

Abstract
(N)-Methanocarba (bicyclo[3.1.0]hexane)-adenosine derivatives were probed for sites of charged sulfonate substitution, which precludes diffusion across biological membranes, e.g. blood brain barrier. Molecular modeling predicted that sulfonate groups on C2-phenylethynyl substituents would provide high affinity at both mouse (m) and human (h) A3 adenosine receptors (ARs), while a N6-p-sulfo-phenylethyl substituent would determine higher hA3AR vs. mA3AR affinity. These modeling predictions, based on steric fitting of the binding cavity and crucial interactions with key residues, were confirmed by binding/efficacy studies of synthesized sulfonates. N6-3-Chlorobenzyl-2-(3-sulfophenylethynyl) derivative 7 (MRS5841) bound selectively to h/m A3ARs (Ki hA3AR 1.9 nM) as agonist, while corresponding p-sulfo isomer 6 (MRS5701) displayed mixed A1/A3AR agonism. Both nucleosides administered i.p. reduced mouse chronic neuropathic pain that was ascribed to either A3 or A1/A3ARs using A3AR genetic deletion. Thus, rational design methods based on A3AR homology models successfully predicted sites for sulfonate incorporation, for delineating adenosine’s CNS vs. peripheral actions.
Keywords: Molecular modeling, G protein-coupled receptor, neuropathic pain, purines, radioligand binding, adenosine receptor
Introduction
G protein-coupled receptors (GPCRs), which constitute ~4% of the human genome’s protein production, remain a fruitful area of pharmaceutical research due to their implication in many pathophysiological processes. The neuromodulator adenosine acts through four closely related GPCRs: A1, A2A, A2B and A3 adenosine receptors (ARs).1 The A3 subtype was the last member of the AR family to be cloned and has become a target in drug discovery for various disease conditions.2 Agonists of A3AR appear to be efficacious, without serious adverse effects, in clinical trials for treatment of hepatocellular carcinoma (1b) and autoimmune inflammatory diseases (1a) such as rheumatoid arthritis, psoriasis, and dry eye disease.3–7 A3AR agonists are also under consideration for treating conditions of the central nervous system (CNS) and peripheral nervous system.3,13 Additionally, A3AR-selective antagonists have been explored in models of glaucoma, septic shock, asthma and other diseases.8–12
Recently, the treatment of chronic neuropathic pain was proposed as a novel potential application of A3AR agonists.13 In mouse models, A3AR agonists blocked the development of mechanically- and chemotherapy-induced neuropathic pain in a dose-dependent fashion and significantly augmented the analgesic effects of currently used analgesics. The ability of adenosine to reduce pain by activating A1 and A2A ARs has been known for decades, but the clinical development of A1AR and A2AAR agonists is largely restricted to local delivery due to the risk of cardiovascular side effects.14 Conversely, the activation of A3AR in humans by potent, selective, and orally bioavailable A3AR agonists, is not associated with cardiac or hemodynamic side effects and could represent a viable therapeutic strategy for chronic pain of distinct etiologies.13,15 There is evidence for both central and peripheral sites of action of A3AR agonists in protection against chronic neuropathic pain, and there is a need for new pharmacological tools to explore this phenomenon.
The structure activity relationship (SAR) of nucleosides at the A3AR has been extensively explored, and modification of the adenosine structure has centered on the adenine C2 and N6 positions and on the ribose moiety (Chart 1).2,16 2-Chloro-N6-(3-iodobenzyl)adenosine-5'-N-methyluronamide (CI-IB-MECA, 1b) was the first potent and highly selective A3AR agonist reported.17 The replacement of the flexible ribose tetrahydrofuryl ring with a conformationally constrained bicyclo[3.1.0]hexane (methanocarba) ring system, which enforces a North (N)-envelope conformation, maintains or enhances affinity at A1 and A3ARs with respect to the ribosides, while decreasing affinity at the A2AAR (compound 2).18,19
Chart 1.

Nucleosides as selective A3AR ligands.
The truncation of nucleosides at the 4′-carbon, i.e. removal of the 5′-group, is another extensively explored modification in various chemical series and generally results in retention of A3AR affinity and selectivity with concurrent reduction in efficacy at A3AR, generating antagonists or low efficacy agonists (compound 3).20–23 In this truncated series, the extent to which the efficacy is reduced to provide either partial agonists or antagonists can depend on the species, the effector system being measured and the level of receptor expression. Such nucleoside derivatives that are classified as antagonists in one effector system (e.g. cAMP) might show greatly increased efficacy by other measures or in the presence of positive allosteric modulators.24
We recently demonstrated that the introduction of rigid, extended C2-arylalkynyl groups in (N)-methanocarba adenosine derivatives, both substituted with a 5'-N-methyluronamide and 4′-truncated, is tolerated at the human (h) A3AR (compound 4).25,26 Previously, C2-phenylalkynyl groups were included in potent A3AR agonists in the relatively flexible 9-riboside series.27 Other groups that enhance affinity and selectivity in the SAR of nucleosides at the A3AR include hydrophobic substituents at the N6 position, such as N6-(3-halobenzyl) groups or small N6-alkyl groups.17,27N6-Methyl derivatives are typically more potent at the hA3AR than rat (r) A3AR, while N6-(3-halobenzyl)adenosines tend to display greater species-independent A3 selectivity.28
Using the known SAR data for nucleosides at the A3AR, this study probes possible sites of insertion of a sulfonate group in the (N)-methanocarba-adenosine series with the aim of developing novel A3AR ligands to be used as pharmacological tools. A negatively charged sulfonate substitution of small molecules is one means of excluding diffusion across biological membranes such as the blood brain barrier.29–31 Such potent and selective A3AR agonists or antagonists would be useful in delineating in vivo effects in the CNS from those in the periphery, depending on the route of administration.
Previously investigated sulfonated nucleosides for ARs include N6-(p-sulfophenyl)adenosine as a moderately potent and selective A1AR agonist and recently reported C2-(p-sulfophenyl)ethylthio-adenosine as an A2AAR agonist.29,30 Moreover, sulfonated derivatives of 1,3-dialkylxanthines (on a C8-phenyl ring) act as peripherally selective A1AR antagonists because they were found not to penetrate into brain after intraperitoneal (i.p.) injection in mice.31 However, our previous attempt to develop sulfonated A3AR ligands through the introduction of an aryl sulfonate group on a N6-benzyl substituent of adenosine-5'-N-methyluronamide was unsuccessful leading to compounds with very low affinity at rARs.32
Consequently, this study uses currently available three-dimensional (3D) information about the AR family to predict sites at which sulfonate substitution is tolerated or even potency enhancing. We apply receptor-based molecular modeling approaches to test in silico sites for the introduction of permanently charged sulfonate groups in both the 5'-N-methyluronamide series and the 4'-truncated series of 2-phenylethynyl (N)-methanocarba adenosines for use as A3AR ligands. We consider not only the binding site features of the hA3AR but also of the mouse (m) A3AR, with the intent of developing compounds with species-independent A3 affinity so that they can be useful in in vivo studies frequently performed in mouse models.8,10,13
In the present study, nucleosides with various substitutions at the C2 and N6 positions were synthesized, guided by insights from molecular modeling. The receptor binding properties and functional effects of these rationally designed sulfonated ligands at both h and mA3ARs have been characterized. Even though their in vivo pharmacokinetics has not been quantified, based on physicochemical considerations, we expect that these permanently anionic compounds will not easily diffuse across biological membranes, resulting in useful probes for delineating the CNS vs. peripheral actions of adenosine. Moreover, two isomeric sulfonate derivatives with different pharmacological profiles have been tested in a well-characterized mouse model of neuropathic pain to determine their efficacy in reversing the development of mechano-allodynia.
Results
Molecular Modeling
In order to design sulfonated nucleoside ligands for the A3AR belonging to the relatively rigid (N)-methanocarba series, we started from the SAR data already available for this class of compounds and from the available 3D information regarding the AR family. So far, the only published crystallographic data for this family of four GPCRs was obtained from the hA2AAR, whose structure has been solved in complex with both antagonists and agonists.33,34 Therefore, to study the putative interactions of (N)-methanocarba nucleosides with the A3AR, we built homology models of the h and mA3AR subtypes based on their close homology to the A2AAR X-ray structures.
As previously reported, the C2-arylalkynyl (N)-methanocarba derivatives were unable to fit inside the binding pocket of the initial hA3AR model, based on the agonist-bound hA2AAR X-ray structure, due to the bulkiness of the substituents at the C2 position.25,26 However, a hybrid model of the A3AR predicted that a reorganization of the helical bundle, mimicking the structures of opsin and the β2 adrenergic receptor through a proposed outward movement of transmembrane helix (TM) 2, was needed to accommodate the inflexible and linearly extended C2-arylalkynyl group.
Therefore, we performed docking studies of several (N)-methanocarba nucleosides (compounds 5–20) using homology models of the h and mA3ARs based on a hybrid template where the extracellular part of TM2 was modeled following the structure of the β2 adrenergic receptor in an active state.35 In these hybrid models as compared to the A3AR models based exclusively on the A2AAR crystal structure, the extracellular terminal of TM2 was shifted outward, which created a larger pocket for the accessibility of C2 substituents of the present (N)-methanocarba nucleosides.25
Alignment of the h and mA3AR sequences (~73% identity) together with an analysis of the docking results revealed that the lower (cytosolic) part of the transmembrane binding site is highly conserved among species, while significant differences are present in the residues of the very upper part of the TMs and of the extracellular loops (ELs, Figure S1, Supporting Information). Notably, a hydrophobic valine residue in EL2 of the hA3AR (Val169) is mutated to a bulkier and positively charged arginine in the mA3AR (Arg170), giving a very different electrostatic and steric character to the entrance of the A3AR binding site (Figure S2, Supporting Information). Residues at this position of the ARs were shown by mutagenesis studies and crystallographic data to be involved in the binding of ligands (e.g. Glu169 in EL2 of the hA2AAR)33,36 and were proposed to determine selectivity among different AR subtypes.37
The performed molecular docking simulations of (N)-methanocarba derivatives showed a common binding mode of the sulfonated compounds inside the h and mA3AR binding pockets (Figures 1 and 2), at least with respect to the adenine and ribose moieties. This binding mode presented most of the key interactions previously observed in the crystal structures and docking poses of nucleosides at the ARs.25,34 In particular, the 3′- and 2′-hydroxyl groups formed H-bonds with the side chains of Ser 7.42 (Ser271 in hA3AR and Ser272 in mA3AR) and His 7.43 (His272 in hA3AR and His273 in mA3AR), respectively; while the 5'-N-methyluronamide chain was interacting through a H-bond with the side chain hydroxyl group of Thr 3.36 (Thr94 in hA3AR and Thr95 in mA3AR). The side chain of conserved Asn 6.55 (Asn250 in hA3AR and Asn251 in mA3AR) strongly interacted with these compounds through two H-bonds involving the 6-amino group and the N7 atom of the adenine ring. Moreover, the adenine core was anchored inside the binding site by a π–π stacking interaction with a phenylalanine in EL2 (Phe168 in hA3AR and Phe169 in mA3AR) and strong hydrophobic contacts with Leu 6.51 (Leu246 in hA3AR and Leu247 in mA3AR) and Ile 7.39 (Ile268 in hA3AR and Ile269 in mA3AR). It has to be noted that the lack of a 5'-methyluronamide chain prevented the 4'-truncated (N)-methanocarba derivatives from interacting with the threonine residue in TM3, resulting in a missing H-bonding interaction (Figure 2).
Figure 1.

Putative binding modes of sulfonated (N)-methanocarba derivatives obtained after docking simulations: (A) compound 6 at the hA3AR; (B) compound 7 at the hA3AR; (C) compound 6 at the mA3AR; (D) compound 7 at the mA3AR. Side chains of some amino acids important for ligand recognition are highlighted. H-bonding and ionic interactions are pictured as dotted lines. Hydrogen atoms are not displayed. Part of TM7 is omitted.
Figure 2.

Putative binding modes of sulfonated (N)-methanocarba derivatives obtained after docking simulations: (A) compound 18 at the hA3AR; (B) compound 16 at the hA3AR; (C) compound 18 at the mA3AR; (D) compound 16 at the mA3AR. Side chains of some amino acids important for ligand recognition are highlighted. H-bonding and ionic interactions are pictured as dotted lines. Hydrogen atoms are not displayed. Part of TM7 is omitted. (E) Per-residue interaction energy graph. Interaction energy values (in kcal/mol) between the ligand and selected key residues of the binding site, calculated from the binding poses reported in panels A–D.
In the binding mode described above, the extended substituents at the N6 and C2 positions were located in the less ordered extracellular portion of the hA3AR binding pocket and therefore were judged to be suitable sites for insertion of a sulfonate group on these (N)-methanocarba derivatives. Thus, we designed N6-3-chlorobenzyl derivatives with a p- or m-sulfo-phenylethynyl group at the C2 position (6 and 7) and N6-p-sulfo-phenylethyl C2-phenylethynyl derivatives (16 and 18) and docked them at our validated h and mA3AR models.
Figure 1 shows the hypothetical binding modes of compounds 6 and 7 inside the h and mA3AR binding pockets. The sulfonated phenylethynyl substituent at the C2 position of these analogs was oriented toward TM2 and EL1 and was anchored by favorable hydrophobic interactions with residues of the upper part of the binding site. Moreover, either the m- or p-sulfonate group on the C2 substituent was predicted to form H-bonds with Gln167 (EL2) and Tyr265 (7.36) at the hA3AR (Figure 1, panels A and B). Both of these residues differed in the mA3AR sequence, and in this case the sulfonate groups of compounds 6 and 7 would interact with Ser74 (EL1) or His168 (EL2) and Arg170 (EL2) (Figure 1, panels C and D). Considering the fact that the binding modes of compounds 6 and 7 presented all the key interactions important for anchoring the (N)-methanocarba series with a good accommodation of the C2 group, both m- and p- insertion of a sulfonate group on the C2-phenylethynyl substituent should be tolerated for the binding at both h and mA3ARs.
On the other hand, in the binding poses of compounds 16 and 18 at the h and mA3ARs (Figure 2), the sulfonated N6-phenylethyl substituent was accommodated in a side pocket surrounded by TM5, TM6 and EL2. Residues delimiting this area diverge between h and mA3ARs, being Val169 (EL2), Met172 (EL2), Arg173 (EL2), Met174 (5.35), Met177 (5.38) and Ile253 (6.58) at the hA3AR, and Arg170 (EL2), Val173 (EL2), Ser174 (EL2), Leu175 (5.35), Met178 (5.38) and Ser254 (6.58) at the mA3AR. At the hA3 subtype, the sulfonated N6 substituent of compounds 16 and 18 fit well in this area and formed ionic interactions with Arg173 (EL2) and a H-bond with the backbone of Met174 (5.35), while the rest of the ligand structure interacted with all the crucial residues in the deeper regions of the binding site (Figure 2, panels A and B). At the mA3AR this sulfonate group formed ionic interactions with Arg170 (EL2) and with another arginine residue located in the upper part of EL2 (Arg152). However, these N6-p-sulfo-phenylethyl derivatives 16 and 18 were unable to strongly interact with Asn251 (6.55) because the adenine core was shifted to accommodate the bulky N6 substituent (Figure 2, panels C and D). The difference in the behavior of these compounds between the h and mA3AR subtypes, seemed to be mainly related to the mutation Val169/Arg170 in EL2 and to the resulting different orientations of ligands in the binding sites. Therefore, we expected the N6-p-sulfo-phenylethyl derivatives to be relative less potent at the mA3AR as compared to the hA3AR. The detrimental effect of the putative missing interaction with Asn251 (6.55) for these derivatives at the mA3AR would be even more relevant for the affinity of the 4'-truncated compound 16, due to the additional missing H-bond with the threonine in TM3 (Figure 2, panel D).
To analyze in a more quantitative way the observed different interactions of these derivatives at the h and mA3ARs, we calculated per-residue interaction energies from the reported ligand-receptor complexes. Figure 2, panel E, highlights the contributions to the interaction energy of selected key residue of the binding sites. In agreement with the analysis of the docking poses, the favorable contribution to the interaction energy of Asn 6.55 was much stronger for the complexes of compounds 16 and 18 with the hA3AR as compared to the mA3AR. While, the contribution of Thr 3.36 was significantly weaker, at both the h and mA3ARs, for the 4'-truncated compound 16 as compared to the 5'-methyluronamide compound 18.
Chemical Synthesis
The synthetic routes used to prepare the new sulfonated and nonsulfonated (N)-methanocarba adenosine analogues 6, 7, 17, and 18 (Scheme 1) and truncated derivative 16 (Scheme 2) are shown. The synthetic approaches followed the general schemes reported by Tosh et al.25,26 Intermediate 19 was subjected to sequential displacement of the 6-Cl group in the presence of appropriate amines at room temperature followed by aminolysis of the 5′ ester with 40% methylamine solution provided the corresponding amide derivatives (23–25). Sonogashira coupling of iodo derivatives (23–25) with sulfonated and non-sulfonated phenylacetylenes in the presence of PdCl2(Ph3P)2, CuI, triethylamine and subsequent acid hydrolysis of the isopropylidene group afforded the final nucleoside derivatives (6, 7, 17 and 18). The intermediate 31 (Scheme 2) in the truncated series, was treated with 4-(2-aminoethyl)benzenesulfonic acid in the presence of triethylamine following a similar sequence including a displacement reaction at 6-Cl as in compound 19 (Scheme 1) to provide compound 32. Sonogashira coupling of 32 with phenylacetylene followed by acid hydrolysis with aqueous trifluoroacetic acid yielded compound 16. The sulfonated phenylacetylenes 38 (p) and 39 (m) used in the preparation of 6 and 7, respectively, were prepared as shown in Scheme S1 (Supporting Information).
Scheme 1.

Synthesis of new sulfonated and nonsulfonated (N)-methanocarba-adenosine analogues containing the 5’-N-methyluronamide group. Reagents: (i) R1NH2, Et3N, MeOH, rt; (ii) 40% MeNH2, MeOH, rt; (iii) HC≡CAr, Pd(PPh3)2Cl2, CuI, Et3N, DMF, rt; (iv) 10% TFA, MeOH, 70°C.
Scheme 2.

Synthesis of new sulfonated, truncated (N)-methanocarba-adenosine analogue 16. Reagents: (i) 4-SO3H-Ph(CH2)2NH2, Et3N, MeOH, rt; (ii) HC°C-Ph, Pd(PPh3)2Cl2, CuI, Et3N, DMF, rt; (iii) 10% TFA, MeOH, 70°C.
Pharmacological Evaluation
The synthesized analogues (Table 1) were assayed using radioligand binding at three hAR subtypes and at three mAR subtypes using standard 3H and 125I-labeled nucleoside ligands of high affinity (40 – 42).25,26 The hA1AR and hA3AR were stably expressed in CHO cells, and the hA2AAR and the mARs were stably expressed in HEK293 cells. The (N)-methanocarba ring modification was previously shown to reduce potency at the A2BAR, therefore these nucleosides were not assayed at this AR subtype.38 The data for hA3AR binding for 5 and 8–15 were already published,26 and the data for these compounds at the mA3AR were determined here. Functional potency and efficacy at the A3AR have been determined for several derivatives including the sulfonated (N)-methanocarba derivatives and for some previously reported analogs (Figure 3).
Table 1.
Potency of 5'-N-methyluronamido and 4'-truncated (N)-methanocarba adenosine derivatives at hARs and mARs and functional efficacy at the A3ARs.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | Structure | Species | Affinity (Ki, nM) or % inhibitiona,b | % Efficacyc | |||
| R1 | R2 | A1 | A2A | A3 | A3 | ||
| 5d | 3-Cl-Bn | H | h | (20%±3%) | (27%±3%) | 1.34±0.30 | 101±5.9 |
| m | (50%±5%) | (2%±1%) | 1.23±0.14 | ND | |||
| 6 | 3-Cl-Bn | 4-SO3H | h | 383±75 | (23%±3%) | 11.1±1.6 | 96.8±5.7 |
| m | 35.1±5.5 | (14%±4%) | 9.68±0.15 | 95.7±19.1 | |||
| 7 | 3-Cl-Bn | 3-SO3H | h | (16%±3%) | (7%±6%) | 1.90±0.03 | 98.2±6.7 |
| m | (15%±2%) | (1%±1%) | 11.3±1.9 | 89.3±7.1 | |||
| 8d | Me | H | h | (18% ± 1%) | (18% ± 3%) | 5.48±1.23 | 12.6±4.0 |
| m | 3800±780 | (8%±3%) | 1530±240 | ND | |||
| 9d | Et | H | h | (36% ± 4%) | (42% ± 4%) | 5.02 ± 2.19 | 0.8±5.2 |
| m | (49% ± 6%) | (49%±2%) | 1480±170 | ND | |||
| 10d | Et | 2-Cl | h | (25%± 11%) | (17% ± 6%) | 5.80 ± 2.08 | −7.0±5.2 |
| m | (47% ± 8%) | (11%±1%) | (50% ± 9%) | ND | |||
| 11d | 3-Cl-Bn | H | h | (37%±4%)g | 680±170 | 39.0±20.0 | 13.8±5.1 |
| 12d | 3-Cl-Bn | 3-Cl | h | (26%±3%) | 1800±310 | 210±40 | 4.5±4.9 |
| 13d | 3-Cl-Bn | 4-Ph | h | (48%±4%) | (12%±7%) | 54.0±7.0 | −3.5±3.2 |
| m | 1110±220 | (0%) | 255±77 | ND | |||
| 14d | Ph(CH2)2 | H | h | (30%±8%) | (22%±5%) | 20.0±6.0 | 4.1±1.2 |
| m | (39%±6%) | (13%±2%) | 480±90 | 14.3±6.1 | |||
| 15d | Ph2CHCH2 | 2-Cl | h | (26%±4%) | (22%±2%) | 140±30 | −2.6±1.3 |
| m | (23% ± 1%) | (16%±%) | (54% ± 3%) | ND | |||
| 16 | 4-SO3H- Ph(CH2)2 | H | h | (10%±5%) | (15%±3%) | 30.2±4.3 | 7.2 |
| m | (18%±2%) | (1%±2%) | 3920±1190 | ND | |||
| 17 | Ph(CH2)2 | H | h | (11%±3%) | (32%±4%) | 1.23±0.57 | 105.3±9.8 |
| m | (25%±5%) | (1%±1%) | 8.75±2.12 | 114.6±14.6 | |||
| 18 | 4-SO3H- Ph(CH2)2 | H | h | (9%±5%) | (1%±1%) | 12.1±1.0 | 93.8±7.1 |
| m | (7%±2%) | (0%) | 71.1±13.0 | ND | |||
Binding in membranes prepared from CHO or HEK293 (A2A only) cells stably expressing one of three hAR subtypes. The binding affinity for A1, A2A and A3ARs was expressed as Ki values (n = 3–4) using agonist radioligands [3H]N6-R-phenylisopropyladenosine 40, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5'-N-ethylcarboxamido-adenosine 41, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5'-N-methyl-uronamide 42, respectively. A percent in parentheses refers to inhibition of binding at 10 µM.
Binding in membranes prepared from HEK293 cells stably expressing one of three mAR subtypes. Radioligand used were [125I]N6-(4-amino-3-iodobenzyl)adenosine-5'-N-methyl-uronamide 43 (A1 and A3ARs) and [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5'-N-ethylcarboxamido-adenosine 41 (A2AAR). The data (n = 3–4) are expressed as Ki values. A percent in parentheses refers to inhibition of binding at 10 µM.
Efficacy, expressed as a percentage of the maximal effect of either 5'-N-ethylcarboxamidoadenosine 43 (hA3ARs) or 2-chloro-N6-(3-iodobenzyl)adenosine-5'-N-methyluronamide 1b (mA3ARs) to inhibit forskolin-stimulated cAMP production, was determined in cAMP assays using hA3AR-transfected CHO cells or mA3AR-transfected HEK cells. In studies with the hA3AR, maximal efficacies of 1b and test compounds were estimated by measuring the extent of inhibition of forskolin-stimulated cAMP accumulation produced each at a concentration of 10 µM. In studies with the mA3AR, maximal efficacies of 1a and test compounds were determined from concentration-effect curves. Data are expressed as mean±SEM (n = 3–5).
Compounds 5 and 8–15 were prepared and tested for binding at the hARs in Ref. 26.
ND – not determined.
Figure 3.
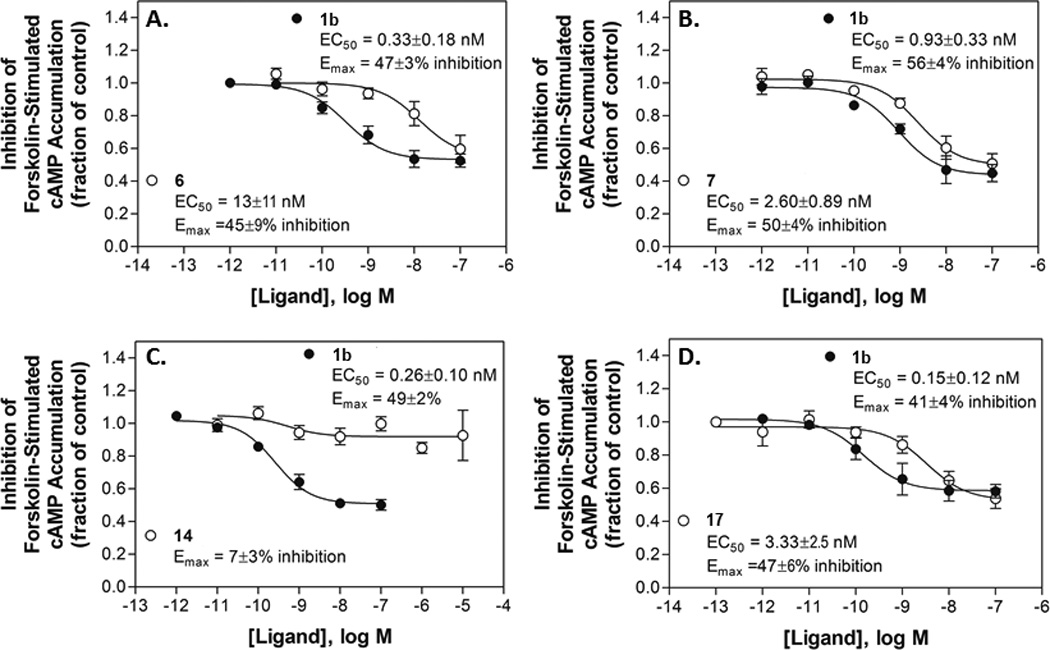
Functional agonism by selected nucleosides, (A) p-sulfonate 6, (B) m-sulfonate 7, (C) truncated N6-phenylethyl derivative 14, (D) N6-phenylethyl 5’-N-methyluronamide derivative 17, in an assay of inhibition of forskolin-stimulated cAMP accumulation with HEK293 cells expressing the mA3AR. Concentration-effect curves with potent A3AR agonist 1b are included for comparison. Data are the mean±SEM, n = 3–5.
The previously reported N6-3-chlorobenzyl C2-phenylethynyl derivative 5 displayed identically high affinity and selectivity at both the h and mA3ARs (Ki hA3AR = 1.3 nM; Ki mA3AR = 1.2 nM), while being nearly inactive at the A1 and A2AARs (h and m). The C2-p-sulfo-phenylethynyl analogue 6 displayed a Ki value of 11.1 nM in binding to the hA3AR and also a comparable affinity at the mA3AR (Ki = 9.7 nM). However, m-sulfo-substitution on the C2-phenylethynyl group was associated with slightly higher affinity at the hA3AR as compared to the mA3AR (compound 7, Ki hA3AR = 1.9 nM; Ki mA3AR = 11.3 nM).
At the A1AR, only 10 to 50% of binding inhibition was seen at 10 µM for the majority of the tested (N)-methanocarba derivatives with few exceptions. In particular, the C2-p-sulfo-phenylethynyl derivative 6 showed enhanced affinity at the hA1AR (Ki = 383 nM) and even higher affinity at the mA1AR (Ki = 35.1 nM).
A N6-p-sulfo-phenylethyl group was tolerated at the hA3AR in both the 5'-N-methyluronamide and 4'-truncated series (uronamide 18, Ki hA3AR = 12.1 nM; truncated compound 16, Ki hA3AR = 30.2 nM). However, this substitution reduced mA3AR affinity, especially in the 4'-truncated series (18, Ki mA3AR = 71.1 nM; 16, Ki mA3AR = 3.92 µM).
Functional data were determined in assays of A3AR-induced inhibition of the forskolin-stimulated production of cAMP in CHO cells stably expressing the hA3AR or in HEK293 cells stably expressing the mA3AR (Table 1, Figure 3). As expected, 4'-truncated derivatives were generally low-efficacy partial agonists or antagonists of the hA3AR, while 5'-N-methyluronamide analogs showed high efficacy at this subtype. In the truncated series the efficacy varied depending on the substitution of the C2-phenylethynyl group and on the N6 substituent. Full curves for functional potency at the mA3AR (EC50 values in nM) were determined for C2 p-sulfonate 6 (13±11), C2 m-sulfonate 7 (2.60±0.89), uncharged truncated N6-phenylethyl derivative 14 and the corresponding 5'-N-methyluronamide N6-phenylethyl derivative 17 (3.33±2.5) in comparison to standard riboside 1b (0.33±0.18). Only 14 displayed significantly less (~14% of the level of 1b) than full efficacy.
For compounds 6 and 7 functional potency and efficacy at the mA1AR were also determined in assays of A1AR-induced inhibition of the production of cAMP in HEK293 cells expressing the mA1AR (Figure 4). The m-sulfo analogue 7 was completely inactive up to 10 µM, and the corresponding p-sulfo isomer 6, the only derivative showing good affinity at the mA1AR, was a moderately potent partial agonist at the mA1AR (EC50 495±52 nM, with maximal efficacy 49% of full agonist 44).
Figure 4.

Functional agonism by mixed A1/A3AR agonist 6 and selective A3AR agonist 7 in an assay of inhibition of forskolin-stimulated cAMP accumulation with HEK293 cells expressing the mA1AR. Concentration-effect curve with reference full agonist 44 is included for comparison. Data are the mean±SEM, n = 4–7.
In vivo studies
The prototypical A3AR agonists N6-(3-iodobenzyl)adenosine-5'-N-methyluronamide (IB-MECA, 1a), 1b and an (N)-methanocarba derivative 2 were shown to reverse the development of neuropathic pain following chronic constriction injury (CCI) in mice via an A3AR-mediated mechanism.13 Using the same model, the p- and m-sulfo derivatives 6 and 7 also reversed mechano-allodynia. When peak mechano-allodynia developed on day 7 (D7) following CCI of the mouse sciatic nerve,39 i.p. administration of either agonist (3 µmol/kg, n=6), but not vehicle (5% DMSO in saline, n=6), reversed mechano-allodynia, with a maximal effect within 1 h (Figure 5A, B). As was demonstrated for the prototypical agonists 1a and 1b,13 the p- and m-sulfo derivatives 6 and 7 lacked effect on Paw Withdrawal Thresholds (PWT, g) in contralateral paws (data not shown). Intrathecal administration of the A3AR selective antagonist 5-propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate (45, MRS1523, 3 nmol, n=5), had no effect on the anti-allodynic actions of systemic agonist 7 (3 µmol/kg, n=5, Figure 5C). The effects of A3AR-selective agonist 7 where completely lost in the A3AR−/− mice (n=4) whereas those of mixed A1AR/A3AR agonist 6 were significantly but not completely abrogated (n=4, Figure 5D).
Figure 5.

Mixed A1/A3AR agonist 6 (n = 6) and selective A3AR agonist 7 (n = 5) reverse CCI-induced neuropathic pain (A, B), measured as described.13 Mechano-allodynia developed by D7 after CCI of the sciatic nerve, which was reversed by i.p. administration (3 µmol/kg. in 5% DMSO in saline) of the sulfonated nucleosides. (C) Intrathecal administration of the selective A3AR antagonist 45 (3 nmol, n=5), did not block the effects of systemic 7, excluding the spinal cord as a site of action of this sulfonate-containing agonist. In male A3AR knock-out mice (D), only 6 (i.p., 3 µmol/kg, n = 2) retained the ability to produce a small protective effect. The protective effect of 7 by (i.p., 3 µmol/kg, n = 3) was completely eliminated. *P< 0.001 vs. D0; †P< 0.01 and ††P< 0.001 vs. D7.
Discussion
This study is an example of applying the new structural insights into GPCRs to substitution of functional groups on existing ligand scaffolds. Previously, such efforts would be performed entirely empirically, and now it is possible to predict interactions and the effects on pharmacological potency using homology modeling and ligand docking.
The previously observed SAR of the (N)-methanocarba series at the A3AR can be rationalized based on steric fitting of the binding cavity and crucial interactions with key binding site residues (Figure 6). The use of hybrid A3AR models helps to explain the high A3AR affinity and selectivity associated with the insertion of a C2-phenylethynyl group that can be accommodated after outward movement of TM2. Different hydrophobic arylalkyl substituents, including substituted phenylethyl groups or longer rigid groups, at the N6 position can be accommodated in a side pocket delimited by EL2 and are tolerated for A3AR affinity. Truncation of the 5’-N-methyluronamide moiety results in a missing H-bonding interaction with a Thr in TM3, key residue for agonist binding, and determines a slight decrease in affinity and a significant efficacy reduction at the A3 subtype.
Figure 6.

General requirements of high affinity binding to the A3AR for the (N)-methanocarba series rationalized towards molecular modeling analysis, showing putative interactions of sulfonate groups.
The present C2-phenylethynyl analogues include both full agonists containing a 5’-N-methyluronamide and the corresponding 4’-truncated antagonists (or low efficacy agonists). In the truncated series the N6-phenylethyl group, in comparison to other N6 substitution, tended to preserve interspecies selectivity at the A3AR. Even the widely used N6-(3-halobenzyl) group in p-biphenylethynyl 13 did not result in high A3AR selectivity in the mouse, as it did in human. Curiously, this compound bound to the mA1AR with nearly as high affinity as at the mA3AR. Similarly, a truncated N6-methyl analogue 8 was relatively weak and nonselective between A1AR and A3AR.
Sulfonated analogues of otherwise uncharged receptor ligands have been shown to restrict diffusion across biological membranes and are invariably associated with increased aqueous solubility.31 Sulfonation inhibits penetration into the brain, and such charged compounds will most likely also have very limited oral bioavailability. There are no previous reports of potent and selective A3AR agonists having a sulfonate group, although such pharmacological tools are needed for the delineation of effects of this receptor in the central and peripheral nervous systems. An earlier attempt to introduce a sulfonate group, which is essentially permanently charged at physiological pH, in A3AR agonists failed.32 Nevertheless, our molecular modeling predicted that an aryl sulfonate group placed on an extended substituent at the C2 position would be tolerated in binding to the h and mA3AR, while a N6-p-sulfo-phenylethyl substituent would determine higher hA3AR affinity as compared to the mA3AR. The present set of analogues contains several aspects of conformational rigidity that increase confidence in the molecular docking: rigidity of the ribose-like moiety and a rigid rod-like arylethynyl group at the C2 position. The precision of the homology modeling, based on the X-ray structure of the closely related A2AAR, allowed predictions to be made for comparing the two species and three different positions of sulfonate substitution, as a result of predicted interactions with amino acid residues in the extracellular regions of the A3AR. The great degree of sequence divergence for the A3AR subtype between rodents and other mammalian species can lead to differences in ligand binding properties. Therefore, the construction of both h and mA3AR models proved to be useful in designing ligands with species-independent A3AR affinity.
In particular, docking results showed how the insertion of a sulfonate group on the C2-phenylethynyl substituent was well tolerated at A3ARs of both species. In fact, despite the presence of different residues in EL2 and EL1 between h and mA3ARs, the region around the C2-substituent is open to the extracellular side in both h and mA3AR models and could easily accommodate a sulfonate group at either the p- or m- position of the phenyl ring.
Much stricter are the requirements regarding the N6 substituents to have compounds with high inter-species A3ARs affinity. Due to the Val 169/Arg 170 substitution in EL2 between h and mA3ARs, respectively, only the hA3AR could tolerate several different N6 substituents without loss in affinity. Conversely, at the mA3AR a good steric fit between the N6 group and the side pocket delimited by EL2 and TMs 5 and 6 seemed to be required to place the ligand in a binding conformation able to strongly interact with all the key residues of the binding site. Therefore, compound 18, bearing a sulfonate group at the p- position of the N6-phenylethyl substituent, showed optimal interactions with the hA3AR but weaker contacts at the mA3AR, due to missing H-bonds of the adenine moiety with Asn251 in TM6. Insertion of a sulfonate group on a N6-phenylethyl substituent, was also examined in the truncated series of nucleosides, which are known to display greatly reduced efficacy at the A3AR. In this case, the effect of the more difficult accommodation of the N6 group and consequent weaker interaction with Asn 6.55 at the mA3AR is even more marked due to the already missing interaction with a Thr in TM3. A truncated nucleoside 16 with a sulfonate on an N6-phenylethyl group was a selective, low efficacy partial agonist at the h but not m A3AR.
Thus, it seems that the N6 substituent can either compensate for the missing interactions of the 4'-truncated series in case of optimal fit with the extracellular region of the binding site, as previously observed for the A1AR,40 or increase the detrimental effect of the truncation on the affinity in case of difficult accommodation in the extracellular region, as observed here.
We have confirmed modeling predictions by chemical synthesis of the designed molecules and binding and efficacy studies. Moreover, two of these sulfonated compounds were tested in a mouse model of neuropathic pain following our initial report of the beneficial effect of A3AR activation in such conditions.13 Compound 6 was found to activate both A1 and A3ARs, while an isomeric nucleoside 7, in which the sulfonate was moved from the p- to m- position on the C2-phenylethynyl group, activated only the A3AR. The A3AR selectivity of 7 in comparison to the A1AR and A2AAR was >1000-fold in the mouse and >10,000-fold in the human. The structural basis for the interaction of 6 with the h or mA1AR has not been probed in this study. However, we can speculate that for some particular C2-arylethynyl derivatives a similar outward movement of TM2, as observed in the A3AR, can occur also in the A1AR. This receptor, in fact, does not present a disulfide bond network constraining the TM2-EL1-TM3 region as observed in the A2AAR crystal structure. Therefore, in particular cases the A1AR could undergo conformational changes when binding to ligands bearing bulkier C2-groups.
The ability of agonists of the A1AR or the A3AR to reduce or prevent neuropathic pain has been studied previously.13,14,41,42 The sulfonated agonists are introduced here as a means of separating the peripheral and central effects of AR agonists, depending on the route of administration. Upon i.p. administration, the mixed agonist 6 elicited a more pronounced reduction in neuropathic pain than the A3AR-selective agonist 7 following CCI. We ascribe the protection by 6 to a combination of A1 and A3AR-dependent effects. Additionally, the selective A3AR antagonist 45, given centrally, was unable to block the anti-allodynic effects of agonist 7, further supporting that the site of action of sulfonated agonist 7 as administered is restricted to the periphery. The same antagonist administered systemically was completely effective in blocking the anti-allodynic effects of A3AR agonist 1a in the identical CCI model.13 However, antagonists of the A1AR and A2AAR were completely ineffective in blocking the protective action of 1a. The attenuation and loss, respectively, of the beneficial actions of agonists 6 and 7 in A3AR−/− mice is consistent with their measured AR selectivity in both binding and functional assays.
The presence and role of the A3AR in the CNS is subject to contradictory findings. Attempts to label the A3AR binding sites autoradiographically in the brain have not succeeded using a potent nonselective labeled agonist in the presence of selective antagonists for other ARs.43 Ravid and coworkers found expression of the A3AR in the mouse brain.44 The A3AR is also known to be expressed in astrocytes and microglial cells.45,46 Using knockout mice, the A3AR was shown to influence heart rate, motor activity and body temperature, even though the level of expression in the brain is likely low.47 It appears that protection against neuropathic pain by A3AR agonists has both central and peripheral components.48 The effects of low doses of 1a in CCI mice to reverse mechano-allodynia had both peripheral and spinal sites of action that were attenuated by intraplantar or intrathecal injection of 45. Thus, the introduction of potent A3AR agonist probes designed to be precluded from crossing the blood brain barrier promises to be useful in delineating the effects of activating central vs. peripheral ARs.
The isomeric compounds 6 and 7 are very close in physicochemical properties and likely in vivo distribution. The cLog P of both is 1.15, and the total polar surface area is 176 Å2, which is outside the range of molecules that readily cross the blood brain barrier. In addition to the exclusion from readily crossing biological membranes, there is the dimension of either very high selectivity for the A3AR in 7 or the feature of dual activation of the mA1AR and mA3AR in 6. This near equivalence of high affinity to the exclusion of the A2AAR is present in the mouse and to a lesser extent in the human. Therefore this pair of isomeric nucleosides can be used together as pharmacological tools.
Conclusions
Molecular modeling has provided a detailed window into the binding site interactions in the ARs. This insight has guided the process of refining the selectivity of A3AR agonists in recent studies, and now we extend the analysis to provide useful pharmacological tools that have increased water solubility and are selective for compartments in the body because they are not expected to diffuse readily across phospholipid bilayers. The ability of molecular modeling now to predict the likely pharmacological effects of functional group substitution on a ligand scaffold is a useful and labor-saving approach in the exploration of SAR at GPCRs.
Experimental Section
Molecular Modeling
hA3AR and mA3AR homology models
Homology models of the hA3 and mA3 ARs were built based on a hybrid template structure following a strategy previously described25 and using the alignment and the homology modeling tools implemented in the MOE suite.49 To build these models, an agonist-bound hA2AAR crystal structure (PDB ID: 3QAK)34 was used as a template for the entire A3AR structure except for the extracellular terminus of TM2 (residues from Val63 to Ser73 at the hA3AR and from Val64 to Ser74 at the mA3AR) and EL1 (residues from Leu74 to Tyr81 at the hA3AR and from Leu75 to Tyr82 at the mA3AR). The X-ray structure of the hβ2 adrenergic receptor in complex with the Gs protein (PDB ID: 3SN6),35 after superimposition with the hA2AAR crystal structure, was used as template to build the extracellular terminus of TM2. No structural template was used for the modeling of EL1.
In particular, h and mA3ARs sequences were retrieved from the UniProt database50 and aligned against the sequence of the hybrid template, taking into account the highly conserved residues in each TM domain and following the numbering scheme by Ballesteros and Weinstein.51 Then, A3AR homology models were built using the automated Homology Modeling protocol implemented in the MOE suite. The previously published docking pose of compound 11 inside the A3AR26 was taken into account during the modeling, following the so called Ligand Based Homology Modeling approach.52 The AMBER99 forcefield has been used for protein modeling and the Protonate 3D methodology has been used for protonation state assignment.53 The final model has been refined through energy minimization until an RMS gradient of 0.1 kcal/mol Å. Model's stereochemical quality was checked using several tools (Ramachandran plot; backbone bond lengths, angles and dihedral plots; clash contacts report; rotameric strain energy report) implemented in the MOE suite.
Molecular docking of (N)-methanocarba derivatives at the A3AR models
Compounds structures were built using the builder tool implemented in the MOE suite49 and subjected to energy minimization using the MMFF94x force field, until a RMS gradient of 0.05 kcal/mol Å. Molecular docking of the ligands at the A3AR models was performed by means of the Glide54 package part of the Schrödinger suite.55 The docking site was defined using key residues in the binding pocket of the A3AR models, namely Phe (EL2), Asn (6.55), Trp (6.48) and His (7.43), and a 20Å × 20Å × 20Å box was centered on these residues. Docking of ligands was performed in the rigid binding site using the XP (extra precision) procedure. The top scoring docking conformations for each ligand were subjected to refinement using the Prime MM-GBSA module implemented in the Schrödinger suite.56 The Prime side-chain sampling was performed on all the residues within a 6Å of the ligand. The refined model for each ligand was chosen as final binding conformation. Finally, to calculate per-residue interaction energies, selected ligands were scored in place in their refined complexes, using the Glide module.
Chemical Synthesis
Materials and Instrumentation
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). 1H NMR spectra were obtained with a Bruker 400 spectrometer using CDCl3 and CD3OD as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Aldrich. All the final sulfonate nucleoside compounds were purified by HPLC with a Luna 5 µmRP-C18(2) semipreparative column (250 mm × 10.0 mm; Phenomenex, Torrance, CA) and using the following conditions: flow rate of 2 mL/min; 10 mM trifluoroacetic acid-water (TFA-H2O)-CH3CN from 80:20 to 30:70 in 20–40 min. The purity of final nucleoside derivatives was checked using a Hewlett–Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 µm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)–CH3CN from 80:20 to 0:100 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6-kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
4-((6-(3-Chlorobenzylamino)-9-((1S,2R,3S,4R,5S)-3,4-dihydroxy-5-(methylcarbamoyl)bicyclo[3.1.0]hexan-2-yl)-9H-purin-2-yl)ethynyl)benzenesulfonic acid (6)
PdCl2(PPh3)2 (2.5 mg, 0.003 mmol), CuI (1.0 mg, 0.004 mmol), 3-ethynyl benzenesulfonic acid (37, 20 mg, 0.11 mmol) and triethylamine (25 µL, 0.18 mmol) were added to a solution of compound 23 (10.8 mg, 0.018 mmol) in anhydrous DMF (1 mL), and stirred at room temperature for overnight. Solvent was evaporated under, vacuum and the residue was roughly purified on flash silica gel column chromatography. The resulting compound was dissolved in methanol (1 mL) and 10% trifluoromethane sulfonic acid (1 mL) and heated at 70 °C for 5 h. Solvent was evaporated under, vacuum and the residue was purified on semi preparative HPLC (retention time 30 min) to give the compound 27 (7.6 mg, 69%) as a syrup. 1H NMR (CD3OD, 400 MHz) d 8.15 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.4 Hz, 2H), 7.46 (s, 1H), 7.38–7.26 (m, 3H), 5.09 (d, J = 6.4 Hz, 1H)), 4.91 (s, 1H), 4.83 (d, J = 6.4 Hz, 1H), 2.84 (s, 3H), 2.14–2.10 (m, 1H), 1.89-1.86 (m, 1H), 1.40–1.36 (m, 1H). HRMS calculated for C28H26N6O6SCl (M + H) +: 609.1323; found 609.1312.
3-((6-(3-Chlorobenzylamino)-9-((1S,2R,3S,4R,5S)-3,4-dihydroxy-5-(methylcarbamoyl)bicyclo[3.1.0]hexan-2-yl)-9H-purin-2-yl)ethynyl)benzenesulfonic acid (7)
Compound 7 (65%) was prepared from compound 23 following the same method for compound 6, except using 38. Semipreparative HPLC retention time 28 min. 1H NMR (CD3OD, 400 MHz) d 8.13 (s, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 7.6 Hz, 1H), 7.54–7.46 (m, 3H), 7.38–7.26 (m, 3), 5.10 (d, J = 6.0 Hz, 1H), 4.91 (s, 1H), 4.05 (d, J = 6.0 Hz, 1H), 2.84 (s, 3H), 2.13–2.10 (m, 1H), 1.89–1.86 (m, 1H), 1.41–1.38 (m, 1H). HRMS calculated for C28H24N6O6SCl (M – H)+: 607.1167; found 607.1161.
4-(2-(9-((1S,2R,3S,4R,5R)-3,4-Dihydroxybicyclo[3.1.0]hexan-2-yl)-2-(phenylethynyl)-9H-purin-6-ylamino)ethyl)benzenesulfonic acid (16)
Compound 16 (93%) was prepared from compound 32 following the same method for compound 17. 1H NMR (CD3OD, 400 MHz) d 8.34 (s, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.69–7.66 (m, 2H), 7.47–7.46 (m, 3H), 7.40 (d, J = 8.0 Hz, 2H), 4.71(t, J = 6.3 Hz, 1H), 4.01 (s, 1H), 3.93 (d, J = 6.4 Hz, 1H), 3.10 (t, J = 6.8 Hz, 2H), 2.02–2.00 (m, 1H), 1.74–1.68 (m, 1H), 1.38–1.32 (m, 1H), 0.82–0.79 (m, 1H). HRMS calculated for C27H24N5O5S (M – H) +: 530.1498; found 530.1495.
(1S,2R,3S,4R,5S)-2,3-Dihydroxy-N-methyl-4-(6-(phenethylamino)-2-(phenylethynyl)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxamide (17)
PdCl2(PPh3)2 (5.8 mg, 0.008 mmol), CuI (1.0 mg, 0.004 mmol), phenylacetylene (27 µL, 0. 25 mmol) and triethylamine (30 µL, 0.41 mmol) were added to a solution of compound 24 (24.6 mg, 0.041 mmol) in anhydrous DMF (1 mL), and stirred at room temperature for overnight. Solvent was evaporated under, vacuum and the residue was roughly purified on flash silica gel column chromatography. The resulting compound was dissolved in methanol (2 mL) and 10% trifluoromethane sulfonic acid (2 mL) and heated at 70 °C for 5 h. Solvent was evaporated under, vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH = 25:1) to give the compound 7 (15 mg, 71%) as a syrup. 1H NMR (CD3OD, 400 MHz) d 8.10 (s, 1H), 6.78–6.75 (m, 2H), 7.47–7.44 (m, 3H), 7.33–7.26 (m, 4H), 7.21–7.17 (m, 1H), 5.05 (d, J = 5.2 Hz, 1H), 4.89 (s, 1H), 4.01 (d, J = 5.2 Hz, 1H), 3.90 (br s, 2H), 3.02 (t, J = 7.2 Hz, 2H), 2.84 (s, 3H), 2.13–2.09 (m, 1H), 1.89–1.87 (m, 1H), 1.41–1.37 (m, 1H). HRMS calculated for C29H29N6O3 (M + H) +: 509.2301; found 509.2291.
4-(2-(9-((1S,2R,3S,4R,5S)-3,4-Dihydroxy-5-(methylcarbamoyl)bicyclo[3.1.0]hexan-2-yl)-2-(phenylethynyl)-9H–purin-6-ylamino)ethyl)benzenesulfonic acid (18)
Compound 18 (63%) was prepared from compound 25 following the same method for compound 6. 1H NMR (CD3OD, 400 MHz) d 8.18 (s, 1H), 7.76 (d, J = 7.6 Hz, 2H), 7.70–7.67 (m, 2H), 7.48– 7.46 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 5.06 (d, J = 6.8 Hz, 1H), 4.90 (s, 1H), 4.03 (d, J = 6.8 Hz, 1H), 3.92 (br s, 2H), 3.09 (t, J = 6.8 Hz, 2H), 2.84 (s, 3H), 2.14–2.11 (m, 1H), 1.89–1.86 (m, 1H), 1.42–1.39 (m, 1H). HRMS calculated for C29H27N6O6S (M – H) +: 587.1713; found 587.1696.
(1S,2R,3S,4R,5S)-Ethyl –(2,3-O-isopropylidene)-4-(2-iodo-6-(phenylethylamino)-9H-purin-9-yl)bicyclo[3.1.0]hexane-1-carboxylate (21)
2-Phenylethylamine (0.14 mL, 1.12 mmol) and triethylamine (0.4 mL, 2.2 mmol) were added to a solution of compound 19 (114 mg, 0.22 mmol) in anhydrous methanol (4 mL) and stirred at room temperature for overnight. Solvent was evaporated under, vacuum and the residue was purified on flash silica gel column chromatography (hexane:ethyl acetate=1:1) to give the compound 21 (109 mg, 82%) as foamy solid. 1H NMR (CD3OD, 400 MHz) d 7.93 (s, 1H), 7.27 (d, J = 4.4 Hz, 4H), 7.21–7.18 (m, 1H), 5.82 (d, J = 7.2 Hz), 4.91 (s, 1H), 4.80 (d, J = 6.4 Hz), 3.79–3.76 (m, 4H), 2.96 (t, J = 7.2 Hz, 2H), 2.25–2.21 (m, 1H), 1.65–1.61 (m, 1H), 1.53–1.49 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H), 1.27 (s, 3H). HRMS calculated for C25H29IN5O4 (M + H) +: 590.1264; found 590.1286.
(1S,2R,3S,4R,5S)-Ethyl –(2,3-O-isopropylidene)-4-{2-iodo-6-(4-(2-aminoethyl) benzene sulfonic acid)-9H-purin-9-yl}bicyclo[3.1.0]hexane-1-carboxylate (22)
Compound 22 (78%) was prepared from compound 19 following the same method for compound 21. 1H NMR (CD3OD, 400 MHz) d 7.91 (s, 1H), 7.78 (d, J = 6.0 Hz, 2H), 7.35 (d, J = 6.0 Hz, 2H), 5.82 (d, J = 7.2 Hz), 4.91 (s, 1H), 4.80 (d, J = 5.6 Hz), 3.77 (br s, 2H), 3.15 (t, J = 6.9 Hz, 2H), 3.05 (t, J = 6.4 Hz, 2H), 2.26–2.22 (m, 1H), 1.65–1.61 (m, 1H), 1.53–1.47 (m, 4H), 1.34 (t, J = 7.2 Hz, 3H), 1.29 (s, 3H). HRMS calculated for C25H29IN5O7S (M + H) +: 670.0832; found 670.0851.
(1S,2R,3S,4R,5S)-(2,3-O-Isopropylidene)-4-(2-iodo-6-(phenylethylamino)-9H-purin-9-yl)-N-methylbicyclo[3.1.0]hexane-1-carboxamide (24)
40% Methylamine solution (2 mL) was added to a solution of compound 21 (65 mg, 0.11 mmol) in methanol (2 mL) and stirred at room temperature for 24 h. Solvent was evaporated under, vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH=40:1) to give the compound 43 (41 mg, 65%) as a syrup.1H NMR (CD3OD, 400 MHz) d 7.95 (s, 1H), 7.27 (d, J = 4.4 Hz, 4H), 7.20–7.17 (m, 1H), 5.72 (d, J = 6.8 Hz, 1H), 4.92 (s, 1H), 4.83 (d, J = 6.0 Hz, 1H), 3.76 (br s, 2H), 2.96 (t, J = 7.2 Hz, 2H), 2.9 (s, 3H), 2.15–2.13 (m, 1H), 1.54–1.49 (m, 4H), 1.40 (t, J = 5.2 Hz, 1H), 1.30 (s, 3H). HRMS calculated for C24H28IN6O3 (M + H) +: 575.1268; found 575.1266.
(1S,2R,3S,4R,5S)-(2,3-O-Isopropylidene)-4-{2-iodo-6-(4-(2-aminoethyl) benzene sulfonic acid)-9H-purin-9-yl}-N-methylbicyclo[3.1.0]hexane-1-carboxamide (25)
Compound 25 (68%) was prepared from compound 22 following the same method for compound 24. 1H NMR (CD3OD, 400 MHz) d 7.93 (s, 1H), 7.78 (d, J = 6.0 Hz, 2H), 7.35 (d, J = 6.0 Hz, 2H), 5.71 (d, J = 7.2 Hz, 1H), 4.92 (s, 1H), 4.83 (d, J = 5.6 Hz, 1H), 3.80 (br s, 2H), 3.12 (t, J = 7.6 Hz, 2H), 2.9 (s, 3H), 2.17–2.12 (m, 1H), 1.54–1.49 (m, 4H), 1.39 (t, J = 5.3 Hz, 1H), 1.30 (s, 3H). HRMS calculated for C24H26IN6O6S (M + H) +: 653.0679; found 653.0677.
(1S,2R,3S,4R,5S)-(2,3-O-Isopropylidene)-4-{2-iodo-6-(4-(2-aminoethyl) benzene sulfonic acid)-9H-purin-9-yl}-bicyclo[3.1.0]hexane (32)
Compound 32 (76%) was prepared from compound 31 following the same method for compound 21.1H NMR (CD3OD, 400 MHz) d 8.02 (s, 1H), 7.78 (d, J = 6.8 Hz, 2H), 7.35 (d, J = 6.8 Hz, 2H), 5.36 (t, J = 6.4 Hz, 1H), 4.95 (s, 1H), 4.69 (d, J = 7.2 Hz, 1H), 3.80 (br s, 2H), 3.16 (t, J = 6.4 Hz, 2H), 2.05–2.02 (m, 1H), 1.72–1.67 (m, 1H), 1.51 (s, 3H), 1.25 (s, 3H), 0.93–0.87 (m, 2H). HRMS calculated for C22H23N5O5SI (M – H) +: 596.0465; found 596.0441.
3-((Trimethylsilyl)ethynyl)benzenesulfonic acid (36)
PdCl2(PPh3)2 (110 mg, 0.15 mmol), CuI (15 mg, 0.15 mmol), trimethylsilylacetylene (0.66 mL, 4.7 mmol) and triethylamine (1.0 mL, 7.8 mmol) were added to a solution of compound 34 (252 mg, 0.78 mmol) in anhydrous DMF (5 mL), and stirred at room temperature for overnight. Solvent was evaporated under, vacuum and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH:TFA = 10:1:0.1) to give the compound 36 (133 mg, 67%) as a syrup. 1H NMR (CD3OD, 400 MHz) d 7.91 (s, 1H), 7.84–7.76 (m, 1H), 7.56–7.42 (m, 2H), 0.21 (s, 9H).. HRMS calculated for C11H13O3SSi (M – H) +: 253.0355; found 253.0361.
4-((Trimethylsilyl)ethynyl)benzenesulfonic acid (37)
Compound 37 (69%) was prepared from compound 35 following the same method for compound 36. 1H NMR (CD3OD, 400 MHz) d 7.80 (d, J = 7.2 Hz, 2H), 7.49 (d, J = 7.2 Hz, 2H), 0.25 (s, 9H). HRMS calculated for C11H13O3SSi (M – H) +: 253.0355; found 253.0355.
3-Ethynylbenzenesulfonic acid (38)
Tetrabutylammonium fluoride (1.9 mL, 1M solution in THF) was added to a solution of compound 36 (406 mg, 1.59 mmol) in THF and stirred at room temperature for 2 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2:MeOH:TFA = 8:1:0.1) to give the compound 37 (235 mg, 81%) as a colorless powder. 1H NMR (CD3OD, 400 MHz) d 7.85–7.76 (m, 2H), 7.48–7.37 (m, 2H), 3.60 (s, 1H). HRMS calculated for C8H5O3S (M -H) +: 180.9959; found 180.9958.
4-Ethynylbenzenesulfonic acid (39)
Compound 39 (84%) was prepared from compound 37 following the same method for compound 38. 1H NMR (CD3OD, 400 MHz) d 7.73 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 3.53 (s, 1H). HRMS calculated for C8H5O3S (M – H) +: 180.9959; found 180.9956.
Binding Studies
[3H]R-N6-Phenylisopropyladenosine (40, [3H]R-PIA, 63 Ci/mmol), [3H](2-[p-(2-carboxyethyl)phenyl-ethylamino]-5'-N-ethylcarboxamido-adenosine) (41, [3H]CGS21680, 40.5 Ci/mmol) and [125I]N6-(4-amino-3-iodobenzyl)adenosine-5'-N-methyluronamide (42, [125I]I-AB-MECA, 2200 Ci/mmol) were purchased from Perkin–Elmer Life and Analytical Science (Boston, MA). Test compounds were prepared as 5 mM stock solutions in DMSO and stored frozen. Pharmacological standards 1b (A3AR agonist), adenosine-5’-N-ethylcarboxamide (43, NECA, nonselective AR agonist) and 2-chloro-N6-cyclopentyladenosine (44, CCPA, A1AR agonist) were purchased from Tocris R&D Systems (Minneapolis, MN).
Cell Culture and Membrane Preparation
CHO cells stably expressing the recombinant hA1 and hA3ARs and HEK293 cells stably expressing the hA2AAR were cultured in Dulbecco’s modified Eagle medium (DMEM) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 µg/mL streptomycin, and 2 µmol/mL glutamine. In addition, 800 µg/mL geneticin was added to the A2A media, while 500 µg/mL hygromycin was added to the A1 and A3 media. After harvesting, cells were homogenized and suspended in PBS. Cells were then centrifuged at 240 g for 5 min, and the pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.5) containing 10 mM MgCl2. The suspension was homogenized and was then ultra-centrifuged at 14,330 g for 30 min at 4 °C. The resultant pellets were resuspended in Tris buffer, incubated with adenosine deaminase (3 units/mL) for 30 min at 37 °C. The suspension was homogenized with an electric homogenizer for 10 sec, pipetted into 1 mL vials and then stored at −80 °C until the binding experiments. The protein concentration was measured using the BCA Protein Assay Kit from Pierce Biotechnology, Inc. (Rockford, IL).57
Binding assays
Into each tube in the binding assay was added 50 µL of increasing concentrations of the test ligand in Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2, 50 µL of the appropriate agonist radioligand, and finally 100 µL of membrane suspension. For the A1AR (22 µg of protein/tube) the radioligand used was [3H]40 (final concentration of 3.5 nM). For the A2AAR (20 µg/tube) the radioligand used was [3H]41 (10 nM). For the A3AR (21 µg/tube) the radioligand used was [125I]42 (0.34 nM). Nonspecific binding was determined using a final concentration of 10 µM 43 diluted with the buffer. The mixtures were incubated at 25 °C for 60 min in a shaking water bath. Binding reactions were terminated by filtration through Brandel GF/B filters under a reduced pressure using a M-24 cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with 3 mL of 50 mM ice-cold Tris-HCl buffer (pH 7.5). Filters for A1 and A2AAR binding were placed in scintillation vials containing 5 mL of Hydrofluor scintillation buffer and counted using a Perkin Elmer Liquid Scintillation Analyzer (Tri-Carb 2810TR). Filters for A3AR binding were counted using a Packard Cobra II γ -counter. The Ki values were determined using GraphPad Prism for all assays.
Similar competition binding assays were conducted using HEK293 cell membranes expressing mARs using [125I]42 to label A1 or A3ARs and [3H]41 to label A2AARs. IC50 values were converted to Ki values as described.58 Nonspecific binding was determined in the presence of 100 µM 43. cAMP accumulation assay: Intracellular cAMP levels in CHO cells expressing the recombinant hA3AR were measured using an ELISA assay.59 Cells were first harvested by trypsinization. After centrifugation and resuspended in medium, cells were planted in 96-well plates in 0.1 mL medium. After 24 h, the medium was removed and cells were washed three times with 0.2 mL DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with the agonist (10 µM 43 for hA3AR) or test compound in the presence of rolipram (10 µM) and adenosine deaminase (3 units/mL). After 30 min forskolin (10 µM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 100 µL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cAMP production, 50 µL of the HCl solution was used in the Amersham cAMP Enzyme Immunoassay following the instructions provided with the kit. The results were interpreted using a SpectroMax M5 Microplate reader (Molecular Devices, Sunnyvale, CA) at 450 nm.
Similar cAMP assays were conducted with HEK293 cells expressing the mA1AR or mA3AR. HEK293 cells were detached from cell culture plates, resuspended in serum-free DMEM containing 25 mM HEPES (pH 7.4), 1 unit/ml adenosine deaminase, 4-(3-butoxy-4-methoxyphenyl)methyl-2-imidazolidone (Tocris, Ro 20,1724, 20 mM) and 300 nM 8-[4-[4-(4-chlorophenzyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine (Tocris, PSB603, 300 nM) inhibit A2BARs expressed endogenously in HEK293 cells, and then transferred to polypropylene tubes (2 × 105 cells/tube). The cells were co-incubated with forskolin (10 µM) and AR ligands for 15 min at 37o C with shaking, after which the assays were terminated by adding 500 µL 1 N HCl. The lysates were centrifuged at 4000 × g for 10 min. The cAMP concentration was determined in the supernatants using a competitive binding assay, as previously described.59 EC50 and Emax values were calculated by fitting the data to: E = Emin + (Emax-Emin)/(1 + 10x-logEC50).
In vivo Studies
Experimental animals
Male CD-1 mice (25–30 g) from Harlan (Indianapolis, IN) were housed 4–5 (for mice) per cage in a controlled environment (12 h light/dark cycles) with food and water available ad libitum. Male A3AR knockout mice (A3AR−/−) (1) were provided by Merck & Co., Inc. (Whitehouse Station, NJ) and used to support A3AR-mediated effects. Sex, age and weight-matched C57BL/6J wild-type (WT; Harlan, Indianapolis, IN) mice were used as controls. The A3AR−/− mice are healthy, fertile and exhibit no differences in the development of any organ systems.60 Experiments were performed in accordance with International Association for the Study of Pain, NIH guidelines on laboratory animal welfare and Saint Louis University Institutional Animal Care and Use Committee recommendations. Experimenters were blinded to treatment conditions in all experiments.
CCI model of neuropathic pain
CCI to the sciatic nerve of the left hind leg in mice was performed under general anaesthesia using the well characterized Bennett model.39 Briefly, mice (weighing 25–30 g at the time of surgery) were anesthetized with 3% isoflurane/100% O2 inhalation and maintained on 2% isoflurane/100% O2 for the duration of surgery. The left thigh was shaved, scrubbed with Nolvasan (Zoetis, Madison, NJ), and a small incision (1–1.5 cm in length) was made in the middle of the lateral aspect of the left thigh to expose the sciatic nerve. The nerve was loosely ligated around the entire diameter of the nerve at three distinct sites (spaced 1 mm apart) using silk sutures (6.0). The surgical site was closed with a single muscle suture and a skin clip. Pilot studies established that under our experimental conditions peak mechano-allodynia develops by D5-D7 following CCI. Test substances or their vehicles were given i.p. at peak mechano-allodynia (D7).
Statistical analysis for in vivo experiments
Data are expressed as mean ± SEM for n animals. Behavioral data were analyzed by two-way ANOVA with Bonferroni comparisons. Significant differences were defined at a P<0.05. All statistical analysis was performed using GraphPad Prism (v5.03, GraphPad Software, Inc., San Diego, CA).
Supplementary Material
Acknowledgment
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the National Institutes of Health (Intramural Research Program of the NIDDK and R01HL077707).
Abbreviations used
- AR
adenosine receptor
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CCI
chronic constriction injury
- CHO
Chinese hamster ovary
- CNS
central nervous system
- DMEM
Dulbecco’s modified Eagle medium
- EL
extracellular loop
- GPCR
G protein-coupled receptor
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HEK
human embryonic kidney
- NECA
5′-N-ethylcarboxamidoadenosine
- PBS
phosphate buffered saline
- PWT
Paw Withdrawal Threshold
- RMS
root-mean-square
- SAR
structure-affinity relationship
- TM
transmembrane helix
Footnotes
Supporting Information Available:
Modeling Figures S1 and S2, reaction Scheme S1, selected spectra of synthesized compounds and 3D coordinates of the modeled hA3AR and mA3AR complexes with 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Mülller C. Nomenclature and classification of adenosine receptors — An update. Pharmacol. Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheong SL, Federico S, Venkatesan G, Mandel AL, Shao YM, Moro S, Spalluto G, Pastorin G. The A3 adenosine receptor as multifaceted therapeutic target: pharmacology medicinal chemistry in silico approaches. Med. Res. Rev. 2012;33:235–335. doi: 10.1002/med.20254. [DOI] [PubMed] [Google Scholar]
- 3.Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Disc. Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, MacLennan S, Feo C, Baraldi S, Borea PA. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J. Cell. Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 5.Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochem. Biophys. Acta. 2011;1808:1400–1412. doi: 10.1016/j.bbamem.2010.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Bar-Yehuda S, Luger D, Ochaion A, Cohen S, Patokaa R, Zozulya G, Silver PB, Garcia Ruiz de Morales JM, Caspi RR, Fishman P. Inhibition of experimental autoimmune uveitis by the A3 adenosine receptor agonist CF101. Int. J. Molecular Med. 2011;28:727–731. doi: 10.3892/ijmm.2011.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madi LL, Rosenberg-Haggen B, Nyska A, Korenstein R. Enhancing pigmentation via activation of A3 adenosine receptors in B16 melanoma cells and in human skin explants. Exp. Dermatol. 2013;22:54–80. doi: 10.1111/exd.12028. [DOI] [PubMed] [Google Scholar]
- 8.Fozard JR. From hypertension (+) to asthma: interactions with the adenosine A3 receptors in muscle protection. In: Borea PA, editor. Chapter 1 in A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics. Dordrecht, Netherlands: Springer; 2010. pp. 3–26. [Google Scholar]
- 9.Bulger EM, Tower CM, Warner KJ, Garland T, Cuschieri J, Rizoli S, Rhind S, Junger WG. Increased neutrophil adenosine A3 receptor expression is associated with hemorrhagic shock and injury severity in trauma patients. Shock. 2011;36:435–439. doi: 10.1097/SHK.0b013e318231ee2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avila MY, Stone RA, Civan MM. Knockout of A3 adenosine receptors reduces mouse intraocular pressure. Invest. Ophthalmol. Vis. Sci. 2002;43:3021–3026. [PubMed] [Google Scholar]
- 11.Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, Kheradmand F, Lee JJ, Blackburn MR. A3 adenosine receptor signaling influences pulmonary inflammation fibrosis. Am. J. Respir. Cell. Mol. Biol. 2008;39:697–705. doi: 10.1165/rcmb.2007-0419OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohsawa K, Sanagi T, Namakura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension migration. J. Neurochem. 2012;121:217–227. doi: 10.1111/j.1471-4159.2012.07693.x. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z, Janes K, Chen C, Doyle T, Tosh DK, Jacobson KA, Salvemini D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012;26:1855–1865. doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zylka MJ. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011;17:188–196. doi: 10.1016/j.molmed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J. Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 16.Yan L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: From basic medicinal chemistry to clinical development. Expert Opin. Emerg. Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 17.Kim HOXD, Ji SM, Siddiqi ME, Olah GL, Stiles KA, Jacobson 2-Substitution of N 6-benzyladenosine-5'-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson KA, Ji XD, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J. Med. Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. Methanocarba 2,N 6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J. Med. Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis o ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleos. Nucleot. Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson KA, Siddiqi SM, Olah ME, Ji XD, Melman N, Bellamkonda K, Meshulam Y, Stiles GL, Kim HO. Structure-activity relationships of 9-alkyladenine and ribose-modified adenosine derivatives at rat A3 adenosine receptors. J. Med. Chem. 1995;38:1720–1735. doi: 10.1021/jm00010a017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Lee SK, Tosh DK, Moon HR. Structure activity relationships of truncated D- and L- 4'-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem. 2008;51:6609–6613. doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg. Med. Chem. 2008;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou X, Majik MS, Kim K, Pyee Y, Lee Y, Alexander V, Chung HJ, Lee HW, Chandra G, Lee JH, Park SG, Choi WJ, Kim HO, Phan K, Gao ZG, Jacobson KA, Choi S, Lee SK, Jeong LS. Structure-activity relationships of truncated C2- or C8-substituted adenosine derivatives as dual acting A2A and A3 adenosine receptor ligands. J. Med. Chem. 2012;55:342–356. doi: 10.1021/jm201229j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao ZG, Verzijl D, Zweemer A, Ye K, GölblyGös A, IJzerman AP, Jacobson KA. Functionally biased modulation of A3 adenosine receptor agonist efficacy potency by imidazoquinolinamine allosteric enhancers. Biochem. Pharmacol. 2011;82:658–668. doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A3 adenosine receptor-selective nucleosides: Combination of 2-arylethynyl and bicyclo[3.1.0] hexane substitutions. J. Med. Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA. Truncated Nucleosides as A3 Adenosine Receptor Ligands: Combined 2-Arylethynyl Bicyclohexane Substitutions. ACS. Med. Chem. Lett. 2012;3:596–601. doi: 10.1021/ml300107e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volpini R, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, Marucci G, Ramadori AT, Klotz KN, Cristalli G. Synthesis biological evaluation of 2-alkynyl-N 6-methyl-5’-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor. J. Med. Chem. 2009;52:7897–7900. doi: 10.1021/jm900754g. [DOI] [PubMed] [Google Scholar]
- 28.Auchampach JA, Gizewski E, Wan TC, de Castro S, Brown GG, Jacobson KA. Synthesis and pharmacological characterization of [125I]MRS5127, a high affinity, selective agonist radioligand for the A3 adenosine receptor. Biochem. Pharmacol. 2010;79:967–973. doi: 10.1016/j.bcp.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson KA, Nikodijevic O, Ji X-D, Berkich DA, Eveleth D, Dean RL, Hiramatsu KI, Kassell NF, van Galen PJM, Lee KS, Bartus R, Daly JW, LaNoue KF, Maillard M. Synthesis and biological activity of N 6 p-sulfophenylalkyl and N 6 p-sulfoalkyl derivatives of adenosine: Water soluble and peripherally selective adenosine agonists. J. Med. Chem. 1992;35:4143–4149. doi: 10.1021/jm00100a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Tayeb A, Michael S, Abdelrahman A, Behrenswerth A, Gollos S, Nieber K, Müller CE. Development of Polar Adenosine A2A Receptor Agonists for Inflammatory Bowel Disease: Synergism with A2B Antagonists. ACS. Med. Chem. Lett. 2011;2:890–895. doi: 10.1021/ml200189u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgold J, Nikodijevic O, Jacobson KA. Penetration of adenosine antagonists into mouse brain as determined by ex vivo binding Biochem. Pharmacol. 1992;43:889–894. doi: 10.1016/0006-2952(92)90257-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu QL, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity relationships of N6-benzyladenosine-5'-uronamides as A3-selective adenosine agonists. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens R. Agonist bound structure of the human adenosine A2A receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor‒Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson KA. Glutamate residues in the second extracellular loop of the human A2A adenosine receptor are required for ligand recognition. Mol. Pharmacol. 1996;49:683–691. [PMC free article] [PubMed] [Google Scholar]
- 37.Lenzi O, Colotta V, Catarzi D, Varano F, Poli D, Filacchioni G, Varani K, Vincenzi F, Borea PA, Paoletta S, Morizzo E, Moro S. 2-Phenylpyrazolo[4,3-d] pyrimidin-7-one as a new scaffold to obtain potent selective human A3 adenosine receptor antagonists: new insights into the receptor-antagonist recognition. J. Med. Chem. 2009;52:7640–7652. doi: 10.1021/jm900718w. [DOI] [PubMed] [Google Scholar]
- 38.Jacobson KA, Ji X-d, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J. Med. Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 40.Tosh DK, Paoletta S, Deflorian F, Phan K, Moss SM, Gao ZG, Jiang X, Jacobson KA. Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking potent anticonvulsant activity. J. Med. Chem. 2012;55:8075–8090. doi: 10.1021/jm300965a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belfrage M, Segerdahl M, Arnér S, Sollevi A. The safety efficacy of intrathecal adenosine in patients with chronic neuropathic pain. Anesth. Analg. 1999;89:136–142. doi: 10.1097/00000539-199907000-00023. [DOI] [PubMed] [Google Scholar]
- 42.Luongo L, Petrelli R, Gatta L, Giordano C, Guida F, Vita P, Franchetti P, Grifantini M, de Novellis V, Cappellacci L, Malone S. 5'-Chloro-5'-deoxy-(±)-ENBA, a Potent and Selective Adenosine A1 Receptor Agonist, Alleviates Neuropathic Pain in Mice Through Functional Glial and Microglial Changes without Affecting Motor or Cardiovascular Functions. Molecules. 2012;17:13712–13726. doi: 10.3390/molecules171213712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rivkees SA, Thevananther S, Hao H. Are A3 adenosine receptors expressed in the brain? Neuroreport. 2000;11:1025–1030. doi: 10.1097/00001756-200004070-00026. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Z, Yaar R, Ladd D, Cataldo LM, Ravid K. Overexpression of A3 adenosine receptors in smooth, cardiac, and skeletal muscle is lethal to embryos. Microvasc. Res. 2002;63:61–69. doi: 10.1006/mvre.2001.2366. [DOI] [PubMed] [Google Scholar]
- 45.Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W. The A3 adenosine receptor mediates cell spreading reorganization of actin cytoskeleton and distribution of Bcl-xL. Studies in human astroglioma cells. Biochem. Biophys. Res. Commun. 1997;241:297–304. doi: 10.1006/bbrc.1997.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohsawa K, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J. Neurochem. 2012;121:217–227. doi: 10.1111/j.1471-4159.2012.07693.x. [DOI] [PubMed] [Google Scholar]
- 47.Yang JN, Wang Y, Garcia-Roves PM, Björnholm M, Fredholm BB. Adenosine A3 receptors regulate heart rate, motor activity and body temperature. Acta Physiol. 2010;199:221–230. doi: 10.1111/j.1748-1716.2010.02091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salvemini D, Janes K, Finley A, Doyle T, Chen Z, Bryant L, Tosh DK, Jacobson KA. FASEB J., Experimental Biology. Boston, MA: 2013. Apr, Adenosine receptor subtype 3 (A3AR) agonists as novel analgesics in chronic neuropathic pain through. Abstract A97 887.1. [Google Scholar]
- 49.Molecular Operating Environment (MOE) version 2012.10. 1255 University St., Suite 1600, Montreal, QC, H3B 3×3 (Canada): Chemical Computing Group Inc.; [Google Scholar]
- 50.The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relationships in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 52.Moro S, Deflorian F, Bacilieri M, Spalluto G. Ligand-based homology modeling as attractive tool to inspect GPCR structural plasticity. Curr. Pharm. Des. 2006;12:2175–2185. doi: 10.2174/138161206777585265. [DOI] [PubMed] [Google Scholar]
- 53.Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins. 2009;75:187–205. doi: 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 55.Schrödinger Suite 2012. New York, NY: Schrödinger, LLC; [Google Scholar]
- 56.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of Crystal Packing Forces in Determining Protein Sidechain Conformations. J. Mol. Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 57.Bradford MM. Arapid sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 58.Cheng YC, Prusoff WH. Relationship between inhibition constant (K1), concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic-reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 59.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal. Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 60.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. USA. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.