

Abstract
We previously showed that in innately resistant tumors, silencing of the estrogen receptor (ER) could be reversed by treatment with a histone deacetylase (HDAC) inhibitor entinostat (ENT). Tumors were then responsive to aromatase inhibitor (AIs) letrozole. Here, we investigated whether ER in the acquired letrozole resistant tumors could be restored with ENT. Ovariectomized athymic mice were inoculated with MCF-7Ca cells, supplemented with androstenedione (Δ4A), the aromatizable substrate. When the tumors reached ~300mm3, the mice were treated with letrozole. After initial response to letrozole, the tumors eventually became resistant (doubled their initial volume). The mice then were grouped to receive letrozole, exemestane (250μg/day), ENT (50μg/day) or the combination of ENT with letrozole or exemestane for 26 weeks. The growth rates of tumors of mice treated with the combination of ENT with letrozole or exemestane were significantly slower than with the single agent (p<0.05). Analysis of the letrozole resistant tumors showed ENT increased ERα expression and aromatase activity but downregulated Her-2, p-Her-2, p-MAPK and p-Akt.
However, the mechanism of action of ENT in reversing acquired resistance did not involve epigenetic silencing, but rather included post-translational as well as transcriptional modulation of Her-2. ENT treatment reduced the association of the Her-2 protein with HSP-90, possibly by reducing the stability of Her-2 protein. In addition, ENT also reduced Her-2 mRNA levels and its stability. Our results suggest that the HDAC inhibitor may reverse letrozole resistance in cells and tumors by modulating Her-2 expression and activity.
Keywords: breast cancer, aromatase inhibitors, HDAC inhibitors, Her-2, preclinical studies of endocrine related cancers
Introduction
Development of aromatase inhibitors (AIs) has significantly improved the treatment outcome of hormone responsive post-menopausal breast cancer. However, not all tumors respond and some eventually acquire resistance. To study the mechanisms of resistance, we have developed a xenograft model that mimics post-menopausal hormone responsive breast cancer (1–4). Ovariectomized (OVX) athymic nude mice are inoculated with human hormone responsive breast cancer cells (MCF-7) stably transfected with the human placental aromatase gene (MCF-7Ca) (2, 5, 6). Aromatase expressed in the tumor cells converts androstenedione (Δ4A) into estrogen and provides a non-ovarian source of estrogen, thus simulating the tumors of post-menopausal patients. Using this model, we have established that as a single agent AI is better than tamoxifen in controlling tumor growth (7–10). Results obtained using this model have been confirmed by several clinical trials (11–15). We also observed that although AI letrozole provides longer control over tumor growth, tumors eventually began to grow and are resistant to further AI letrozole treatment. A cell line was isolated from these Long-Term Letrozole Treated tumors and designated as LTLT-Ca (3, 4). These cells and the tumors had decreased expression of ERα and aromatase compared to parental MCF-7Ca cells. On the other hand, growth factor receptor Her-2 and downstream kinases such as MAPK and Akt were increased. Inhibition of Her-2 using trastuzumab (a humanized antibody against the extracellular domain of Her-2) to block the Her-2 pathway resulted in reversed resistance and restoration of sensitivity to estrogens, antiestrogens and AIs (16).
Breast cancers that are innately resistant to AIs or antiestrogens (AEs) lack expression of the ER protein, which is thought to be due to gene silencing (17). HDAC inhibitors have been shown to induce ERα expression in an ERα-negative cell line such as MDA-MB-231 (18–20). We recently reported that ER-negative tumors pretreated with HDAC inhibitors such as entinostat (ENT) became responsive to endocrine therapy with aromatase inhibitors (21). Thus, the ENT + letrozole combination resulted in marked reduction of tumor growth compared to either ENT or letrozole treatment alone. Analysis of tumors and cells (MDA-MB-231) revealed induction of ER and aromatase by ENT. These results were consistent with ENT’s actions to inhibit histone deacetylase and allow expression of ERα and aromatase, rendering the tumors sensitive to hormones and hormonal therapy. Since the ERα and aromatase are reduced in letrozole resistant tumors by long-term treatment with the drug, we hypothesized that ENT may also increase ERα and aromatase in AI resistant tumors by the same mechanism, HDAC inhibition at the ERα and aromatase promoters. However, the results of the current study show that in acquired resistance, ENT acts by a different mechanism, which results in reduction of Her-2 protein and mRNA.
Several studies have shown that HDAC actions are not limited to histone modifications, as some members of HDAC family acetylate and modulate non-histone proteins, thereby regulating their stability and subcellular localization (22, 23). Hsp 90 has been shown to be one of the targets of HDACs. HDAC6 has been shown to induce acetylation of hsp90 and lead to degradation of its client proteins such as Bcr-Abl (24). More recently, inhibition of HDAC 1 has been shown to be responsible for inhibition of hsp90 activity, leading to degradation of client proteins such as FLT3 and DNMT1 (25, 26). Our results suggest that inhibition of Her-2 due to inhibition of hsp90 may be one of the mechanisms responsible for reversal of resistance to AIs in the xenograft model. It has been shown that HDAC inhibitors can also induce the decay of mature Her-2 transcript, leading to growth inhibition in Her-2 positive cell lines and tumors (27). In our model, we observed that ENT treatment reduced the half-life of Her-2 mRNA, which could be another mechanism by which letrozole resistance is reversed. In summary, the mechanism by which ENT reverses letrozole resistance is through modulation of Her-2.
Materials and Methods
Materials
Dulbecco’s Modified Eagle Medium (DMEM), Modified Improved Minimum Essential medium (IMEM), penicillin/streptomycin solution (10,000IU each), 0.25% trypsin–1 mM EDTA solution, Dulbecco’s phosphate-buffered saline (DPBS), and geneticin (G418) were obtained from Invitrogen (Carlsbad, CA). Androstenedione (Δ4A), and Matrigel were obtained from Sigma Chemical Company (St. Louis, MO). Antibodies against Her-2 and p-Her-2 were purchased from Millipore (Billerica, MA); antibodies against p-MAPK, MAPK, p-Elk-1, and p-p90RSK were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against ERα and aromatase (CYP 19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Radioactive ligand for aromatase assay 3H-Δ4A (23.5 Ci/mmole) was purchased from Perkin Elmer (Boston, MA). MCF-7 human breast cancer cells stably transfected with the human aromatase gene (MCF-7Ca) were kindly provided by Dr. S. Chen (City of Hope, Duarte, CA) (6).
Cell Culture
MCF-7Ca cells were routinely cultured in DMEM supplemented with 5% FBS, 1% penicillin/streptomycin, 700μg/mL G418. LTLT-Ca cells were developed from MCF-7Ca cells as described earlier (3) from tumors of mice treated with letrozole for 56 weeks and cultured in steroid-depleted medium, containing 1μM of letrozole. Anastrozole resistant (AnR) cells were developed as described earlier (28).
Cell line authentication
All cell lines used in this study were authenticated in August 2013, using CellCheck™ service (microsatellite marker (STR) analysis) provided by Idexx Radil (Columbia, MO). The cell lines were found to be identical to the genetic profile reported for the MCF-7 cell line (ATCC# HTB-22).
Tumor Growth in Ovariectomized Female Athymic Nude Mice
All animal studies were performed according to the guidelines and approval of the Animal Care Committee of the University of Maryland, Baltimore. Female ovariectomized (OVX) athymic nude mice 4–6 weeks of age were obtained from the National Cancer Institute - Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed in a pathogen-free environment under controlled conditions of light and humidity; received food, and water ad libitum.
The tumor xenografts of MCF-7Ca cells were grown in the mice as previously described (2, 3, 29). Each mouse received subcutaneous (sc) inoculations in one site per flank with 100μL of cell suspension containing ~ 2.5×107 cells/mL in Matrigel. The mice were injected daily with supplemental Δ4A (100μg/day). Weekly tumor measurements and treatments began when the tumors reached ~ 300 mm3. Mice were assigned to groups for treatment so that there was no statistically significant difference in tumor volume among the groups at the beginning of treatment. Letrozole and Δ4A for injection were prepared using 0.3% HydroxyPropylCellulose (HPC) in 0.9% NaCl solution. ENT was prepared in 30% HydroxyPropyl β-Cyclodextrin (HPBC) solution to obtain the required concentration. Mice were then injected sc 5 times weekly with the indicated drugs (except ENT was administered orally (po) with Δ4A being given sc). The doses of ENT (50μg/day), letrozole (10μg/day) and Δ4A (100μg/day) used are as previously determined and reported (3, 18)
Western blotting
The protein extracts from tumor tissues were prepared by homogenizing the tissue in ice-cold DPBS containing protease and phosphatase inhibitors. Total 50μg of protein from each sample was analyzed by SDS-PAGE as described previously (16, 18, 29, 30). Bands were quantitated by densitometry using ImageJ (NIH). The densitometric values are corrected for β-actin loading control and reported as fold-change compared to control underneath each band. The blots shown are representative blot from at least 3 separate experiments (n=3).
3H2O release assay for aromatase activity measurement
For measuring aromatase activity in tumor samples, the tumors were homogenized in ice-cold DPBS containing protease and phosphatase inhibitors. The resulting homogenate was used for aromatase activity assay. The radiometric 3H2O release assay was performed as described previously (16, 18, 29–31) using [1-β3H] Δ4A as substrate in presence of molecular oxygen. The activity of the enzyme is corrected for protein concentration in the tumor homogenates.
ChIP Assay
For in vitro ChIP assay, the treated cells were washed with DPBS and fixed with 1% formaldehyde/DPBS for 10 minutes at 37°C after which the cells were washed with ice-cold DPBS containing protease and phosphatase inhibitors. The cells were collected into 1ml DPBS and pelleted by centrifugation at 6000rpm for 5minutes at 4°C. The cell pellet was re-suspended in nuclear lysis buffer (ChIP Kit, Millipore, Billerica, MA) and incubated on ice for 10 minutes. Samples were sonicated on ice for 10 × 10 sec cycles, with 20 sec pauses between each cycle. The sonicated samples were centrifuged at 14000rpm for 10 minutes at 4°C. Sonicated samples were diluted 1:10 with dilution buffer (ChIP kit) before being immunocleared in a solution containing of Protein A or G Sepharose slurry - salmon sperm DNA for 2 h at 4°C. Immunocleared supernatants incubated overnight at 4°C with pan-acetyl H3 (Millipore, Billerica, MA) and total H3 antibody (Cell Signaling Technologies). Protein A or G Sepharose beads and salmon sperm DNA were then added and incubated for 1 h at 4°C. The beads were then washed sequentially with 1 ml each of wash buffers. The protein-DNA complexes were then eluted by twice incubating beads in elution buffer for 10 min at room temperature with vigorous mixing. To separate immunoprecipitated protein and DNA, the pooled elutes were incubated at 65°C overnight. The DNA was purified using the Qiaquick PCR purification kit (Qiagen, Valencia, CA).
The yield of target region DNA in each sample after ChIP was analyzed by conventional PCR and real time qPCR as described earlier (16, 32). The promoter that was analyzed was I.3/II, which is the main aromatase promoter utilized in breast cancer cells lines such as MCF-7 (16, 33) and thus measures the effect of ENT and trastuzumab on endogenous aromatase in MCF-7 cells. The MCF-7Ca cells are transfected with human placental aromatase cDNA (placental CYP-19 uses promoter I.1 and I.2).
RNA Extraction and Reverse Transcription (RT) and PCR
RNA was extracted and purified using the RNeasy Mini Kit (Qiagen, Valencia, CA) as per manufacturer’s protocol. Analysis of ERα, aromatase (CYP-19), pS2, progesterone receptor (PgR) and Her-2 mRNA expression was carried out by real time qRT-PCR as described earlier (16, 32) using a Bio-Rad CFX Connect real time system (Bio-Rad). Total RNA was diluted 1:10 before amplification of 18S ribosomal RNA (rRNA) for 18 cycles. An annealing temperature of 60°C was used in all cases. Each 20μl reaction mixture included 2 μl cDNA, 10 μl SsoAdvanced SYBR green qPCR mix (Bio-Rad), 0.8 μl primer mix (5 μM each primer), and 7.2 μl molecular grade water. Each sample was assayed in duplicate. A standards curve was generated by serially diluting control cDNA. The yield of product for each unknown sample was calculated by applying its threshold cycle, or C(T), value (the cycle at which the sample’s fluorescence trace exceeds background noise and begins to increase linearly) to the standard curve using the CFX Connect real time system software. Values were normalized to corresponding 18S rRNA values and expressed as the fold-increase relative to control.
Co-immunoprecipitation (Co-IP)
The association between proteins was measured with co-IP. Cell lysates were pre-cleared with either Protein A or G agarose beads. The lysates were then incubated with primary antibody as per manufacturer’s protocol. Next, 30μL of agarose beads (Millipore) were added and the samples were incubated at room temperature for 1hr with rotation. The immunocomplex bound beads were precipitated by centrifugation, washed with TBS (with 0.02% Tween 20) three times. The immunocomplex was eluted from the beads by adding Laemmli Sample Buffer (Bio-rad, Hercules, CA) and boiling for 5 minutes at 100°C. The IP complex was analyzed by western blotting.
Statistics
For in vivo studies, mixed-effects models were used. The tumor volumes were analyzed with S-PLUS (7.0, Insightful Corp.) to estimate and compare an exponential parameter (βi) controlling the growth rate for each treatment groups. The original values for tumor volumes were log transformed. The spline model with a single knot at time = week-15 was used to accommodate the non-linearity with a piece-wise linear model (16, 18, 29, 30). For in vitro studies, western blots and IP were performed at least 3 times and a representative blot is shown. For real-time RT-PCR studies, the fold-change values were analyzed using One-Way ANOVA with Tukey-Kramer multiple comparison test. All p values less than 0.05 were considered statistically significant. The graphs are represented as mean ± standard error of the mean (SEM).
Results
Treatment of letrozole resistant MCF-7Ca xenografts with the combination of ENT and letrozole or exemestane
To examine whether the mechanism of acquired resistance to letrozole was also due to gene silencing, we utilized the MCF-7Ca xenograft model. MCF-7Ca xenografts were grown as previously described (3, 16, 29, 30). We inoculated OVX athymic nude mice with MCF-7Ca (engineered to express aromatase) cells. All the mice received Δ4A supplement, which was converted to estrogen by aromatase in the tumor cells. This provides a non-ovarian source of estrogen that stimulates tumor growth. When the tumors reached ~300mm3, the mice were divided into 2 groups such that the average tumor volume was not significantly different between the two groups (p=0.93).
One group (Control, n=10) received only supplemental Δ4A (100μg/day) and the second group (n=50) received Δ4A (100μg/day) plus letrozole (10μg/day). The mice were treated with letrozole for 16 weeks. During this time the tumors regressed, but eventually began to grow. Over the first 10 weeks of treatment, the growth rate of tumors in the letrozole treated group was significantly slower than that of the control group (p<0.0001). The tumor volumes of the control and letrozole groups were also significantly different at week 10 (p=0.01). However, the tumors of letrozole treated mice then grew and had doubled in size by week 16. Previously, we have shown that tumors adapt to low estrogen environment that ensues upon letrozole treatment by upregulating alternative signaling pathways such as Her-2. During this time, the tumors first regress and then start to re-grow, becoming resistant as they continue to grow in the presence of the drug (3, 29, 34). At this time, the mice were grouped to receive second line treatment with entinostat (ENT) 50μg/day, exemestane 250μg/day, ENT + exemestane or ENT + letrozole and one group was continued on letrozole (n=10 each). The average tumor volumes at the start of second-line treatment were not statistically significantly different among the groups (p values for pair wise comparisons ranging from 0.35 to 0.89). The mice were treated till week 26. The addition of ENT to letrozole or exemestane treatment significantly reduced the growth rate of tumors compared to each agent alone (p=0.009 ENT vs. ENT + exemestane; p=0.048 ENT vs. ENT + letrozole) or continued letrozole treatment (p<0.0001 letrozole vs. ENT + letrozole). As shown in Figure 1A, the combination of ENT with letrozole or exemestane was significantly better in inhibiting tumor growth compared to single agents. This suggested that ENT overcame the acquired resistance of tumors to AIs (Figure 1A).
Figure 1.

Figure 1A: Effect of ENT alone or in combination with letrozole or exemestane on the growth of letrozole resistant MCF-7Ca xenografts:
Ovariectomized athymic nude mice were inoculated with MCF-7Ca cells. Tumors were allowed to form in the presence of androstenedione (Δ4A), aromatizable substrate for estrogen. When the tumors reached ~300mm3, mice were treated with letrozole (10μg/day) for total 15 weeks, during this time the tumors regressed, but eventually began to grow. When the tumors had reached double the initial volume, they were randomized again into 5 groups; letrozole continued, exemestane (250μg/day), ENT (50μg/day) or the combination of ENT with letrozole or exemestane. The mice were treated till week 26. The growth rates of tumors of mice treated with the combination of ENT with letrozole or exemestane was significantly better than single agent alone (p=0.0009 ENT vs ENT+letrozole; p=0.048 ENT vs ENT+exemestane; p<0.0001 ENT vs ENT+letrozole)).
Figure 1B: Effect of ENT alone or in combination with letrozole or exemestane on the tumor protein expression in letrozole resistant MCF-7Ca xenografts:
Protein expression in the tumors was examined by western immunoblotting as described in “materials and methods”. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 1C: Effect of ENT alone or in combination with letrozole or exemestane on the tumor aromatase activity in letrozole resistant MCF-7Ca xenografts:
The aromatase activity in the homogenized tumors was analyzed as described in “materials and methods”. Tumors of mice treated with letrozole for 15 weeks followed by ENT (in presence of Δ4A) have significantly higher aromatase activity than the letrozole treated tumors (*a p<0.01) and control tumors (*b p<0.05).
HDAC inhibitor ENT upregulates ERα and aromatase while reducing Her-2/MAPK pathway activation in letrozole resistant tumors
Tumors of the mice shown in Figure 1A were examined for changes in protein expression. Consistent with our previous finding, tumors of the mice treated with letrozole exhibited downregulation of ERα and aromatase along with upregulation of Her-2 and MAPK. When treated with ENT, Her-2, p-Her-2, p-MAPK, p-c-Raf, p-MEK1/2 and p-Akt were downregulated along with increase in ERα and aromatase protein expression (Figure 1B). These changes were seen in tumors of mice treated with ENT alone and in combination with letrozole or exemestane. Furthermore, aromatase activity in the tumors of mice treated with ENT was significantly greater than those treated with letrozole (*a p<0.01) or control (*b p<0.05) (Figure 1C).
HDAC inhibitor ENT increases aromatase expression in an estrogen dependent manner
Letrozole resistant tumors of mice treated with ENT (supplemented with aromatizable Δ4A), exhibited ~3.5-fold increase in aromatase activity (Figure 1C) and ~1.7-fold increase in aromatase protein expression (letrozole versus letrozole to ENT) (Figure 1B). To elucidate the mechanism of upregulation of aromatase, we performed an in vitro ChIP assay using LTLT-Ca cells. To examine whether the promoter region of the ERα and aromatase (CYP-19) genes were activated following ENT treatment, we immunoprecipitated chromatin using acetyl histone H3 antibody. The aromatase promoter that was analyzed was I.3/II, which is the endogenous aromatase promoter in breast cancer cell lines such as MCF-7 (33). The MCF-7Ca cells are transfected with human placental aromatase cDNA (placental CYP-19 uses promoter I.1 and I.2) with a β-actin promoter. (6, 35). It is known that HDAC inhibitors upregulate silenced genes by acetylating histones to allow transcription. However, in LTLT-Ca cells, ERα promoter was active to the same level as that in the MCF-7Ca cells (Figure 2). Furthermore, treatment with ENT alone did not cause any change in promoter activation. However, when supplemented with E2 or Δ4A, ENT treatment caused an increase in CYP-19 promoter activation. A similar finding was seen with activation of a known estrogen inducible gene, pS2. This suggests that ENT activates aromatase and pS2 in an estrogen dependent manner. These effects were similar to those observed with TRZ (a humanized monoclonal antibody against Her-2) and consistent with our previous findings that inhibition of Her-2 results in increased ER expression (16). Based on these observations, we hypothesized that ENT modulates Her-2, which in turn results in reversal of resistance to letrozole.
Figure 2. ChIP analysis of LTLT-Ca cells to examine the activation of ERα, pS2 and aromatase (PI.3/II) promoter.
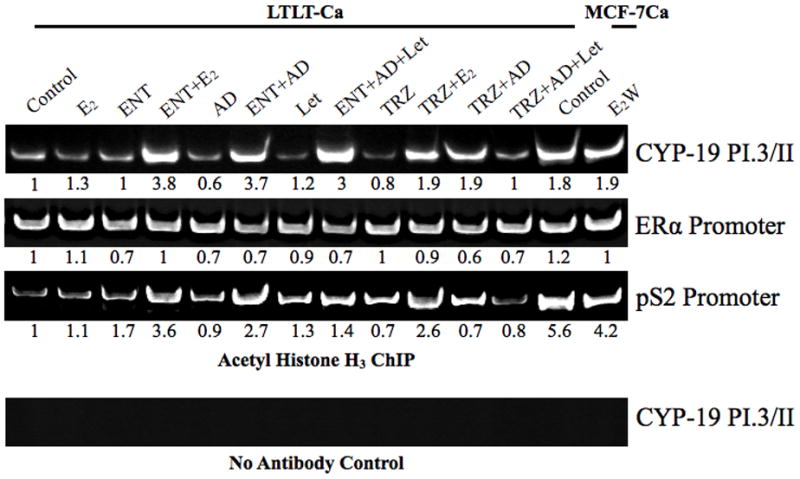
Activation of ERα and aromatase promoter was measured by immunoprecipitating chromatin bound acetylated histone H3. Blot shows conventional PCR analysis and the numbers below the blot show real-time qPCR analysis.
HDAC inhibitor ENT induced ERα mRNA and protein expression in LTLT-Ca cells but not in MCF-7Ca cells
ENT exhibited differential effects in MCF-7Ca versus LTLT-Ca cells. ENT treatment did not change ERα mRNA expression in MCF-7Ca cells (data not shown), but caused reduction in ERα protein levels in MCF-7Ca cells (Figure 3A, middle row, center panel) and reduced ERα half-life (middle row right panel compared to middle row left panel). These results are consistent with other reports showing attenuation of ERα levels and transcriptional activity by HDAC inhibitors (36) in ER-positive cells. Conversely, in LTLT-Ca cells treatment with ENT increased the low levels of ERα protein (Figure 3B, center panel, middle row) and mRNA expression (*p<0.001) (Figure 3C). ENT alone does not cause activation of estrogen responsive gene such as pS2. However, when followed by E2 treatment, pS2 mRNA is significantly increased († p<0.001). A similar trend is also observed in CYP-19 mRNA.
Figure 3.

Western blotting analysis of (A) MCF-7Ca and (B) LTLT-Ca cells treated with entinostat: MCF-7Ca and LTLT-Ca cells were treated with ENT + letrozole. Cycloheximide (5μM) treatment was added to measure protein half-life.
Figure 3C: RT-PCR analysis of MCF-7Ca and LTLT-Ca cells treated with entinostat in presence or absence of E2, Δ4A and letrozole: MCF-7Ca and LTLT-Ca cells were treated with ENT (1μM) in presence or absence of E2 (1nM), Δ4A (25nM) or Δ4A+ letrozole (1μM). Changes in the mRNA levels were measured with real-time qRT-PCR. Numbers are corrected for the expression of housekeeping gene (18s ribosomal RNA) and expressed as fold change over control (fixed as 1).
HDAC inhibitor ENT reduced Her-2 in LTLT-Ca cells
Treatment of LTLT-Ca cells with ENT significantly reduced Her-2 protein expression (Figure 3B, top row). Her-2 mRNA was also significantly (*p<0.05) reduced with ENT (Figure 3C). ENT treatment followed by E2, produced a further reduction in Her-2 mRNA (**p<0.001). Previously, we have shown that letrozole treatment increases stability of Her-2 protein in MCF-7Ca cells (29). However, Her-2 stability remained unchanged when MCF-7Ca cells were treated with ENT + letrozole (Figure 3A). On the other hand, Her-2 half-life was reduced in LTLT-Ca cells when treated with ENT (Figure 3B, top row, right panel compared to top row left panel).
ENT causes Her-2 degradation via the proteosomal pathway
To elucidate whether ENT was degrading Her-2 via the proteasomes or the lysosomes, we performed a rescue experiment with MG-132 (proteosomal inhibitor) or ammonium chloride (lysosomal inhibitor). LTLT-Ca cells were treated with ENT in presence or absence of MG-132 or NH4Cl or the combination of MG-132 with NH4Cl and Her-2 protein levels were measured by western blotting. ENT treatment reduced Her-2 protein; MG-132, NH4Cl or the combination of MG-132 with NH4Cl reversed this reduction, although, MG-132 was more effective in this rescue. These results suggest that ENT degrades Her-2 protein mainly via the proteosomal pathway, but also via the lysosomal pathway (Figure 4A). The results were also confirmed by flow-cytometry analysis (Supplemental Figure 1). This finding is consistent with ENT treatment increasing the degradation of Her-2 protein and leading to its downregulation through increased ubiquitination (Supplemental Figure 2). This suggests that post-translational modifications could be responsible for the rapid effect of ENT on the protein expression. ENT also reduced Her-2 protein levels and activation of its downstream signaling molecules (MAPK and Akt) in two other Her-2 positive cell lines, SKBr3 and BT474 (Figure 4B). However, the reduction in Her-2 was modest in these cell lines, which could be due to the fact that both SKBr3 and BT-474 cells have Her-2 gene amplification that is not seen in LTLT-Ca cells. On the other hand, two other drug resistant cell lines; namely anastrozole resistant (AnR) and exemestane resistant (ExR) cells showed marked reduction in Her-2 and downstream signaling pathway upon ENT treatment. (Figure 4C).
Figure 4.
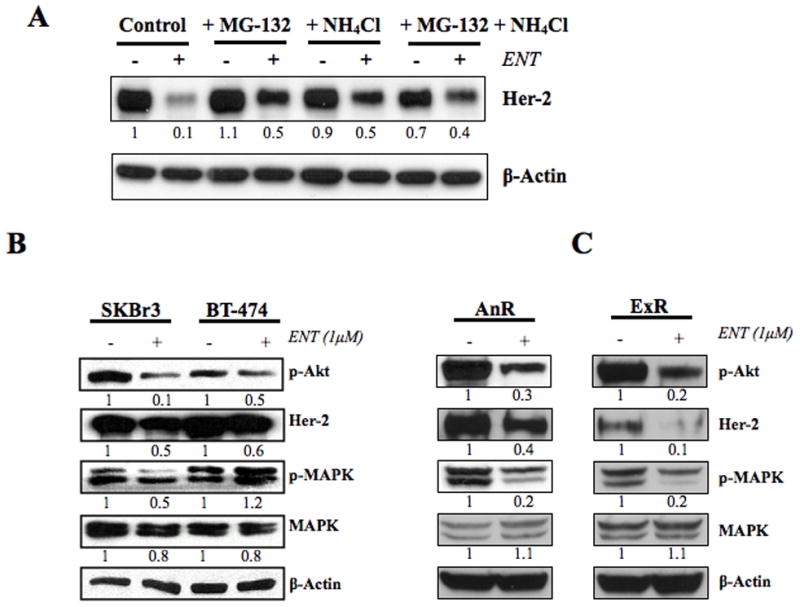
Figure 4A: Western blotting analysis of LTLT-Ca cells treated with entinostat alone or in presence of MG-132 (proteosomal inhibitor) or NH4Cl (lysosomal inhibitor): LTLT-Ca cells were treated with ENT (1μM) alone or in presence of MG-132 (5μM) or NH4Cl (100μM) or both. Protein expression in the cells was examined by western immunoblotting as described in “materials and methods”. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 4B: Western blotting analysis of SKBr3 and BT-474 cells treated with entinostat: Her-2 positive SKBr3 and BT-474 cells were treated with ENT (1μM). Protein expression in the cells was examined by western immunoblotting as described in “materials and methods”. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 4C: Western blotting analysis of AnR and ExR cells treated with entinostat: Her-2 positive anastrozole resistant (AnR) and exemestane resistant (ExR) cells were treated with ENT (1μM). Protein expression in the cells was examined by western immunoblotting as described in “materials and methods”. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Role of hsp90 in modulating Her-2 expression
The above results suggest that ENT modulates Her-2 through post-translational modulation. Her-2 is a known client protein maintained in the hsp90 chaperone complex. In addition, HDAC inhibitors are known to hyperacetylate hsp90, leading to degradation of its client proteins (24, 36). We investigated the association of hsp90 with Her-2 in the letrozole resistant tumors treated with ENT in combination with letrozole or exemestane (Figure 1A–C). There was increased association of hsp90 with Her-2 in letrozole resistant tumors compared to control tumors. This association was reduced after treatment with ENT alone or in combination with letrozole or exemestane (Figure 5A). Although treatment with ENT alone caused a marked decrease in Her-2-hsp90 association, the resulting increase in ERα and aromatase may be responsible for lower efficacy of the single agent in inhibiting tumor growth. As such, inhibition of both ERα and Her-2 pathways are required for enhanced inhibition of tumor growth (16). The increased association of Her-2 with hsp90 was also observed in LTLT-Ca cells compared to parental MCF-7Ca cells in vitro (Figure 5B). In addition, LTLT-Ca cells have a higher level of hsp90 compared to MCF-7Ca cells (Figure 5C). Similar results were also obtained in anastrozole (AnR) and exemestane resistant (ExR) cells (Figure 5D). These results suggest that ENT can reduce the hsp90 association with Her-2, leading to degradation of Her-2 protein. As a result of lower Her-2 stability and reduced protein levels, letrozole resistance could be reversed.
Figure 5.
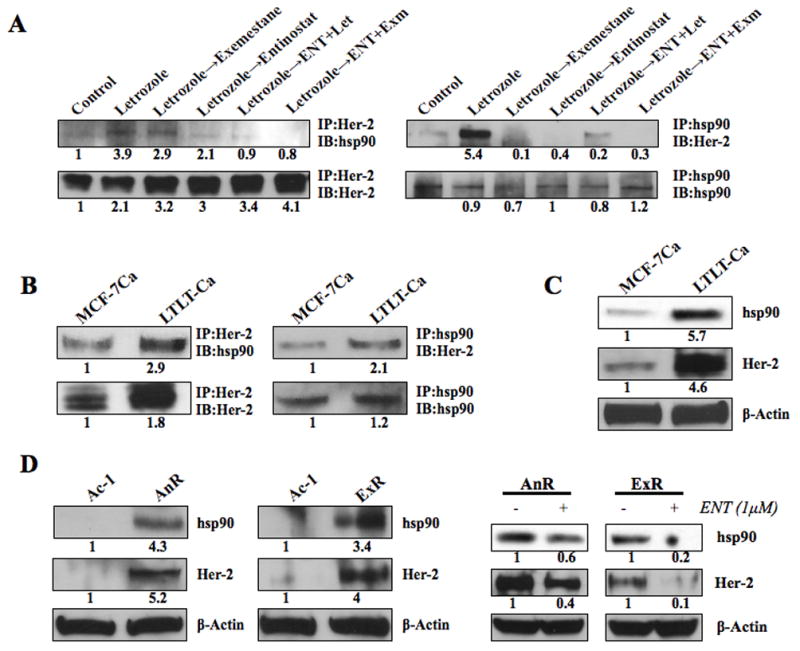
Figure 5A: Co-immunoprecipitation of Her-2 and hsp-90 in MCF-7Ca xenografts: Tumor lysates (500μg) of MCF-7Ca xenografts (from fig 1) were immunoprecipitated with either anti-Her-2 or anti-hsp90 antibody. The blots were then probed for hsp90 or Her-2 to measure association between the two proteins. Blots were probed for IP antibody to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 5B: Co-immunoprecipitation of Her-2 and hsp-90 in MCF-7Ca and LTLT-Ca cells: Lysates (500μg) of MCF-7Ca and LTLT-Ca cells were immunoprecipitated with either anti-Her-2 or anti-hsp90 antibody. The blots were then probed for hsp90 or Her-2 to measure association between the two proteins. Blots were probed for IP antibody to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 5C: Western blotting analysis of MCF-7Ca and LTLT-Ca cells: Relative levels of Her-2 and hsp90 proteins in MCF-7Ca and LTLT-Ca cells were detected by western blotting and measured by densitometry. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
Figure 5D: Western blotting analysis of AnR and ExR cells: Relative levels of Her-2 and hsp90 proteins in AnR and ExR cells were detected by western blotting and measured by densitometry. Blots were probed for β-actin to verify equal loading. The numbers below the blots show densitometric values that are corrected for loading.
ENT reduces Her-2 mRNA stability
Lastly, we examined the effect of ENT on Her-2 mRNA and its stability in LTLT-Ca cells using real-time qRT-PCR. HDAC6 inhibition has been shown to increase the degradation of Her-2 transcript (27). LTLT-Ca cells have a significantly (*p<0.001) higher basal level of Her-2 mRNA than MCF-7Ca cells (Figure 6A), however, the half-life of Her2 mRNA is not significantly different in LTLT-Ca cells versus MCF-7Ca (Supplemental Figure 3). To examine the effect of ENT on reduction in mRNA levels, LTLT-Ca cells were treated with actinomycin D (5μM) to inhibit all transcription and RNA was collected at indicated time points. Her-2 mRNA has a half-life of approximately 8 hours in LTLT-Ca cells. ENT treatment (1μM) alone reduced Her-2 mRNA in LTLT-Ca cells in a time dependent manner, with 50% reduction seen between 4 and 8 hours. However, when all new mRNA synthesis was inhibited with actinomycin-D and the cells were treated with ENT, 50% reduction in the Her-2 mRNA was seen as early as 4 hours (Figure 6A). These findings suggest that ENT can also increase Her-2 mRNA degradation along with protein degradation.
Figure 6.
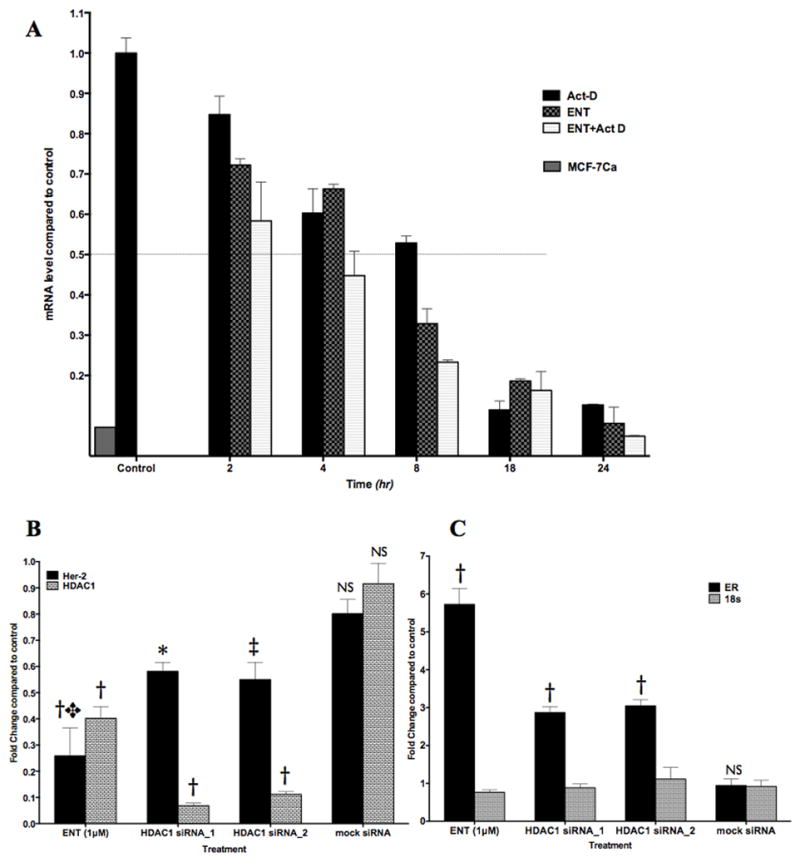
Figure 6A: RT-PCR analysis of Her-2 mRNA levels in LTLT-Ca cells: Relative levels of Her-2 mRNA after treatment with ENT were analyzed by real-time RT-qPCR. Cells were treated with ENT (1μM) alone or in presence of actinomycin D (5μM) and cells were collected at indicated time points. RNA was isolated, quantified and diluted to 0.08μg/μl. Total 0.64Mg of RNA was reverse transcribed and Her-2 was amplified using real-time qPCR as described in Materials and Methods. Graph represents relative levels (±SEM) of Her-2 mRNA compared to LTLT-Ca control (set at 1).
Figure 6B, C: RT-PCR analysis of Her-2, HDAC1, ERα and 18s mRNA levels in LTLT-Ca cells: Relative levels of Her-2, HDAC1, ERα and 18s mRNA after treatment with ENT, or siRNA against HDAC1 were analyzed by real-time RT-qPCR. Cells were treated with ENT (1μM) or siRNAs against HDAC1 (5nM) or mock siRNA. The cells were collected after 48-hour incubation at 37°C. RNA was isolated, quantified and diluted to 0.08μg/μl. Total 0.64Mg of RNA was reverse transcribed and cDNA was amplified using real-time qPCR as described in Materials and Methods. Graph represents relative levels (±SEM) of (B) Her-2 and HDAC1 mRNA and (C) ERα and 18s mRNA. The relative mRNA levels (±SEM) are reported compared to LTLT-Ca control (set at 1). P-values: *p=0.0024, ‡p=0.0021, †p<0.0001 versus control. NS: not significant.
Previously, investigators have shown that HDAC inhibitors reduce Her-2 protein through hyperacetylation of hsp90 and increased Her-2 mRNA degradation. However, both of these effects were modulated through inhibition of a class IIb HDAC6 (24, 27, 36). However, ENT is a class I selective HDAC inhibitor. To confirm that the effects seen with ENT treatment were due to inhibition of a class I HDAC, we treated LTLT-Ca cells with siRNA against HDAC1 (Figure 6). We compared the effect of HDAC1 siRNA on the expression of Her-2, HDAC1 (Figure 6B) and ERα, 18s ribosomal RNA was used as housekeeping gene (Figure 6C). HDAC1 siRNA treatment reduced Her-2 levels significantly (~40% reduction; *p=0.0024, ‡p=0.0021) compared to control. ENT was however, more potent in reducing the Her-2 mRNA levels (>70% reduction compared to control; †p<0.0001) compared to HDAC1 siRNAs (✥p=0.026 vs HDAC1 si_1 and p=0.049 vs HDAC1 si_2), suggesting that HDAC1 alone may not be involved in Her-2 regulation, although inhibition of HDAC1 by ENT is responsible for reduction is Her-2.
Treatment of MCF-7Ca xenografts with the combination of ENT and letrozole does not delay resistance to letrozole
Since, upregulation of Her-2 is seen as soon as 4 weeks after beginning treatment with letrozole (3) due to increased stability of Her-2 (29), we postulated that addition of entinostat (which causes degradation of Her-2 and reduces its stability) to letrozole may help delay resistance and prolong responsiveness of MCF-7Ca xenografts to letrozole (Figure 7). Mice bearing MCF-7Ca xenografts were grouped such that the mean tumor volumes were not different across the groups on week 0 (p=0.67). Mice were treated with letrozole (10μg/day) or ENT (50μg/day) or the combination. All mice received Δ4A (100μg/day); control group received only Δ4A. As observed previously, letrozole treated tumors had significantly lower growth rate than control (p=0.0001). Similarly, ENT+letrozole had significantly lower growth rate than control (p<0.0001). However, growth rates of tumors of mice treated with ENT+letrozole and letrozole were not significantly different over 17 weeks (p=0.28). These results suggest that although, ENT is effective in reversing resistance to letrozole, addition of ENT to letrozole for treating hormone sensitive tumors may not provide any additional benefit. MCF-7Ca xenografts are very hormone dependent and inhibition of estrogen synthesis by AIs can result in marked tumor inhibition. However, once they become resistant to AIs, other pathways are activated and until that time inhibition of those pathways does not impede the growth of these tumors.
Figure 7. Effect of ENT alone or in combination with letrozole on the growth of MCF-7Ca xenografts.
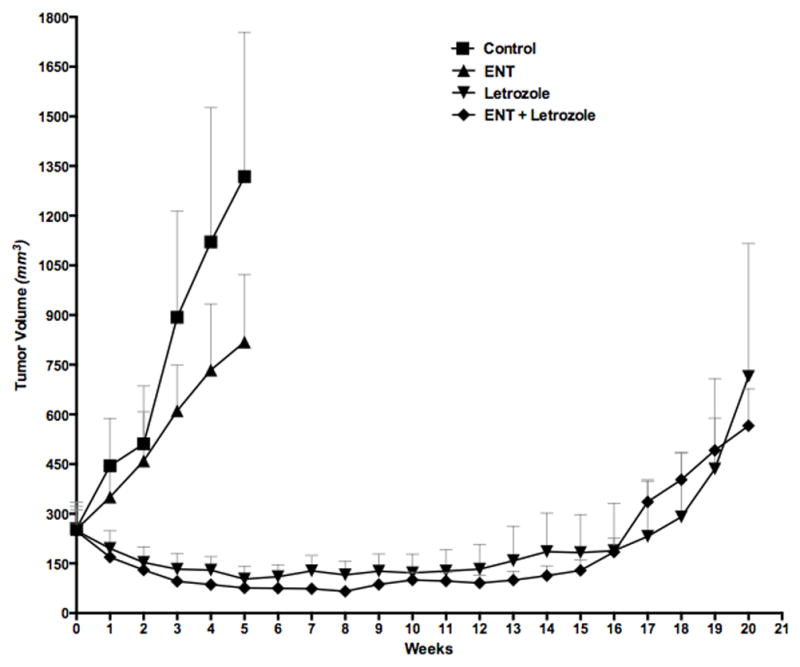
Ovariectomized athymic nude mice were inoculated with MCF-7Ca cells. Tumors were allowed to form in the presence of androstenedione (Δ4A), aromatizable substrate for estrogen. When the tumors reached ~300mm3, mice were treated with either letrozole (10μg/day), ENT (50μg/day) or the combination of letrozole and ENT for 20 weeks, The growth rates of tumors of mice treated with the combination of ENT plus letrozole was not significantly different than letrozole alone (p=0.28).
Discussion
It has been suggested that epigenetic silencing of ERα could be responsible for de novo resistance to endocrine therapy (17). In our previous study using a triple negative breast cancer model, we showed that treatment of ER-negative MDA-MB-231 cells and tumors resulted in increased expression of ERα and aromatase (21), which resulted in response of the cells and tumors to the growth inhibitory effects of AI letrozole and mitogenic effects of E2. To evaluate the effect of HDAC inhibition on reduced ERα expression, which results from long-term letrozole treatment and acquired resistance, we treated letrozole resistant cells and tumors with HDAC inhibitor ENT. Although the HDAC inhibitor reversed the acquired resistance to letrozole, the mechanism of this reversal was found to be different than that observed in the ER-negative MDA-MB-231 cells, which exhibit de novo resistance. Although ERα protein levels in the LTLT-Ca cells are significantly lower than in the parental MCF-7Ca cells, ERα was not epigenetically repressed in LTLT-Ca cells. Chromatin Immunoprecipitation (ChIP) analysis showed that in the presence of letrozole, the ERα promoter was still active but did not require E2 for activation, suggesting ligand independent activation of ERα. In addition, ENT activated the aromatase (PI.3/II) promoter in an E2 dependent manner. The results obtained with ENT were similar to those obtained with trastuzumab (TRZ); a humanized monoclonal antibody against extra-cellular domain of Her-2 (16). Since ENT exhibited such similar effects on LTLT-Ca cells and tumors as TRZ, we hypothesized that ENT may modulate Her-2 and thus restore responsiveness of LTLT-Ca cells and tumors to AIs.
Our previous study showed that upregulation of Her-2 in LTLT-Ca cells is the result of longer half-life of Her-2 protein and not gene amplification (29). In this study, we show that ENT reduced the stability of the Her-2 protein in the LTLT-Ca cells. The results of this study suggest that HDAC inhibitors can overcome resistance to AI letrozole through modulation of Her-2. The treatment of LTLT-Ca cells or letrozole resistant tumors with ENT resulted in downregulation of Her-2 along with up-regulation of ERα and aromatase. These molecular changes rendered the tumors responsive to AIs such as letrozole and exemestane. The combination of ENT with letrozole or exemestane was significantly better at controlling the growth of letrozole resistant tumors compared to single agents. Analysis of the tumors revealed that the Her-2 protein in letrozole resistant tumors associates more with hsp90, suggesting increased stabilization. ENT treatment reduced this association. HDAC 1 and 6 are shown to have hsp90 deacetylase activity and inhibition of these HDACs is responsible for acetylation of hsp90, which leads to inactivation of its chaperone activity, leading to ubiquitination of the client proteins (24, 25). The potential mechanism of Her-2 degradation by HDAC inhibitor ENT could involve the capacity of HDAC inhibitors to cause disassociation of client protein Her-2 from the hsp90 chaperon complex leading to its destabilization and degradation. This was confirmed by the reduced half-life and increased ubiquitination of the Her-2 protein in LTLT-Ca cells treated with entinostat. In addition, it has been shown that HDAC inhibitors are also capable of rapidly destabilizing mature Her-2 transcript (27). Our studies show that ENT was also able to reduce Her-2 stability along with its transcription.
These results suggest that non-nuclear effects of HDAC inhibition on the Her-2 protein as well as nuclear effects on Her-2 transcription are responsible for the ability of ENT to reverse the resistance to letrozole treatment.
ENT is being investigated in a phase II open-label (ENCORE-303) clinical trial for women with ER-positive breast cancer that are progressing on AI therapy. This preliminary proof-of-concept study showed that the combination provided some benefit, resulting in disease stabilization in heavily hormonally pre-treated and relatively hormone resistant breast cancer patients (37). The disease stabilization achieved with the addition of entinostat supported the hypothesis that entinostat has the ability to restore sensitivity to AIs. Furthermore, a double blind, randomized, placebo-controlled phase 2 study of entinostat in combination with the aromatase inhibitor exemestane (ENCORE 301), showed 27% reduction in the risk of disease progression, translating into a 2-month improvement in progression free survival (38). This study provides further evidence supporting the clinical benefit and tolerability of entinostat in combination with aromatase inhibitors.
Based on these findings we hypothesize that the increased half-life of Her-2 protein and increased activation of Her-2 gene, following estrogen deprivation (ensuing letrozole treatment) provides the cells an alternative pathway to escape growth inhibition due to estrogen withdrawal (29). This may result in the cells adapting to growth factor signaling pathways and overcoming growth inhibition by letrozole. Consequently, agents that interfere with the stability of the Her-2 protein and/or mRNA may also extend the responsiveness of hormone dependent tumors to AIs. Since HDAC inhibitor ENT can modulate expression of Her-2, it may provide additional benefit in the treatment of Her-2 positive or breast cancers that may express ERα (at a lower level), but exhibit resistance to aromatase inhibitors.
Supplementary Material
Acknowledgments
This work was supported by grants to G Sabnis (KG10037 from Susan G Komen) and to A. Brodie (CA-62483 from NCI/NIH and SAC100010 from Susan G Komen).
Abbreviations used
- Δ4A
Androstenedione
- AIs
aromatase inhibitors
- CYP-19
aromatase
- HDACi
Histone Deacetylase Inhibitors
- ER
Estrogen Receptor
- Let
Letrozole
- ENT
entinostat
- E2
17β-Estradiol
Footnotes
Note: Syndax Pharmaceuticals (MA, USA) provided entinostat and letrozole used in this study. This work was also presented in part at the Annual AACR meeting, Washington DC, April 2013.
Conflict: Syndax Pharmaceuticals partly provided financial support for the study.
References
- 1.Yue W, Brodie A. MCF-7 human breast carcinomas in nude mice as a model for evaluating aromatase inhibitors. J Steroid Biochem Mol Biol. 1993;44:671–3. doi: 10.1016/0960-0760(93)90278-5. [DOI] [PubMed] [Google Scholar]
- 2.Yue W, Zhou D, Chen S, Brodie A. A new nude mouse model for postmenopausal breast cancer using MCF-7 cells transfected with the human aromatase gene. Cancer Res. 1994;54:5092–5. [PubMed] [Google Scholar]
- 3.Jelovac D, Sabnis G, Long BJ, Macedo L, Goloubeva OG, Brodie AM. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 4.Long BJ, Jelovac D, Handratta V, Thiantanawat A, MacPherson N, Ragaz J, et al. Therapeutic strategies using the aromatase inhibitor letrozole and tamoxifen in a breast cancer model. Journal of the National Cancer Institute. 2004;96:456–65. doi: 10.1093/jnci/djh076. [DOI] [PubMed] [Google Scholar]
- 5.Yue W, Wang J, Savinov A, Brodie A. Effect of aromatase inhibitors on growth of mammary tumors in a nude mouse model. Cancer Res. 1995;55:3073–7. [PubMed] [Google Scholar]
- 6.Zhou DJ, Pompon D, Chen SA. Stable expression of human aromatase complementary DNA in mammalian cells: a useful system for aromatase inhibitor screening. Cancer Res. 1990;50:6949–54. [PubMed] [Google Scholar]
- 7.Brodie A, Jelovac D, Macedo L, Sabnis G, Tilghman S, Goloubeva O. Therapeutic observations in MCF-7 aromatase xenografts. Clin Cancer Res. 2005;11:884s–8s. [PubMed] [Google Scholar]
- 8.Brodie A, Lu Q, Liu Y, Long B. Aromatase inhibitors and their antitumor effects in model systems. Endocrine-related cancer. 1999;6:205–10. doi: 10.1677/erc.0.0060205. [DOI] [PubMed] [Google Scholar]
- 9.Jelovac D, Macedo L, Handratta V, Long BJ, Goloubeva OG, Ingle JN, et al. Effects of exemestane and tamoxifen in a postmenopausal breast cancer model. Clin Cancer Res. 2004;10:7375–81. doi: 10.1158/1078-0432.CCR-04-0565. [DOI] [PubMed] [Google Scholar]
- 10.Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:2378–88. [PubMed] [Google Scholar]
- 11.Buzdar A. Anastrozole as adjuvant therapy for early-stage breast cancer: implications of the ATAC trial. Clinical breast cancer. 2003;4 (Suppl 1):S42–8. doi: 10.3816/cbc.2003.s.014. [DOI] [PubMed] [Google Scholar]
- 12.Goss PE. Preventing relapse beyond 5 years: the MA.17 extended adjuvant trial. Seminars in oncology. 2006;33:S8–12. doi: 10.1053/j.seminoncol.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 13.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. Journal of the National Cancer Institute. 2005;97:1262–71. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- 14.Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med. 2003;349:1793–802. doi: 10.1056/NEJMoa032312. [DOI] [PubMed] [Google Scholar]
- 15.Swain SM. Aromatase inhibitors--a triumph of translational oncology. N Engl J Med. 2005;353:2807–9. doi: 10.1056/NEJMe058273. [DOI] [PubMed] [Google Scholar]
- 16.Sabnis G, Schayowitz A, Goloubeva O, Macedo L, Brodie A. Trastuzumab reverses letrozole resistance and amplifies the sensitivity of breast cancer cells to estrogen. Cancer Res. 2009;69:1416–28. doi: 10.1158/0008-5472.CAN-08-0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–5. [PubMed] [Google Scholar]
- 18.Sabnis GJ, Goloubeva OG, Chumsri S, Nguyen N, Sukumar S, Brodie AH. Functional activation of ER-{alpha} and aromatase by the HDAC inhibitor entinostat increases the sensitivity of ER-negative tumors to letrozole. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-10-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66:6370–8. doi: 10.1158/0008-5472.CAN-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keen JC, Yan L, Mack KM, Pettit C, Smith D, Sharma D, et al. A novel histone deacetylase inhibitor, scriptaid, enhances expression of functional estrogen receptor alpha (ER) in ER negative human breast cancer cells in combination with 5-aza 2′-deoxycytidine. Breast Cancer Res Treat. 2003;81:177–86. doi: 10.1023/A:1026146524737. [DOI] [PubMed] [Google Scholar]
- 21.Sabnis GJ, Goloubeva O, Chumsri S, Nguyen N, Sukumar S, Brodie AM. Functional Activation of the Estrogen Receptor-alpha; and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011;71:1893–903. doi: 10.1158/0008-5472.CAN-10-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–8. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 23.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Molecular and cellular biology. 2006;26:2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 25.Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leukemia research. 2008;32:1382–92. doi: 10.1016/j.leukres.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res. 2008;6:873–83. doi: 10.1158/1541-7786.MCR-07-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott GK, Marx C, Berger CE, Saunders LR, Verdin E, Schafer S, et al. Destabilization of ERBB2 transcripts by targeting 3′ untranslated region messenger RNA associated HuR and histone deacetylase-6. Mol Cancer Res. 2008;6:1250–8. doi: 10.1158/1541-7786.MCR-07-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabnis GJ, Kazi AA, Goloubeva O, Zhang B, Schech A, Shah P, et al. Effect of selumetinib and AZD8055 on the growth of anastrozole resistant tumors. AACR Meeting Abstracts. 2012:Abstract 2919. [Google Scholar]
- 29.Sabnis G, Goloubeva O, Gilani R, Macedo L, Brodie A. Sensitivity to the aromatase inhibitor letrozole is prolonged after a “break” in treatment. Molecular cancer therapeutics. 2010;9:46–56. doi: 10.1158/1535-7163.MCT-09-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabnis GJ, Macedo LF, Goloubeva O, Schayowitz A, Brodie AM. Stopping treatment can reverse acquired resistance to letrozole. Cancer Res. 2008;68:4518–24. doi: 10.1158/0008-5472.CAN-07-5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long BJ, Tilghman SL, Yue W, Thiantanawat A, Grigoryev DN, Brodie AM. The steroidal antiestrogen ICI 182,780 is an inhibitor of cellular aromatase activity. J Steroid Biochem Mol Biol. 1998;67:293–304. doi: 10.1016/s0960-0760(98)00122-8. [DOI] [PubMed] [Google Scholar]
- 32.Kazi AA, Jones JM, Koos RD. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Molecular endocrinology. 2005;19:2006–19. doi: 10.1210/me.2004-0388. [DOI] [PubMed] [Google Scholar]
- 33.Zhou C, Zhou D, Esteban J, Murai J, Siiteri PK, Wilczynski S, et al. Aromatase gene expression and its exon I usage in human breast tumors. Detection of aromatase messenger RNA by reverse transcription-polymerase chain reaction. J Steroid Biochem Mol Biol. 1996;59:163–71. doi: 10.1016/s0960-0760(96)00100-8. [DOI] [PubMed] [Google Scholar]
- 34.Sabnis G, Brodie A. Trastuzumab sensitizes ER negative, Her-2 positive breast cancer cells (SKBr-3) to endocrine therapy. Endorine Society’s Annual Meeting; 2009; Washington DC. p. Abstract OR38–04. [Google Scholar]
- 35.Sebastian S, Bulun SE. A highly complex organization of the regulatory region of the human CYP19 (aromatase) gene revealed by the Human Genome Project. J Clin Endocrinol Metab. 2001;86:4600–2. doi: 10.1210/jcem.86.10.7947. [DOI] [PubMed] [Google Scholar]
- 36.Fiskus W, Ren Y, Mohapatra A, Bali P, Mandawat A, Rao R, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13:4882–90. doi: 10.1158/1078-0432.CCR-06-3093. [DOI] [PubMed] [Google Scholar]
- 37.Wardley A. Phase II data for entinostat, a class 1 selective histone deacetylase inhibitor, in patients whose breast cancer is progressing on aromatase inhibitor therapy. ASCO; Chicago, IL: 2010. [Google Scholar]
- 38.Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, et al. Randomized Phase II, Double-Blind, Placebo-Controlled Study of Exemestane With or Without Entinostat in Postmenopausal Women With Locally Recurrent or Metastatic Estrogen Receptor-Positive Breast Cancer Progressing on Treatment With a Nonsteroidal Aromatase Inhibitor. J Clin Oncol. 2013;31:2128–35. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.