRNAs? | Binds to and localizes MLL complex and H3K4me3 via chromosomal looping for gene activation. | (95, 96) |
| NF-YA | PANDA | p53 inducible and titrates away NF-YA to favor survival over cell death during DNA damage. | (47) |
| PRC1 | ANRIL, Xist | Targets PRC1 in cis for gene silencing. ANRIL influences p16INK4a expression and cell senescence. | (9, 91) |
| PRC2 | Xist, HOTAIR, ANRIL, COLDAIR, Gtl2, Kcnq1ot1, many others | Targets PRC2 either in cis or trans to mediate H3K27 methylation and gene silencing for dosage compensation, imprinting, and developmental gene expression. | (49, 81, 83, 88, 109) |
| SR splicing factors | MALAT1 | Sequesters SR splicing factors to regulate alternative splicing. | (125) |
| Staufen | ½ SBS RNAs | Pairs with mRNAs vis Alu repeast and targets them into non-sense mediated decay pathway. | (126) |
| Set1 and Hda1/2/3 HDACs | CUTs, XUTs | Antisense RNAs repress sense transcription via control of H3K4me3 and histone deacetylation | (127–130) |
| TERRA | hnRNP-A | Controls telomerase access to telomeres in cell cycle phase-specific manner. | (131) |
| TFIIB | DHFR minor | Titrates away TFIIB during cell quiescence to decrease DHFR transcription | (99) |
| TLS | CCND1 promoter ncRNA | Allosterically binds TLS to inhibit CBP and p300 activity, leads to repression of CCND1 gene. | (132) |
| YY1 | Xist | YY1 binding nucleates Xist on the inactive X chromosome. | (100) |
LncRNAs can target several chromatin modification complexes involved in gene silencing (Table 1). One of the most dramatic examples of lncRNA-mediated chromatin regulation occurs during X chromosome dosage compensation in mammals. Briefly, dosage compensation refers to the process whereby the gene expression level of the two X chromosome in female cells is made equal to the single X in male cells. The lncRNA Xist is expressed from one of the two X chromosomes in female cells, and results in altering the chromatin structure of an entire chromosome– the inactive X (Xi)—where most genes are transcriptionally silenced [reviewed in (80)]. Importantly, Xist physically associates with the Polycomb repressive complex 2 (PRC2) through a structured domain termed Repeat A (RepA), resulting in the localization of PRC2 and its cognate histone mark histone H3 lysine 27 trimethylation (H3K27me3) to the inactive X chromosome (81). In an analogous fashion, plants control the seasonal timing of flowering (a process termed vernalization) by a cold-inducible intronic lncRNA termed COLDAIR; COLDAIR recruits PRC2 in cis to silence the flowering regulator gene FLC (82). LncRNA-mediated PRC2 recruitment can also regulate distantly located genes throughout the genome. Human lncRNA HOTAIR is the first such RNAs recognized: HOTAIR physically associate with PRC2, and modulates PRC2 and H3K27me3 localization of hundreds of sites throughout the genome (83, 84). Several additional studies have identified HOTAIR and Xist interfacing with PRC2 via the catalytic methytransferase subunit EZH2 (81, 85), although other proteins are likely also involved (86). The precise molecular interactions between lncRNAs and the Polycomb complex have yet to be defined.
LncRNAs can target additional chromatin regulators. In imprinting, the paternally and maternally inherited alleles are differentially expressed, and lncRNAs are often involved in distinguishing the two alleles. Both Air and Kcnq1ot1 are lncRNAs that are transcribed from the silenced paternal allele, and specifically bind to and recruit the histone H3 lysine 9 methylase G9a in cis to mediate H3K9me3 and transcriptional silencing of Kcnq1 or Igf2r loci, respectively (87, 88). LncRNA can also regulate DNA methylation. In plants, the interplay of small interfering RNAs and nascent lncRNAs to target DNA methylation are well known (89), but a different mechanism, apparently independent of small regulatory RNAs also operates in mammalian cells. Transcriptional repression of the repetitive ribosomal RNA gene loci (rDNA) depends in part on an ncRNA termed pRNA, which recruits DNMT3b to mediate cystosine methylation (90). Additionally, ANRIL, a lncRNA that is associated with cardiac disease, associates with CBX7 of the PRC1 complex facilitating the H3K27me3 based silencing of the INK4a locus (91). Two p53 regulated lncRNAs, linc-p21 and PANDA, have been recently identified as interfacing with DNA binding proteins such as hnRP-K and NF-Y, and these interactions result in transcriptional repression at specific genomic loci (47, 48). Finally beyond chromatin modifications, the lncRNA SRA can interact with and enhance the function of the insulator protein CTCF (92); CTCF can control higher order chromosomal looping and “insulate” specific genes from the effects of long range enhancers and regulatory elements. Based on these numerous examples, it is clear that many chromatin regulatory complexes moonlight as RNA binding proteins; the ability to bind lncRNAs endows them with condition- or allele-specific recognition of target gene chromatin.
Enhancer RNAs
For historical reasons, many of the initial studies focused on RNAs associated with repressive chromatin modifying complexes. Yet several other studies have also demonstrated that active chromatin states are associated with lncRNAs. Genome-scale mapping of histone modifications and enhancer binding proteins have provided an additional layer of information to identify lncRNAs involved in gene activation. ChIP-seq analysis of H3K4me1, H3K27ac, and p300 –several marks associated with gene activating enhancers—showed these regions also produce lncRNA transcripts. Many such enhancer RNAs (eRNAs) were bidirectional, lacked polyA tail, and had very low copy number (93, 94). Although many of these transcripts were initially thought to be byproducts of Pol II transcription or enhancer-promoter interaction, more evidence is pointing to functional roles of the lncRNAs. A recent study performed LOF experiments and found 7 of 12 lncRNAs affected expression of their cognate neighboring genes (95). The authors continued to demonstrate it was not the act of transcription rather the RNA itself that was important for gene “enhancer” activation. Although this trend of lncRNAs affecting transcription of neighboring genes not a universal phenomena (47, 52), these studies clearly demonstrates a functional role for the RNA molecule beyond a simple byproduct of transcription in enhancer regions.
More recently, an enhancer-like lncRNA termed HOTTIP has been discovered to directly interact with WDR5 protein, a key component of the MLL/Trx complex that catalyzes the activating H3K4me3 mark (96). HOTTIP is encoded on the distal 5′ end of the HOXA gene cluster, and chromosomal looping of the 5′ end of the HOXA in an enhancer like manner brings HOTTIP into spatial proximity multiple HOXA genes, enforcing the maintenance of H3K4me3 and gene activation. Remarkably, in vivo HOTTIP LOF experiments silenced HOXA expression and altered limb morphology, consistent with its role in activating HOXA genes. Collectively, these studies demonstrate the critical importance of lncRNAs interfacing with chromatin modifying machinery resulting in enhancer based gene activation, and raises the possibility that many other enhancer-like RNAs may operate via similar mechanisms. Thus, a critical path forward for understanding the functions of lncRNAs is to understand the repertoire of lncRNA binding proteins.
The RNA-Chromatin Interface
How does a lncRNA interface with selective regions of the genome? Several hypotheses that have been forwarded, including (i) formation of a RNA: DNA:DNA triplex; (ii) RNA binding to a sequence-specific DNA binding protein; (iii) RNA: DNA hybrid that displaces a single-strand of DNA (so called R-loop); and (iv) RNA:RNA hybrid of lncRNA with a nascent transcript (97, 98). Mechanisms (i) and (ii) have been experimentally demonstrated in several systems (Fig. 4).
Figure 4. Models of lncRNA mechanisms of action.
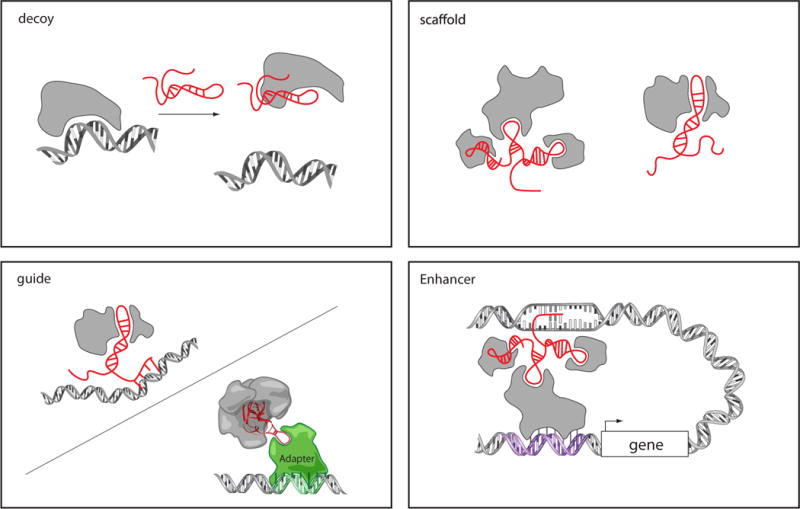
LncRNA can act as decoys that titrate away DNA-binding proteins such as transcription factors. LncRNAs may act as scaffolds to bring two or more proteins into a complex or spatial proximity. LncRNAs may also act as guides to recruit proteins, such as chromatin modification enzymes, to DNA; this may occur through RNA-DNA interactions or RNA interaction with a DNA binding protein. LncRNA guidance can also be exerted through chromosome looping in an enhancer-like model, where the looping defines the cis nature and spread of the lncRNA effect.
Two studies have demonstrated that the association of lncRNAs and chromatin complexes can also include recruitment to DNA. The first of such studies demonstrated that a lncRNA encoded upstream of DHFR gene forms a triplex structure with the promoter which binds to and sequesters the general transcription factor IIB (TFIIB) and prevent transcription of DHFR (99). More recently, another study identified 150–250 nt species of ncRNA, termed pRNA, that also forms a triplex at rDNA loci to recruit DNMT3b to this location through the DNA-RNA triplex (90). In each of these cases, purified ncRNA is able to bind to the cognate DNA sequence to form triplex structure in vitro, but it is difficult to demonstrate that such triplex forms in living cells. Nonetheless, based on these precedents, it is likely that many more DNA-RNA interactions will be identified that serve as molecular beacons to recruit specific protein complexes.
In contrast, the ability of Xist to localize to the inactive X chromosome depends on the ability of Xist to bind to the sequence-specific transcription factor, YY1 (100). When nascent Xist is transcribed, the interaction of Xist repeat C region with YY1 on the Xi captures and nucleates Xist. Conversely, ectopic insertion of multiple copies of YY1 motif can mobilize Xist from the Xi to the ectopic sites. Why Xist is not captured by the numerous YY1 sites on autosomes remains a mystery.
In addition to the four hypothesized targeting strategies, the recent example of HOTTIP introduced a new concept for lncRNA targeting via chromosomal looping (Fig. 4) (96). Mature HOTTIP RNA appears to have no ability to seek out the HOXA locus if HOTTIP is ectopically produced elsewhere in the genome. But endogenous, nascent HOTTIP RNA is brought to its target genes via chromosomal looping. In this way, the lncRNA can serve as a faithful conduit to transform spatial information in chromosome conformation into chemical information in histone modifications.
Emerging Mechanistic Themes: Decoys, Scaffolds, and Guides
The ability of lncRNAs to bind to protein partners endows them with several regulatory capacities. Despite our limited knowledge from just dozens of characterized examples, several mechanistic themes of lncRNAs function have emerged (101). Three main themes that encompass many of the examples discussed thus far (Fig. 4).
Decoys: First and at the simplest level, lncRNAs can serve as decoys that preclude the access of regulatory proteins to DNA. For example, the lncRNA Gas5 is induced upon growth factor starvation; Gas5 contains a hairpin sequence motif that resembles the DNA binding site of glucocorticoid receptor (102). Thus, upon starvation conditions, GAS5 is induced and serves as a decoy to release the receptor from DNA to prevent transcription of metabolic genes. A more recent lncRNA decoy example was identified termed PANDA, which associates with the transcription factor NF-YA to prevent p53-mediated apoptosis (47). NF-YA transactivates several key genes for apoptosis, but PANDA binding to NF-YA titrates NF-YA away from target gene chromatin.
Scaffold: lncRNAs can serve as adaptors to bring two or more proteins into discrete complexes (103). The telomerase RNA TERC is a classic example of a RNA scaffold that assembles the telomerase complex (104). A prime example of lncRNA scaffolds is HOTAIR, which can simultaneously bind both PRC2 and LSD1 -CoREST complex via specific domains of RNA structure (84). This combination of interactions ensures H3K27 methylation and H3K4me2 demethylation, ensuring gene silencing. Additional examples include ANRIL, which combines PRC2 and PRC1 (91, 105); Kcnq1ot1 interacts with both PRC2 and G9a to promote H3K27me3 and H3K9me3, two different silencing histone marks (88). Both of these combinations are likely to reinforce the transcriptionally silent state. Importantly, the concept of RNA as molecular scaffold is likely to generalize more globally as hundreds of lncRNAs have been identified to form ribonucleic-protein interactions with multiple protein partners (49, 52).
Guides: As described above (Table 1), many lncRNA are individually required for the proper localization of specific protein complexes. LncRNAs involved in dosage compensation and imprinting (Xist, Kcnq1ot1, Air) serve as guides to target gene silencing activity in an allele-specific fashion. HOTAIR also serves as a guide to localize PRC2 in developmental and cancer-related gene expression (74, 83). As another example, lincRNA-p21 is directly induced by p53 upon DNA damage, and in turn physically associates with nuclear factor hnRNP-K to reroute this protein to specific promoters (48). Guide lncRNAs thereby combine two basic molecular functions—binding of a protein partner plus a mechanism to interface with selective regions of the genome.
The concept of guide lncRNAs has previously been parsed by whether the guidance occur in cis (on neighboring genes) or in trans (on distantly located genes). The cis actor have been assumed to occur in a co-transcriptional manner, leading to the analogy of lncRNAs as tethers (106). But recent experiments where ectopically supplied lncRNAs are shown to seek out their cognate target sites show that even cis-acting lncRNAs have the capacity to act in trans (90, 99, 100). Cis-action also do not simply correlate with distance from the site of lncRNA synthesis (87, 96), perhaps reflecting the important role of chromosomal looping in so-called cis effects. Interestingly, all of the classic long and small ncRNAs that have been well characterized also work in trans through interactions with proteins (i.e. rRNA, tRNA, snoRNA, RNase P, TERC). Future studies that allow global mapping of lncRNA sites of action may better define the cis vs. trans nature of lncRNA actions.
These three modes of action encompass many of the recently discovered lncRNA mechanistic themes; yet there are likely many other mechanisms to be uncovered. It is clear from the above examples that as additional protein partners and targeting mechanisms (not just to DNA but perhaps other cellular structures) are discovered, it is possible to build complex regulatory scripts out of lncRNAs (107). Essential to understanding the meaning of such scripts will be a systematic understanding of the individual parts of lncRNA and their relevant interactions—akin to deciphering the codons of messenger RNAs.
THE GLOBAL RNA-CHROMATIN NETWORK
RIP-seq and CLIP-seq
Could these simply be a few quirky examples of lncRNAs interacting with chromatin modifying complexes, or lncRNAs a more global phenomenon? Several recent studies point to the latter. These studies employ protein immunoprecipitation followed by microarray or deep-sequencing to enumerate all RNAs associated with a protein complex of interest (RIP-seq) (108) (49, 109); often times crosslinking is performed to trap the relevant interactions in living cells (CLIP-seq) (110). For example, human PRC2 is associated with approximately 20% of lincRNAs expressed in a given cell type. Moreover, LOF of PRC2 bound lincRNAs resulted altered expression from PRC2 regulated gene loci in trans (49). Several of these PRC2 bound lincRNAs were further identified as bound to chromatin fraction (111), and a subset of these same lncRNAs are also associated with LSD1 complexes (49), raising the possibility of numerous lncRNA scaffolds. A similar study in mouse embryonic stem cells (mESCs) identified approximately 9,000 lncRNAs associated with PRC2 (109). Thus, numerous lncRNAs are associated with PRC2, and may serve similar mechanistic themes as HOTAIR, Xist, Kcnq1ot1 and Air.
To address lncRNA-protein interactions at a more global level, a recent study systematically integrated RIP of multiple chromatin complexes with LOF experiments by depleting the lincRNAs bound to a given chromatin-modifying complex as well as the chromatin modifying complex itself (52). Interestingly, depletion of the lincRNAs associated with a given complex collectively phenocopied the depletion of the complex itself for PRC2 and several other complexes. These results strongly suggesting that lincRNAs serve to modulate the targeting of chromatin modifying complex to specific genomic loci as an emerging mechanistic theme. Yet, future studies will need to investigate the directness of these interactions, and determine how lncRNAs confer specificity to highly dynamic chromatin modifying complexes.
lncRNA Structure and Function
One of the most the intriguing features of RNA is the malleable adoption of secondary and tertiary structures that relate to function. Classic chemical probing and structural studies have resulted in a structural understanding of several lncRNAs, including the atomic structure of the largest known RNP complex–the ribosome (112). Several recent studies have developed genome-scale approaches to measure RNA secondary structures, which also applies to lncRNAs (113). These studies use either chemical probing to acylate flexible RNA bases that are not participating in structural interactions, or using specific enzymes that cleave structured and unstructured regions of RNAs. For example, a large scale application of chemical probing followed by sequencing of RT products, termed SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension), revealed the secondary structure of the entire RNA genome of the human immunodeficiency virus (114). More recently, deep sequencing of RNA fragments generated by enzymes that cleave single stranded and double stranded regions of RNA mapped the secondary structure of the entire budding yeast transcriptome, revealing several global structural properties (115). Notably, this study observed a triplet based structural motif across gene bodies that correlates with translational efficiency. In contrast, 5′ and 3′ UTRs were observed to be much more lowly structured. In addition, to the S. cerevisiae transcriptome, this study was also able to confirm known structural motifs and structural properties of the HOTAIR lncRNA. Two other studies successfully mapped the secondary structures of mouse small nuclear RNAs and compared wild type and mutant RNase P (116, 117). With these new technologies in hand, it will be possible to gain a much needed understanding of the relationship between lncRNA structure and function. Perhaps revealing common motifs of RNA structure that result in specific protein interactions or other functional properties.
lncRNAs and Disease
Underscoring the importance of lncRNAs’ regulatory roles is their emergence as key players in the etiology of several disease states (118). The strongest connection at present is with cancer (119). Dozens of lncRNAs have been documented to have altered expression in human cancers, and are regulated by specific oncogenic and tumor-suppressor pathways such as p53, MYC, and NF-κB (44, 47, 48). Hung et al. recently described a class of lncRNAs that show periodic expression during the human cell cycle, and many of these are dysregulated in expression in human cancer samples (47). The lncRNA HOTAIR is highly induced in a approximately one quarter of human breast cancers, and HOTAIR expression is strongly predictive of eventual metastasis and death (74). HOTAIR overexpression in fact drives breast cancer metastasis in vivo, in part by relocalizing Polycomb occupancy patterns genome-wide to alter the positional identity of cancer cells (74). Elevated HOTAIR level is also predictive of metastasis or progression in colon and liver cancers, suggesting a general oncogenic trait (120, 121). In effect, cancer cells reprogram themselves to act as if they belong in other anatomic sites (74). The concept of lncRNAs as disease markers is strongly bolstered by the notable discovery that lncRNAs, perhaps due to their secondary structures, are stable in body fluids and enable non-invasive diagnoses (122). Chinnaiyan and colleague discovered a large set of lncRNAs in human prostate cancers by RNA-seq, and further identified PCAT-1, a lncRNA involved in gene repression that can identify poor prognosis patients based on its level in urine (122). The human lncRNA ANRIL is located upstream of the CDKN2A tumor suppressor locus encoding the p16 CDK inhibitor. Mutations in ANRIL are associated with cancer and cardiovascular disease, and lead to aberrant ANRIL transcripts and loss of p16 repression (123). Together, these examples illustrate diverse pathogenic mechanisms–from altering the epigenetic landscape (HOTAIR and ANRIL), modulation of the p53 pathway (linc-p21 and PANDA), alternative splicing that increases an oncogenic protein production (Zeb2 antisense RNA) (124), to controlling the DNA damage response (CCND1 promoter transcript).
Although cancer has been the most studied, it is likely that lncRNAs are involved in the pathogenesis of many other diseases. Consistent with this notion, hundreds of genomic regions that do not contain protein coding genes are strongly associated with a wide spectrum of human diseases. Future studies will need to pinpoint potential lncRNA transcripts in these regions and discern whether and how the noncoding genome contributes to human diseases.
Summary Points.
Functional lncRNAs: Several examples of functional lncRNAs have been identified that play key roles in maintaining pluripotency to cancer.
lncRNA-RNPs: A common emerging theme of lncRNAs forming ribonucleic acid-protein interactions to carry out their functions. Either by modulating chromatin modifying complexes, preventing transcriptional activation, and likely many additional mechanisms.
eRNAs: The emerging theme of enhancers being transcribed. Two classes have been identified so far: those that are byproducts of transcription versus lncRNAs that play a role in forming enhancer contacts to promote gene-expression.
Future Issues.
LncRNA targeting to the genome: Very little is understood about how specific lncRNAs seek out selective sites in the genome for interaction, and nature of lncRNA-chromatin interactions, and their possible functional roles in lncRNA biology.
Complete lncRNA annotation: Numerous annotation resources are available that will need to be compiled into parsimonious transcript databases.
Defining “families” of lncRNA through structure and function: A deeper understanding of sequence and structural elements that relate to function will allow classification, or even prediction, of lncRNA families similar to protein families with similar structural domains.
Cis and trans: Idnetifying potential ways of determining cis acting enhancer-like lncRNAs from lncRNAs that function in trans.
Global perturbation analyses: The need for large-scale LOF or GOF studies to causally demonstrate lncRNA functions.
Principles of Protein-RNA and RNA-DNA interactions: The need for detailed mapping and structural studies to understand the sequence and or structural basis of RNA-Protein and RNA-DNA interactions.
Evolution across species: The clear difference in conservation between protein-coding and lncRNA genes raises the question of how are these lncRNAs rapidly evolving.
lncRNAs and Disease: Genomic regions genetically associated with disease containing only lncRNAs pointing to their genetic importance to disease yet functional roles largely unresolved
Genetics: There is a clear need to develop genetic model systems towards understanding of lncRNAs function in vivo.
Acknowledgments
We apologize to colleagues whose work could not be discussed or cited due to space limitations. J.L.R is supported by NIH (1DP2OD00667-01, 1P01GM099117-01). J.L.R is a Damon Runyon-Rachleff, Searle, Smith Family and Merkin Foundation Scholar. H.Y.C. is supported by NIH (R01- HG004361, R01-CA118750) and California Institute for Regenerative Medicine. H.Y.C. is an Early Career Scientist of the Howard Hughes Medical Institute
Mini Glossary
- Scaffold
The formation of lncRNA-RNPs where the RNA joins several proteins together in a complex.
- Guide
The formation of a lncRNA-RNP that imparts specificity to genomic locations. This can be through DNA-protein or RNA-DNA recognition rules.
- Signal
Similar to the classic idea of RNA serving as a signal for translation, lncRNAs could signal numerous other cellular processes.
- Decoy
The notion that lncRNAs can associate with DNA binding proteins to prevent or their binding to DNA recognition elements.
- Pervasive Transcription
The phenomena recognized in the early 21st century describing that a vast majority of the genome is transcriptionally active.
- Guilt By Association
Hypothesis generation of a given lncRNA function by the co-expression of lncRNAs with protein coding mRNAs. Can group lncRNAs into the pathways they may regulate.
Define Acronyms
- lncRNA
long non-coding RNA that functions as a large RNA gene.
- lincRNA
long intergenic non-coding RNA that does not overlapping protein coding genes.
- lncRNA-RNP
lncRNA- Ribonucleic acid Protein complexes.
- hnRNA
heterogeneous nuclear RNA: Discovered as rapidly transcribed population of RNA species believed to be pre-mRNA but may contain many lncRNAs.
- CSF
Codon Substitution Frequency determines the evolutionary pressure to preserve synonymous amino acid content.
- GOF/ LOF
Gain of function and Loss of function approaches to define lncRNA functional roles.
- CUTs
Cryptic unstable transcripts;
- XUTS
Xrn1-sensitive unstable transcripts;
- MLL
Mixed lineage leukemia protein;
- PRC1 or PRC2
Polycomb repressive complex 1 or 2;
- TLS
translocated in liposarcoma
Contributor Information
John L. Rinn, Email: john_rinn@harvard.edu.
Howard Y. Chang, Email: howchang@stanford.edu.
LITERATURE CITED
- 1.Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. Journal of molecular biology. 1961;3:318–56. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 2.Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–9. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 3.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg RA, Penman S. Small molecular weight monodisperse nuclear RNA. J Mol Biol. 1968;38:289–304. doi: 10.1016/0022-2836(68)90387-2. [DOI] [PubMed] [Google Scholar]
- 5.Paul J, Duerksen JD. Chromatin-associated RNA content of heterochromatin and euchromatin. Mol Cell Biochem. 1975;9:9–16. doi: 10.1007/BF01731728. [DOI] [PubMed] [Google Scholar]
- 6.Salditt-Georgieff M, Harpold MM, Wilson MC, Darnell JE., Jr Large heterogeneous nuclear ribonucleic acid has three times as many 5′ caps as polyadenylic acid segments, and most caps do not enter polyribosomes. Mol Cell Biol. 1981;1:179–87. doi: 10.1128/mcb.1.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salditt-Georgieff M, Darnell JE., Jr Further evidence that the majority of primary nuclear RNA transcripts in mammalian cells do not contribute to mRNA. Mol Cell Biol. 1982;2:701–7. doi: 10.1128/mcb.2.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nickerson JA, Krochmalnic G, Wan KM, Penman S. Chromatin architecture and nuclear RNA. Proc Natl Acad Sci U S A. 1989;86:177–81. doi: 10.1073/pnas.86.1.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006;26:2560–9. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maison C, Bailly D, Peters AH, Quivy JP, Roche D, et al. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat Genet. 2002;30:329–34. doi: 10.1038/ng843. [DOI] [PubMed] [Google Scholar]
- 11.Warner JR, Soeiro R, Birnboim HC, Girard M, Darnell JE. Rapidly labeled HeLa cell nuclear RNA. I. Identification by zone sedimentation of a heterogeneous fraction separate from ribosomal precursor RNA. Journal of molecular biology. 1966;19:349–61. doi: 10.1016/s0022-2836(66)80009-8. [DOI] [PubMed] [Google Scholar]
- 12.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagano T, Fraser P. No-nonsense functions for long noncoding RNAs. Cell. 2011;145:178–81. doi: 10.1016/j.cell.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein E, Allis CD. RNA meets chromatin. Genes Dev. 2005;19:1635–55. doi: 10.1101/gad.1324305. [DOI] [PubMed] [Google Scholar]
- 15.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 16.Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–62. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 17.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–4. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 18.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 19.Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 20.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 21.Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–9. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- 22.Rinn JL, Euskirchen G, Bertone P, Martone R, Luscombe NM, et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003;17:529–40. doi: 10.1101/gad.1055203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponjavic J, Ponting CP, Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007;17:556–65. doi: 10.1101/gr.6036807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, et al. Experimental annotation of the human genome using microarray technology. Nature. 2001;409:922–7. doi: 10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- 26.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 27.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 28.Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409:685–90. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- 29.Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, et al. A gene map of the human genome. Science. 1996;274:540–6. [PubMed] [Google Scholar]
- 30.Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–95. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- 31.Volkin E, Astrachan L. Intracellular distribution of labeled ribonucleic acid after phage infection of Escherichia coli. Virology. 1956;2:433–7. doi: 10.1016/0042-6822(56)90001-0. [DOI] [PubMed] [Google Scholar]
- 32.Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–9. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 34.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–70. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 35.Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, et al. The reality of pervasive transcription. PLoS biology. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14:103–5. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 37.van Bakel H, Nislow C, Blencowe BJ, Hughes TR. Most “dark matter” transcripts are associated with known genes. PLoS biology. 2010;8:e1000371. doi: 10.1371/journal.pbio.1000371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Bakel H, Nislow C, Blencowe BJ, Hughes TR. Response to “the reality of pervasive transcription”. PLoS biology. 2011;9:e1001102. doi: 10.1371/journal.pbio.1000625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 41.Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu Rev Biochem. 2009;78:245–71. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–33. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guttman M, Amit I, Garber M, French C, Lin MF, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–7. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011 doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koziol MJ, Rinn JL. RNA traffic control of chromatin complexes. Current opinion in genetics & development. 2010;20:142–8. doi: 10.1016/j.gde.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rinn JL, Huarte M. To repress or not to repress: this is the guardian’s question. Trends in cell biology. 2011;21:344–53. doi: 10.1016/j.tcb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011 doi: 10.1038/nature10398. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 54.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 2008;18:1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature biotechnology. 2010;28:503–10. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–77. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 58.Cabili MNTC, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative Annotation of Human Large Intergenic Non-Coding RNAs Reveals Global Properties and Specific Subclasses. Genes Dev. 2011 doi: 10.1101/gad.17446611. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beck AH, Weng Z, Witten DM, Zhu S, Foley JW, et al. 3′-end sequencing for expression quantification (3SEQ) from archival tumor samples. PloS one. 2010;5:e8768. doi: 10.1371/journal.pone.0008768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jan CH, Friedman RC, Ruby JG, Bartel DP. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature. 2011;469:97–101. doi: 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–8. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin MF, Carlson JW, Crosby MA, Matthews BB, Yu C, et al. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Res. 2007;17:1823–36. doi: 10.1101/gr.6679507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin MF, Deoras AN, Rasmussen MD, Kellis M. Performance and scalability of discriminative metrics for comparative gene identification in 12 Drosophila genomes. PLoS Comput Biol. 2008;4:e1000067. doi: 10.1371/journal.pcbi.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–9. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pueyo JI, Couso JP. Tarsal-less peptides control Notch signalling through the Shavenbaby transcription factor. Dev Biol. 2011;355:183–93. doi: 10.1016/j.ydbio.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci U S A. 2007;104:20454–9. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leygue E. Steroid receptor RNA activator (SRA1): unusual bifaceted gene products with suspected relevance to breast cancer. Nucl Recept Signal. 2007;5:e006. doi: 10.1621/nrs.05006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jenny A, Hachet O, Zavorszky P, Cyrklaff A, Weston MD, et al. A translation-independent role of oskar RNA in early Drosophila oogenesis. Development. 2006;133:2827–33. doi: 10.1242/dev.02456. [DOI] [PubMed] [Google Scholar]
- 70.Kloc M, Wilk K, Vargas D, Shirato Y, Bilinski S, Etkin LD. Potential structural role of non-coding and coding RNAs in the organization of the cytoskeleton at the vegetal cortex of Xenopus oocytes. Development. 2005;132:3445–57. doi: 10.1242/dev.01919. [DOI] [PubMed] [Google Scholar]
- 71.Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, et al. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol. 2008;10:1098–105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 72.Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A. 2008;105:716–21. doi: 10.1073/pnas.0706729105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dinger ME, Amaral PP, Mercer TR, Pang KC, Bruce SJ, et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008;18:1433–45. doi: 10.1101/gr.078378.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Broadbent KM, Park D, Wolf AR, Van Tyne D, Sims JS, et al. A global transcriptional analysis of Plasmodium falciparum malaria reveals a novel family of telomere-associated lncRNAs. Genome Biol. 2011;12:R56. doi: 10.1186/gb-2011-12-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165:349–57. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 77.Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- 78.Barlow DP, Stoger R, Herrmann BG, Saito K, Schweifer N. The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature. 1991;349:84–7. doi: 10.1038/349084a0. [DOI] [PubMed] [Google Scholar]
- 79.Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–5. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 80.Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12:542–53. doi: 10.1038/nrg3035. [DOI] [PubMed] [Google Scholar]
- 81.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–6. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heo JB, Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2010;331:76–9. doi: 10.1126/science.1197349. [DOI] [PubMed] [Google Scholar]
- 83.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, et al. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science. 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kaneko S, Li G, Son J, Xu CF, Margueron R, et al. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–20. doi: 10.1101/gad.1983810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maenner S, Blaud M, Fouillen L, Savoye A, Marchand V, et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010;8:e1000276. doi: 10.1371/journal.pbio.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 88.Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–46. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 89.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmitz KM, Mayer C, Postepska A, Grummt I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010;24:2264–9. doi: 10.1101/gad.590910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–74. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yao H, Brick K, Evrard Y, Xiao T, Camerini-Otero RD, Felsenfeld G. Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev. 2010;24:2543–55. doi: 10.1101/gad.1967810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hung T, Chang HY. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2010;7:582–5. doi: 10.4161/rna.7.5.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–6. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445:666–70. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- 100.Jeon Y, Lee JT. YY1 Tethers Xist RNA to the Inactive X Nucleation Center. Cell. 2011;146:119–33. doi: 10.1016/j.cell.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular Cell. 2011 doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spitale RC, Tsai MC, Chang HY. RNA templating the epigenome: Long noncoding RNAs as molecular scaffolds. Epigenetics. 2011;6:539–43. doi: 10.4161/epi.6.5.15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zappulla DC, Cech TR. RNA as a flexible scaffold for proteins: yeast telomerase and beyond. Cold Spring Harb Symp Quant Biol. 2006;71:217–24. doi: 10.1101/sqb.2006.71.011. [DOI] [PubMed] [Google Scholar]
- 105.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2010;30:1956–62. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–42. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Organization of intracellular reactions with rationally designed RNA assemblies. Science. 2011;333:470–4. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 108.Gerber AP, Herschlag D, Brown PO. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004;2:E79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–53. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–9. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mondal T, Rasmussen M, Pandey GK, Isaksson A, Kanduri C. Characterization of the RNA content of chromatin. Genome Res. 2010;20:899–907. doi: 10.1101/gr.103473.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steitz TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol. 2008;9:242–53. doi: 10.1038/nrm2352. [DOI] [PubMed] [Google Scholar]
- 113.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–55. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009;460:711–6. doi: 10.1038/nature08237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–7. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, et al. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, et al. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci U S A. 2011;108:11063–8. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–61. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 119.Tsai MC, Spitale RC, Chang HY. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71:3–7. doi: 10.1158/0008-5472.CAN-10-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, et al. Long non-coding RNA HOTAIR regulates Polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 121.Yang Z, Zhou L, Wu LM, Lai MC, Xie HY, et al. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol. 2011;18:1243–50. doi: 10.1245/s10434-011-1581-y. [DOI] [PubMed] [Google Scholar]
- 122.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–9. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008;22:756–69. doi: 10.1101/gad.455708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–8. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.van Dijk EL, Chen CL, d’Aubenton-Carafa Y, Gourvennec S, Kwapisz M, et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–7. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- 128.Berretta J, Pinskaya M, Morillon A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev. 2008;22:615–26. doi: 10.1101/gad.458008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, Stutz F. Trans-acting antisense RNAs mediate transcriptional gene cosuppression in S. cerevisiae. Genes Dev. 2009;23:1534–45. doi: 10.1101/gad.522509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell. 2007;131:706–17. doi: 10.1016/j.cell.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 131.Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, et al. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature. 2011;471:532–6. doi: 10.1038/nature09772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang X, Arai S, Song X, Reichart D, Du K, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–30. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]