

Abstract
Using the repeat finding algorithm FT-Rep, we have identified 154 pentatricopeptide repeat (PPR) proteins in nine fully sequenced genomes from green algae (with a total of 1201 repeats) and grouped them in 47 orthologous groups. All data are available in a database, PPRdb, accessible online at http://giavap-genomes.ibpc.fr/ppr. Based on phylogenetic trees generated from the repeats, we propose evolutionary scenarios for PPR proteins. Two PPRs are clearly conserved in the entire green lineage: MRL1 is a stabilization factor for the rbcL mRNA, while HCF152 binds in plants to the psbH-petB intergenic region. MCA1 (the stabilization factor for petA) and PPR7 (a short PPR also acting on chloroplast mRNAs) are conserved across the entire Chlorophyta. The other PPRs are clade-specific, with evidence for gene losses, duplications, and horizontal transfer. In some PPR proteins, an additional domain found at the C terminus provides clues as to possible functions. PPR19 and PPR26 possess a methyltransferase_4 domain suggesting involvement in RNA guanosine methylation. PPR18 contains a C-terminal CBS domain, similar to the CBSPPR1 protein found in nucleoids. PPR16, PPR29, PPR37, and PPR38 harbor a SmR (MutS-related) domain similar to that found in land plants pTAC2, GUN1, and SVR7. The PPR-cyclins PPR3, PPR4, and PPR6, in addition, contain a cyclin domain C-terminal to their SmR domain. PPR31 is an unusual PPR-cyclin containing at its N terminus an OctotricoPeptide Repeat (OPR) and a RAP domain. We consider the possibility that PPR proteins with a SmR domain can introduce single-stranded nicks in the plastid chromosome.
Keywords: pentatricopeptide repeat, green algae, chloroplast, mitochondrion, evolution, cyclin, small MutS-related, tRNA methyltransferase
Introduction
PentatricoPeptide Repeat (PPR) proteins are major players in organelle gene expression.1 They interact with mitochondrial and plastidial RNAs at all steps of their biogenesis, including stabilization of 5? and 3? ends, splicing and trans-splicing, editing, and translation initiation. PPR proteins are characterized by the presence of a repeated 35-amino acid motif. The motif is highly degenerated, i.e., all positions are variable, but it can be recognized by virtue of its repetitiveness. It shows some similarity to the 34-amino acid TetratricoPeptide Repeat (TPR) motif to which it may be related.2 Based on this similarity, it was thus postulated that the repeat folds as a pair of ?-helices, which was confirmed by the structural analysis of a PPR domain found in mitochondrial RNA polymerase.3Key hydrophilic residues are predicted to point toward the super-helical groove, thus determining RNA sequence binding specificity. Recently, two slightly different versions of the ?PPR code? linking the sequence of the repeat to that of the target RNA have been published.4,5
PPR proteins are found in all Eukaryotic lineages, but the family is most expanded in photosynthetic organisms. The nuclear genome of land plants contains up to 450 different PPR genes, many of which are essential to plant development, and most studies in the field have been performed in Arabidopsis and maize. Little is known on the PPRs of the Chlorophyte green algae (which together with Streptophytes form the Viridiplantae, or green lineage), and even less on those of the Rhodophytes (red algae) and Glaucophytes, the other two groups of Archaeplastida (algae with primary endosymbiotic plastids). Similarly, algae with secondary endosymbiotic plastids have not been examined for their PPR content. Still, it should be remembered that PPRs have been functionally described in non-plant systems such as yeast and human even before their description in Arabidopsis. Recently, a survey of yeast PPRs allowed derivation of a more appropriate scoring matrix, leading to a dramatic expansion of the PPR catalog in this group of fungi.6 Almost all are somehow linked to mitochondrial function. Clearly, there is more to PPRs than what studies on land plants have thus far revealed.
One of the most fascinating aspects of the PPR family is its intricate evolutionary history. In spite of its diversity, the land plant PPR family is extremely conserved, with many orthology relationships extending across the entire Embryophyta lineage.7 It is postulated that colonization of terrestrial environments was accompanied by a burst of mutations and rearrangements in the plastidial and mitochondrial genomes. This posed a threat to the continued expression of organellar genes, as new sequences were generated that needed new RNA-binding proteins with the corresponding sequence specificity (and in the case of editing, the ability to change the nucleotide base). However, once established, the relationship between an RNA sequence and the repeat protein that binds to it appears rather stable,8 and loss of the protein is prevented by the absolute necessity to maintain tight binding to the target to allow its proper editing, splicing, stabilization, etc.
In addition to land plants, Streptophyta harbor several groups of aquatic algae in which multi-cellularity has started to emerge, but for which sequence information is sparse. In contrast, several nuclear genomes of Chlorophyte algae have been fully sequenced. These genomes represent the most primitive sub-group, the Mamiellophyceae, also known as ?Prasinophytes? (five species from the genera Ostreococcus and Micromonas), as well as the more evolved Trebouxiophyceae (Chlorella, Coccomyxa), and the most derived group, the Chlorophyceae (Chlamydomonas, Volvox). No genome sequence is yet available for the fourth major group, the Ulvophyceae. Up to now, the question of how PPR proteins have evolved in Chlorophyta has not been addressed, except as part of a study on the MRL1 gene of Chlamydomonas.9 Here, we present a systematic exploration of green algal PPRs, centered on but not limited to those of the most studied species, Chlamydomonas reinhardtii. In particular, we have tried to trace the evolutionary history of green algal PPRs, in search for conserved targets and conserved functions. We find that the analysis of additional domains found at their C terminus often sheds light on the possible functions of algal PPRs, and of their relatives in other groups.
Results and Discussion
Overview of green algal PPRs
Several computational tools are available to identify PPR proteins.6,10,11 Here, we have used FT-Rep, a newly developed repeat-finding algorithm that combines a classical motif search with a Fourier-transform analysis of its repetitiveness. The code was kindly communicated to us by the developer, Lorenzo Cerutti (Swiss-Prot, Geneva). Using FT-Rep with rather relaxed cutoffs, we have built lists of candidate PPR proteins from the publicly available genomes of nine green algae, and for comparison of one Glaucophyte and three red algae (Table 1). Table S1 describes all PPRs identified in green algae, with details on their length, repeat number, additional domains, and targeting predictions. The sequences of these 154 PPR proteins and 1201 repeats were organized into a relational database, PPRdb, available online at http://giavap-genomes.ibpc.fr/ppr. The website allows searching by keywords and BLAST, retrieval of the protein and repeat sequences, and running FT-Rep on user-entered queries. It also allows browsing of maximum likelihood phylogenetic trees generated from these sequences (see below). These trees were used to examine the evolutionary relationship between PPRs, assign them to 47 orthology groups, and name them accordingly.
Table 1. Distribution of PPR proteins in algae.
| Genus | Species/Strain | Number of PPRs | Average repeat/protein | |||
|---|---|---|---|---|---|---|
| Viridiplantae |
Chlorophyta |
Mamiellophyceae |
Ostreococcus |
O. tauri |
17 |
8.0 |
|
O. lucimarinus |
20 |
8.1 |
||||
|
O. sp. RCC809 |
16 |
8.1 |
||||
|
Micromonas |
M. pusilla CCMP1545 |
18 |
7.2 |
|||
|
M. pusilla RCC299 |
15 |
7.7 |
||||
| Trebouxiophyceae |
Chlorella |
C. sp. NC64A |
25 |
7.4 |
||
|
Coccomyxa |
C. subellipsoidea C169 |
19 |
8.8 |
|||
| Chlorophyceae |
Volvox |
V. carteri |
10 |
7.8 |
||
|
Chlamydomonas |
C. reinhardtii |
14 |
7.0 |
|||
| Rhodophyta |
|
Florideophyceae |
Chondrus |
C. crispus |
17 |
13.8 |
| Bangiophyceae |
Galdieria |
G. sulphuraria |
18 |
9.3 |
||
|
Cyanidioschyzon |
C. merolae |
8 |
9.9 |
|||
| Glaucocystophyceae | Cyanophoraceae | Cyanophora | C. paradoxa | many | ? |
For C. reinhardtii, gene models have been carefully checked and modified if necessary (for example, a new model was generated for PPR9). For other species, we relied entirely on the current structural annotation, which may lead to an underestimation of the real number of genes or to an inaccurate description of their sequence, as automatic annotations are never perfect. For example, there is no gene model for PPR15 in the current annotation of Ostreococcus sp. RCC809, so it does not appear in our database, but we have verified that the gene is present, and it was annotated in a previous version. For PPR20, the models in Ostreococcus tauri and Micromonas pusilla RCC299 were clearly inaccurate, leading to absence of several repeats. All this can lead to offsets between the positional numbering (rank) of orthologous repeats in orthologous PPR proteins (see example in Fig. 10B).
We feared that even when the gene model was correct, we could be missing some repeats if the motif itself had evolved in algae and become too divergent from the mostly higher plant-derived Pfam consensus. We therefore built a sequence logo-plot (which gives a graphical representation of sequence conservation in a multiple alignment) based on our entire set of algal repeats, and compared it to that obtained from 503 Arabidopsis PPRs identified using the same procedure. As can be seen from Figure 1, the logos are highly similar (including at positions 1 and 6 deemed important for sequence specificity), suggesting an excellent conservation of the motif and of its intrinsic variability between land plants and algae. Indeed, all our attempts to generate a more specific similarity matrix using our set of algal repeats led to a loss of sensitivity and accuracy (not shown), and we therefore stuck to the original one.
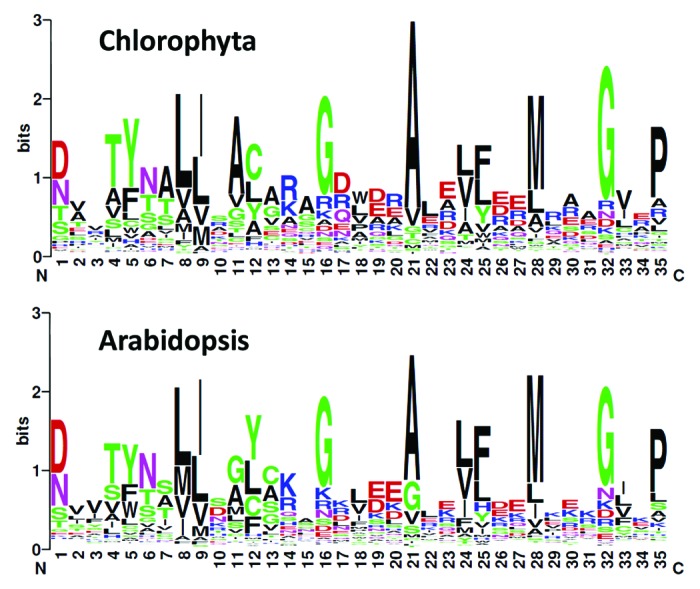
Figure 1. Sequence logos of the PPR repeats in Chlorophyta (top panel, based on 1201 sequences) and Arabidopsis (lower panel; 5000 sequences).
Our analysis reveals a few interesting general trends in the evolution of PPRs in algae. The number of PPR genes is lower than in land plants, but the family clearly is present in all algal groups, with eight to 25 members per genome. Interestingly, the number of repeats per protein appears to be lower in algae than in land plants. For Chlorophyta, the average is 7.7, far below the 12.5 we find in Arabidopsis PPRs, or the 14.9 we find for a set of 129 PPRs from the moss Physcomitrella patens. Note that we probably underestimated the average repeat number in land plants, since based on references 10 and 12 our lists must include some false positives (with fewer repeats). We conclude that algal PPRs overall have fewer repeats, hence, lower sequence specificity. Because of that we did not systematically attempt to predict a target sequence, except for MCA1 (see below). Red algae such as Chondrus, Galdieria, and Cyanidioschyzon show a number of PPRs comparable to those of Chlorophyta (with more repeats per protein in Chondrus), but no clear orthology relationship could be identified with the PPRs of green algae or land plants (data not shown). Concerning the Glaucophyte Cyanophora, sequence conservation with the other groups was difficult to judge because of the fragmented nature of the genome assembly,13 but here again, orthology to PPRs in other taxa was not evident. We conclude that although PPRs were clearly present in the ancestral Archaeplastida, sequence divergence has been too rapid to allow tracing of their evolution history across large evolutionary distances. Within Chlorophyta, sequence diversification is also rapid, as will be detailed below. Note that the number of PPR genes tends to be lower in the most evolved Chlorophyceae (Chlamydomonas and Volvox) compared with the other groups. This results from a complex pattern of gene losses, combined with gain of new PPR genes within specific clades.
Our attempts to generate full-scale multiple alignments of the entire set of PPR proteins were not very informative, beyond the most evident orthology relationships. This is probably because of the presence of unrelated regions/domains that can fortuitously align with the repeats, and because many different possibilities exist for matching divergent repetitive sequences. We therefore decided to restrict our analysis to the PPR domain and to use the repeats as independent sequence units to calculate evolutionary distances. We started by aligning the repeats themselves, based on the FT-Rep output (which shows insertions and deletions). Using the program Prot-Test, we then identified the evolution model that was most consistent with our data. Based on the calculated distances between the repeats, we built a maximum likelihood tree using RAxML. Finally, we generated trees for the proteins themselves, based on that of their repeats: we computed the distance between two proteins as the average distance between each of their repeats and its best match in the other protein. Orthology relationships were derived from this tree, with help from the repeat tree. We also used an additional protein tree computed using as inter-protein distance the minimum (rather than average) distance between any of the repeats, which gave largely congruent but noisier results. All these trees can be examined on our PPRdb website using the Archaeopteryx visualization software. Note that this method identifies similarity between proteins even if the repeats order is not conserved, which makes it more sensitive than multiple alignment of the entire sequences, but also more prone to artifactual grouping of unrelated proteins.
The vast majority (if not all) of PPR proteins in land plants are directed to either the mitochondrion (Mt) or the chloroplast (Cp), and we have set out to predict the intracellular location of green algal PPRs. We have used the popular program TargetP,14 as well as the newly developed PredAlgo tool,15 which was tailored to Chlamydomonas proteins and was found superior to TargetP for Chlorophyceae and Trebouxiophyceae. To try and screen out the gene models that may be truncated at the N terminus (usually the weak spot of structural annotation), we have examined multiple alignments of all the orthologous groups and marked as probably N-terminally truncated (?N? in Table S1) all those proteins for which the sequence started within a conserved part of the protein. We were thus able to predict intracellular location for 97 of the 154 PPRs (see individual predictions in Table S1). Of these, 46 were predicted by both programs to reside in an organelle (14 in Cp, 13 in Mt, 19 in one or the other), vs. 27 predicted as cytosolic and two as secreted. The others yielded contradictory results. Orthologs of MCA1 and MRL1, the only two established Cp PPRs,9,16 were almost always predicted as either Cp- or Mt-targeted. TargetP addressed more proteins to the Mt than to the Cp, while it was the reverse for PredAlgo, as already observed on larger data sets.15 We must stress, however, that these are only computer predictions which should not be taken at face value, if only because gene models of PPRs present in a single or two species could not be assessed for gene model quality. Experimental work will be needed to establish the intracellular location of algal PPRs.
Distribution of PPRs in the main branches of Chlorophyta
In order to understand the evolution of the PPR family in algae, we first examined the distribution of the various orthology groups across the different clades that constitute the Chlorophyta. Only a fraction of the genes are found in all groups. This includes the rbcL M- (mRNA stabilization) factor MRL1, as described earlier, as well as the petA M-factor MCA1 and two other PPRs of unknown function in algae: PPR1 and PPR7. This does not mean that the common ancestor of green algae encoded only this reduced set of PPRs: as will be detailed below, gene losses have obviously occurred during the evolution of Chlorophyta. Some groups also invented new PPRs, i.e., probably evolved them from a pre-existing gene, the relationship to which is now obscured by sequence divergence. For example PPR8, PPR10, PPR11, PPR12, and PPR13 are present only in Chlamydomonas and Volvox, and so presumably represent recent additions, specific to Chlorophyceae. Note that the annotation of the Volvox genome available at Phytozome has missed PPR8, 10, and 12, which are thus absent from our database, but BLAST searches unambiguously identifies the genes. Relatively decent gene models can be found in the version 1 of the genome (at http://genome.jgi-psf.org/Volca1 with protein IDs 96380, 119157, and 99237, respectively). In addition, the PPR9 gene is found in Chlamydomonas, Volvox, and Coccomyxa, but not in Chlorella. Again, no good gene model is available for Volvox PPR9, which is found near position 6862000 on scaffold_1. All our efforts to uncover a PPR9 ortholog in Chlorella remained vain, indicating that the gene either has been lost in this alga, or lies in an unsequenced fraction of the genome. Based on this analysis, it seems that PPR8 to 13 have appeared during the evolution of the most evolved groups of green algae.
In contrast, PPR17, 18, 19, 20, 21, 22, 23, 24, 25, and 26 are specific of the most ancestral group, the Mamiellophyceae, with an ortholog found in each of the five genomes but not in other algae. These genes may have arisen during the evolution of Mamiellophyceae, or be ancestral in Chlorophyta but lost in the other branches. Note that PPR22 of Ostreococcus and Micromonas branch at different positions on the average distance tree, but this is due to annotation errors in Micromonas, and orthology can be inferred from comparison of alternative gene models (not shown). Some of the Mamiellophyceae-specific PPRs are clearly paralogous, i.e., generated by gene duplication in a common ancestor of the group. For example, PPR19 and PPR26 clearly form a clade, possibly related to PPR20 and PPR1 (Fig. 2). Not far away in the tree, PPR17 and PPR18 are also clearly related (Fig. 3A). Similarly, PPR24 and PPR25 form a separate clade, apparently related to MCA1 (Fig. 3B). The Trebouxiophyceae also have their group-specific PPRs. PPR27, 28, 29, 30, and 31 are found in both Chlorella and Coccomyxa, but not in the other Chlorophytes. In addition, PPR32 to 42 are found only in Chlorella, and PPR43 to 46 only in Coccomyxa, indicating that differentiation of PPR genes has continued after the two genera diverged. Based on the protein tree, PPR31 and PPR33 are clearly derived form a recent gene duplication event, as are PPR32 and PPR41. Coccomyxa PPR46 is not far in the tree from Chlorella PPR34 and PPR42, suggesting a common origin. Finally, a few cases of possible gene disappearance were encountered. For example, PPR5 is found in Mamiellophyceae and Chlorophyceae, but not in Trebouxiophyceae. But this could be due to a fast evolution of PPR5, rather than a simple loss in Trebouxiophyceae: based on the repeat tree, the repeat order is scrambled between Mamiellophyceae and Chlorophyceae. In fact, PPR5 probably has changed function with time: the Chlamydomonas protein has been found in the flagella,17 which are lacking in Ostreococcus. A special case is that of PPR15 and PPR16, which are found in all the genomes under study except for Chlamydomonas and Volvox. This suggests that these genes are ancestral in Chlorophyta, but were lost in the Chlorophyceae. In the Mamiellophyceae, PPR16 shows a C-terminal Asp-/Glu-rich region absent in Trebouxiophyceae, which instead carry a SmR domain (see below).

Figure 2. Sub-tree extracted from the phylogenetic tree of PPR proteins computed from average inter-repeat distances, showing the relationship between the MT4-domain containing PPR19 and PPR26, and the relationship to PPR20 and PPR1/HCF152. PPR repeats are indicated by red boxes, Methyltransferase_4 domains by blue boxes, Zn-finger CCCH domains in PPR19 by white dots.

Figure 3. Sub-trees from the average-distance tree of PPR proteins showing the relationship between PPR17 and PPR18 (A) and between PPR24 and PPR25 (B). (B) also shows PPR14_MCA1. For clarity, Ostreococcus proteins have been indicated in green. Note the C-terminal domains in PPR18 (CBS) and PPR17/24 (SmR).
In the following, we will review the various PPRs of green algae and their functions, starting with those that have been studied experimentally, and trying for the others to infer from the analysis of their sequences clues as to their possible functions in the cell.
MCA1 controls the petA mRNA level
MCA1 was the first PPR protein to be functionally characterized in Chlorophyta. In C. reinhardtii, mca1 mutants transcribe the petA mRNA normally but fail to accumulate it.18 MCA1 acts on the very first 21 nucleotides of the petA 5? UTR to protect the whole transcript from 5??3' degradation.16 Expression of the petA gene also depends on another factor, TCA1, which is required for its translation. MCA1 and TCA1 recognize adjacent but distinct targets at the 5? end of the petA 5?UTR, and they display partially overlapping functions in its stabilization and translation. Indeed, a mutated petA transcript whose stability does not require MCA1 shows decreased TCA1-dependent rates of translation in the absence of MCA1. MCA1, although not strictly required for translation, thus behaves as a translational enhancer, presumably by assisting the binding of TCA1 to its own target. Conversely, TCA1 contributes to the full stabilization of the transcript through its interaction with MCA1.16 Direct interaction between the two proteins was shown in yeast by two-hybrid experiments. They associate in vivo in high molecular mass complexes of about 600 kDa that also contain the petA mRNA.19
C-terminal to the PPR domain, MCA1 contains a short well-conserved region, which we propose to call the MCA1-C domain as it is specific to this protein (Fig. S1). The yeast two-hybrid experiments have revealed that the full-length MCA1 can interact with TCA1, but a C-terminal truncated version lacking the last 290 residues (hence, the last five PPR repeats and the MCA1-C domain) cannot, suggesting that this motif could be involved in the interaction with TCA1. Note, however, that TCA1 is poorly conserved between Chlamydomonas and Volvox, and that we have failed to identify a TCA1 ortholog in other algae.
In transformed strains expressing variable amounts of MCA1, the accumulation level of petA mRNA is controlled by the concentration of MCA1. While TCA1 is quite stable, with a half-life longer than the doubling time of the culture (8 h), MCA1 was found to be short-lived with a half-life of 2?3 h. Its abundance varies rapidly with physiological conditions that deeply affect expression of the petA gene in vivo, for instance, in aging cultures or upon changes in nitrogen availability, while TCA1 shows more limited abundance changes under the same conditions. Thus, MCA1, a short-lived protein showing rapid variations in abundance and acting as a translation enhancer, appears central in the regulation of cytochrome f expression.20
Major clues on the regulatory role of MCA1 came from the study of the signal leading to its degradation. The proteolysis of MCA1 is triggered by its interaction with unassembled cytochrome f that transiently accumulates during the biogenesis of the cytochrome b6f complex. In Chlamydomonas, cytochrome f is a typical ?CES? protein (controlled by epistasy of synthesis),21 whose rate of synthesis decreases 10-fold in the absence of its assembly partners, cytochrome b6, or subunit IV. This regulation of cytochrome f synthesis results from a negative feedback mediated by a regulatory motif?the tetrapeptide cluster K305QFE308 exposed to the stroma, together with two upstream residues, K302 and Q297.22 This motif, exposed by unassembled cytochrome f, but shielded upon assembly of the cytochrome b6f complex, inhibits the translation of the petA mRNA. Strikingly, the interaction between MCA1 and unassembled cytochrome f relies on the same residues that form the CES repressor motif: mutations that disrupt the CES regulatory motif also prevent degradation of MCA1.19 The CES process for cytochrome f synthesis thus results from the regulatory function of MCA1 as a translation enhancer.
With the exception of Mamiellophyceae, the target of MCA1 seems to be highly conserved among Chlorophyta. Indeed, as detailed in Figure 4 and Table 2, the first 10 nucleotides of the Chlamydomonas petA transcript (5?GAGAAGAAAA3?) are found upstream of the petA coding sequence?and in this location only?in most organisms of the UTC (Ulvophyceae, Trebouxiophyceae, Chlorophyceae) clade. In Ulvophyceae and Chlorophyceae, this sequence is often found a few nucleotides downstream of a -10 box promoter consensus sequence, suggesting that it corresponds, as in C. reinhardtii, to the primary 5?end of the petA transcript.

Figure 4. Multiple sequence alignment of the putative targets of MCA1, as listed in Table 2. Nucleotides matching the sequence GAGAAGAAAA are written in red and putative -10 box promoter consensus are written in blue. The petA initiation codon, when close enough, is boxed.
Table 2. Occurrences of the putative MCA1 binding site in available chloroplast genome of Chlorophytes and Streptophyte algae.
| Organism | Loc.a | Occ.b | |||
|---|---|---|---|---|---|
| core chlorophytes |
Ulvophyceae |
Bryopsidales |
Bryopsis hypnoides (NC_013359) |
57 |
1 |
| Ulotrichales |
Pseudendoclonium akinetum (NC_008114) |
62 |
1 |
||
| Oltmannsiellopsidales |
Oltmannsiellopsis viridis (NC_008099) |
37 |
1 |
||
| Chlorophyceae |
Oedogonales |
Oedogonium cardiacum (NC_011031) |
no petA |
0 |
|
| Chaetophorales |
Stigeoclonium helveticum (NC_008372) |
no petA |
35 |
||
|
Schizomeris leibleinii (NC_015645) |
no petA |
6 |
|||
| Chaetopeptidales |
Floydiella terrestris (NC_014346) |
no petA |
19 |
||
| Chlamydomonales |
Chlamydomonas reinhardtii (NC_005353) |
260 |
2 |
||
|
Chlamydomonas raudensis (AY039799) |
207 |
na |
|||
|
Gonium pectoral (NC_020438) |
390 |
1 |
|||
|
Pleodorina starrii (NC_021109) |
494 |
1 |
|||
|
Volvox carterii (GU084820) |
3207 |
1 |
|||
|
Dunialliela salina (NC_016732) |
none 352 |
17 CAGAAGAAAA (5) |
|||
|
Haematococcus pluvialis |
32 |
na |
|||
| Sphaeroplerales |
Scenedesmus obliquus (NC_008101) |
119 |
11 |
||
| Trebouxiophyceae |
Chlorellales |
Chlorella variabilis (NC_015359) |
62 |
3 |
|
|
Chlorella vulgaris (NC_001865) |
360 |
5 |
|||
|
Parachlorella kessleri (NC_012978) |
40 |
3 |
|||
|
Pedinomonas minor (NC_016733) |
none 139 |
1 AGGAGAAAA (1) |
|||
| Oocystaceae |
Oocystis solitaria (FJ968739) |
26 |
1 |
||
| |
Helicosporidium sp. (NC_008100) |
no petA |
|
||
| Cnetocladales |
Leptosira terrestris (NC_009681) |
none 44 |
0 GAGAAGACAA (1) |
||
| Trebouxiales |
Trebouxia aggregata (EU123973) |
38 |
na |
||
|
Trebouxiophyceae sp. MX-AZ01 (NC_018569) |
none 43 |
0 GAGAAGAGAA (1) |
|||
|
Parietochloris incisa |
150 |
na |
|||
| |
Coccomyxa sp. C-169 (NC_015084) |
58 |
1 |
||
| Prasinophytes |
|
Pyraminonadales |
Pyramimonas parkeae (NC_012099) |
none |
2 |
| Mamiellophyceae |
Mamiellales |
Micromonas pusilla CCMP1545 (NC_012568) |
no petA |
0 |
|
|
Micromonas pusilla RCC299 (NC_012575) |
none |
0 |
|||
|
Ostreococcus tauri (NC_008289) |
none |
0 |
|||
| Monomastigales |
Monomastix sp. OKE-1 (NC_012101) |
none |
4 |
||
| |
Pycnococcaceae |
Pycnococcus provasolii (NC_012097) |
none 42 |
0 AGAAGAAAA (6) |
|
| Nephroselmidophyceae |
Nephroselmis olivacea (NC_000927) |
none 37 |
3 CAGAAGAAAA (2) |
||
| Charophytes | Chlorokybophyceae |
|
Chlorokybus atmophyticus (NC_008822) |
1693c |
1 |
| Mesostigmatophyceae |
|
Mesostigma viride (NC_002186) |
none |
1 |
|
| Zygnematophycee | Staurastrum punctulatum (NC_008116) | none | 5 |
a Distance (in nucleotide) between the first nucleotide of the target sequence (GAGAAGAAAA) and the A of the petA initiation codon. ?None? indicates that the target sequence was not found upstream of petA within 5 kb. When a slightly divergent sequence was found upstream of the petA gene, its distance to the initiation codon is indicated in the lower line. bNumber of occurrence of the target sequence in the whole chloroplast genome (na chloroplast genome not available). When a slightly divergent sequence was found upstream of the petA gene, it is shown in the lower line with its number of occurrences indicated between parentheses. cIn Chlorokybus atmophyticus, the target sequence, present only once in the chloroplast genome is located upstream of the cemA gene, the gene immediately upstream of the petA gene.
As illustrated in Figure 3B, MCA1 is conserved in all Chlorophyta whose sequence is available, including in Mamiellophyceae that lack a GAGAAGAAAA sequence upstream of petA. In Mamiellophyceae, MCA1 contains 11 PPR repeats in tandem whereas in Trebouxiophyceae and Chlorophyceae it comprises 14 repeats organized in two blocks of 9 and 5. Phylogenetic analysis of the repeats suggests that this is due to a block duplication of the first two repeats, plus the acquisition of a new one at the N terminus (Fig. 5A). In Trebouxiophyceae and Chlorophyceae, the intervening sequence separating the two blocks is not conserved between the various organisms and contains long stretches of A, S, Q, H, or P residues. It is predicted by PsiPred23 to adopt mainly a random coil structure.

Figure 5. (A) Schematic comparison of the distribution of the PPR repeats in the MCA1 protein of Chlorophyceae vs. Trebouxiophyceae. The phylogenetically related repeats are connected by arrows. Key residues critical for the nucleotide recognition are indicated, as well as the putative target sequence (in red) as deduced from the ?PPR code.? (B) Alignment of the petA 5?UTRs from O. tauri and M. pusilla RCC299. The arrow points to the 3? end of the trnQ, located immediately upstream of petA and the petA initiation codon is boxed. A region conserved between the two organisms is written in bold and underlined. The MCA1 binding site in Chlamydomonas is shown below for comparison.
Attempts to correlate the putative RNA target of MCA1 with the sequence of the PPR repeats, using the recently established ?PPR code,?4,5 gave ambiguous results (Fig. 5A). The main reason is that MCA1 proteins often present at the positions that determine specificity (1 and 6) residues that are not frequently found in the higher plants repeats used to infer the code, leading to a number of undetermined positions (Fig. 5A). In spite of these limitations, the deduced target of MCA1 in Chlorophyceae and Trebouxiophyceae was compatible with the conserved sequence found upstream of the petA gene in these organisms (compare Figs. 4 and 5). In Mamiellophyceae, however, the result was too ambiguous to deduce a binding site. In these algae, the petA 5?UTR most likely is not a primary 5? end, but is generated by cleavage of the upstream trnQ. Alignment of the petA 5?UTRs of O. tauri and M. pusilla RCC299 highlights a short stretch of nucleotides conserved between the two organisms, which is distantly related to the target of MCA1 in the UTC clade (Fig. 5B).
MRL1, an ancient PPR targeting rbcL
MRL1 was the second Chlamydomonas PPR for which molecular function was unraveled.9 Mutants fail to accumulate the rbcL mRNA because of its destabilization and, thus, show a non-phototrophic and light-sensitive phenotype typical of RuBisCO mutants. Subsequently, a new series of mrl1 alleles was characterized, and complemented strains with various levels of restoration were used to study phototrophic growth and ROS production as a function of RuBisCO level.24 The Chlamydomonas MRL1 protein is part of a high MW complex of approximately 800 kDa, whose size was shifted to 550?600 kDa after Rnase I treatment or in a ?rbcL mutant, indicating that it includes the target mRNA as well as probably other proteins.9 MRL1 is conserved across the entire Viridiplantae lineage, and Arabidopsis mutants, although able to carry out photosynthesis normally, show a specific disturbance of rbcL transcript processing: the shorter mRNA, generated by processing from the longer primary transcript, fails to accumulate. While it was impossible to distinguish between failure to generate the transcript and lack of stabilization, the latter seems more likely, because the primary transcript does not over accumulate and because this appears to be the general mode of action of mRNA stabilizing PPRs.25 A mild deficiency in polysome loading of the long transcript suggested an additional role of MRL1 as an enhancer of rbcL translation. RbcL is also a CES protein26,27 and it is not impossible that mRNA stabilization factors in general play a role in the CES process.
Our analysis of algal PPRs confirms our previous findings on the conservation of MRL1. It is not found in Rhodophyta, Cyanophora, nor any secondary endosymbionts, but is present in all Viridiplantae examined. Three paralogs are present in Physcomitrella, and MRL1A and MRL1B probably result from the whole genome duplication that occurred in the moss ~45 million years ago.28 In all MRL1 proteins, the PPR domain is followed by a long C-terminal region, which is not recognized by Pfam as a specific domain, but that we have characterized previously and named MRL1-C domain.9 It is predicted by PsiPred23 to be composed entirely of ?-helices, so we may speculate that it is derived from additional repeats that are no longer recognized as PPRs, but participate in RNA-binding. In Chlamydomonas and Volvox, The MRL1-C domain is followed by an ill-conserved ?C-tail? not found in other organisms. The tail is not necessary for functional complementation, whereas the MRL1-C domain is.9 Identification of MRL1 orthologs is facilitated by the MRL1-C domain, but the PPR domain itself is conserved, not only across Chlorophyta (as evidenced by their grouping in our protein tree, not shown) but also with Streptophyta. The exact number of repeats is slightly higher than that deduced from FT-Rep analysis, probably because some of the repeats are slightly degenerated, with insertions and deletions.9
PPR1/HCF152 and its conserved C-terminal motif
PPR1 is conserved across all Chlorophyta, and BLAST searches in Viridiplantae with the Mamiellophyceae PPR1 sequences consistently hit Arabidopsis HCF152 and its land plant orthologs. In reciprocal BLAST searches, algal PPR1 and land plant HCF152 proteins consistently appeared as reciprocal best matches when only the PPR domain was used as a query, indicating good conservation of the RNA-binding domain (even though the number of repeats can vary, from 12 in Chlamydomonas to 14 in Trebouxiophyceae, vs. 12 in Arabidopsis). The only exceptions were Chlamydomonas and Volvox PPR1, which hit other land plant PPRs before HCF152, but their grouping within the PPR1 clade is evident (Fig. 2).
Interestingly, a short conserved domain is found in all HCF152 and PPR1 sequences (including the Chlorophyceae), downstream of the PPR domain (Fig. 6). We propose to call it the HCF152-C domain, as it appears to be specific of this group of PPRs. It is characterized by the conserved core sequence Ex(W/F)KxWLGLPNxYY. Based on the good conservation of both the PPR region and the HCF152-C domain, we are confident that all green algal PPR1 proteins are true HCF152 orthologs, even if the PPR domain of Chlamydomonas and Volvox appears to have more extensively diverged. The situation is in fact similar to that of MRL1, with the presence of a conserved C-terminal domain specific to the gene confirming the orthology inferred from the alignment of PPR domains in spite of variations in the number and sequence of repeats. Like MRL1, PPR1/HCF152 appears to be specific to the Viridiplantae: no ortholog could be found in Cyanophora, red algae, or secondary endosymbiotic algae.

Figure 6. Multiple sequence alignment of the HCF152-C domain of PPR1/HCF152 proteins. The number of residues trimmed from the alignment at the C terminus is indicated in parentheses. For sequences marked by a *, we used manually curated gene models because those in the official annotation were C-terminally truncated. The Pro residue mutated in hcf152-2 is marked by an arrow.
HCF152 is one of the best studied PPRs in land plants. Mutants in this gene show reduced splicing of petB and reduced accumulation of the petB and psbH mRNAs.29 Its binding site between psbH and petB was defined in vitro and found to correspond to a small RNA footprint.25 Interestingly, one of the mutant alleles, hcf152-2 is a Pro?Leu substitution at the fully conserved P residue of the HCF152-C domain (arrowhead in Fig. 6). Compared with the insertion allele hcf152-1, it has a less severe phenotype, with reduced accumulation of cytochrome b6f content but not of PSII, and while petB splicing is impaired, the psbH 3? and petB 5? ends appear to be almost fully protected (Meierhoff et al., 2003). In vitro, the mutation severely reduces the ability of the protein to form a dimer (Nakamura et al., 2003). Altogether, these results suggest that the HCF152-C domain promotes dimerization, which would be essential for splicing activity but not for binding to the psbH-petB intergenic region nor stabilization of the processed transcripts. In this respect, it is worth noting that the petB gene of Chlorophytes does not contain an intron, and is not located downstream of psbH. Thus, in spite of a significant conservation of the PPR1 protein sequence, both in its PPR and C-terminal (dimerization) domains, its function has probably changed after the divergence of algae and land plants. This contrasts with MRL1, where the target (the 5? region of rbcL) has been conserved, even though its sequence has changed.
PPR7 is found in all green algae, but not in land plants
PPR7 is a short PPR, with no more than four repeats, showing an N-terminal extension usually recognized as an organellar-targeting peptide. It lacks any additional domain or remarkable sequence features, but is clearly present in all green algae. BLAST searches failed to identify any ortholog in land plants or in other Archaeplastida or in secondary endosymbionts. Its function has not been uncovered yet, but ongoing work (Joerg Nickelsen, personal communication) indicates that it is a chloroplast protein, which binds to at least seven different plastid transcripts in vivo, based on RIP-CHIP data. PPR7-knockdown lines show a mild phenotype suggesting that PPR7 is involved in the stabilization/processing of some of these transcripts. However, its precise working mode remains to be elucidated.
PPR18 has a C-terminal CBS domain
Many green algal PPRs contain additional domains, always found C-terminal to the PPR domain (which is also where additional domains of land plant PPRs are usually found). In some cases, these additional domains show no similarity to domains of known function, and not much can be inferred as to their function (except when experimental data are available as for the MCA1-C, MRL1-C, and HCF152-C domains described above). For example, PPR23 carries a C-terminal extension rich in charged residues, similar but apparently unrelated to that found in Mamiellophycean PPR16. More interestingly, Chlorella PPR33 carries a DNA-binding SAP domain, like the Arabidopsis PPR pTAC3, and a CBS domain is found in all PPR18 proteins (including in the unannotated ortholog that we found in M. pusilla RCC299). While CBS domains (Pfam PF00571, Interpro IPR000644) usually occur in pairs and form a dimerization interface able to bind regulatory ligands,30 plant CBS-containing proteins tend to contain a single CBS domain,31 and this is the case for PPR18. The exact role of this domain is unclear, but based on their expression patterns, plant CBS proteins were postulated to play a role in signaling. Interestingly, this study31 has described a plant PPR protein containing a single CBS domain, named CBSPPR1 (At5g10690 in Arabidopsis, Os09 g26190 in rice, GRMZM2G019901_P01/ AC206761.3_FGP002 in maize). The maize ortholog has been detected in nucleoids32 but the function of this protein is unknown. At5g10690 was not the best BLAST hit of PPR18 in Arabidopsis when only the PPR domain was used as a query, so we will refrain from inferring a common origin and calling the two proteins true orthologs, but the coincidence is striking. Actually, the PPR domain of At5g10690 is close to that of several Arabidopsis PPR-SmR proteins (see below). We speculate that the SAP and CBS domains of PPR33/pTAC3 and PPR18/CBSPPR1 play a role similar to that of the SmR domain in PPR-SmR, possibly in regulating transcription or replication within the nucleoid.
The PPR-SmR proteins
PPR-SmR proteins also appear linked to nucleoid functions. Pfam search identifies a large series of Chlorophyta PPRs containing at their C terminus an SmR domain (for ?Small MutS Related;? Pfam PF01713).33 These include PPR17 and PPR24, specific to Mamiellophyceae (see Fig. 3), plus the Trebouxiophycean PPR16, PPR29, PPR37, and PPR38. Based on their sequence and organization, three subfamilies of SmR domains have been distinguished:34 the domain can be found at the C terminus of a MutS2 protein (subfamily 1), as a standalone protein (subfamily 3), or at the C terminus of a protein with other domains (subfamily 2, in eukaryotes only). PPR-SmR proteins obviously fall into subfamily 2, and multiple sequence alignment of their SmR regions with that of other representative SmR domains (Fig. 7; Fig. S2) confirms this assignment as phylogenetically relevant. For example, they all lack the HGxG motif diagnostic of subfamilies 1 and 3 (H is replaced by a T, or the motif is altogether missing as in PPR16), while showing the subfamily 2-specific LDxH motif at the N terminus.

Figure 7. Phylogenetic tree of the SmR and SmR-like domains of PPR-SmR and PPR-cyclins (names in green and yellow, respectively), along with representative SmR domains from subfamilies 1 (white), 2 (blue), and 3 (purple). E. coli YdaL was used as an outgroup.
Seven land plants PPRs can be found within subfamily 2, of which three have been functionally characterized. SVR7 appears to play a classical role of translational activation of the atpB/E and rbcL transcripts.35,36 In contrast, pTAC2 has been identified as a component of the transcriptionally active plastid chromosome, and necessary for transcription from PEP promoters.37 GUN1, a major player in retrograde signaling, is co-localized with pTAC2, and its SmR domain binds DNA.38 Still, its exact role remains enigmatic.39,40 Based on the alignment and phylogenetic tree, the SmR domains of the plant PPRs clearly show similarity with those of algal PPR-SmR, with PPR24 appearing as the most closely related (Fig. 7).
Based on the available data, we can only speculate as to the role of the PPR-SmR proteins in green algae, in particular because sequence analysis does not allow us to ascertain whether their SmR domains are catalytically active. The SmR domain of MutS2 has nicking endonuclease activity on branched DNA structures such as Holliday junctions or D-loops, supporting the activity of MutS2 as a suppressor of homologous recombination by destroying its early intermediates.41 The residues involved in catalysis have not been experimentally defined, but comparison with Rnase E points to a few residues which are conserved in subfamilies 1 and 3, but not in subfamily 2. Yet, catalytic activity has been demonstrated for a subfamily 2 protein, human N4BP2, which has been shown to nick supercoiled plasmid DNA.42 In the following, we will assume that at least some of the PPR-SmR proteins of algae and plants can create single-stranded nicks in genomic DNA. In the case of land plants, for which a Cp location is ascertained, the substrate would probably be the nucleoid. Since algal plastidial DNA is also organized in nucleoids, this is where we expect their PPR-SmR proteins to act. What the consequences of DNA-nicking would be regarding transcription or replication of the plastid chromosome remains a matter of speculation. We note that the plastid chromosome in nucleoids is believed to be largely structured as linear branched multimeric molecules due to its mode of replication,43 with ?open? structures, similar to those recognized by SmR domains, expected at the branching points. As will be detailed below, PPR-cyclins also show a SmR-like domain, and we expect to find them also somehow related to nucleoid functions.
The PPR-cyclin proteins
Four clades of algal PPRs show a C-terminal cyclin domain (Pfam PF00134, PF02984, or PF08613). The combination PPR+cyclin appears to be specific to green algae, as we have failed to uncover it in other taxa. Three PPR-cyclin genes, called PPR3, PPR4, and PPR6, are shared by Chlorophyceae and Trebouxiophyceae. The genome sequence of Chlorella contains gaps around PPR4 and the gene model is poor, but BLAST analysis indicates that the protein is very well conserved. Based on the trees generated from the PPR domains (Fig. 8A), PPR3, PPR4, and PPR6 clearly have a common origin, with PPR3 and PPR4 showing the closest relationship, probably as a result of a more recent differentiation. Several Trebouxiophycean PPRs without a cyclin domain also cluster in this region of the tree, suggesting that they have evolved from a PPR-cyclin by loss of the latter domain. The repeats of same rank of all PPR-cyclins tend to group together in the repeat tree (not shown), suggesting good conservation of the target binding properties. In the tree generated from the cyclin domains (Fig. 8B), the PPR-cyclins also group together, and far away from all other cytoplasmic cyclins of Chlamydomonas or Arabidopsis. As a result we cannot infer which particular type of cyclin was recruited to form the PPR-cyclins.

Figure 8. (A) A section of the average-distance PPR protein tree showing the grouping of PPR-cyclins. (B) Phylogenetic tree generated using only the cyclin domains of PPR-cyclins. Note that they form a clade distinct from those of cytoplasmic cyclins.
As mentioned above, we found that all the PPR-cyclins possess a region with similarity to the SmR domain, sandwiched between the PPR and the cyclin domain. We should call these intervening sequences ?SmR-like? because they are not recognized as SmR domains at normal stringency cutoffs, and the PPR-cyclins thus are not listed as having a SmR in our PPRdb database. Still, they always hit in PSI-BLAST searches the SmR domains of plant and algal PPR-SmR proteins, just after the other algal PPR-cyclins. We thus considered them as derived SmR domains, and included them in our alignment (Fig. S2) and phylogenetic tree (Fig. 7). These SmR-like domains group within the subfamily 2 SmR domains, forming a distinct clade sister to the one that groups the Arabidopsis PPR-SmR and PPR24, and not far from PPR17 and the Trebouxiophycean PPR-SmR proteins. Even though the PPR domains of PPR-cyclins do not seem especially related to those of PPR-SmR, the similarity of the SmR domains suggest that PPR-cyclins have evolved from one of the PPR-SmR proteins initially present in green algae, by recruitment of a C-terminal cyclin domain.
In addition to these, the Trebouxiophyceae contain another type of PPR-cyclin, with a peculiar structure: PPR31 is made up of an OctotricoPeptide Repeat (OPR) domain,44 followed by a RAP domain (as is often the case in this family of repeat proteins), followed by a PPR, a SmR-like and a Cyclin domain. The Coccomyxa gene model is split in the middle, with the OPR-RAP part annotated as a different gene, but we have verified that a gene model can be constructed that combines the two repeat domains. Even though there is no EST support for these genes in Coccomyxa or Chlorella, we believe that this combination of two major RNA-binding repeat modules is not an annotation artifact fusing two distinct genes: we have obtained evidence for a similar organization of the PPR31 ortholog in another Trebouxiophyceae, Parietochloris incisa, with the OPR and PPR repeat domains being part of a single transcript based on genome and transcriptome sequence data (Vallon, Tourasse, unpublished). In PPR31, the conservation of the PPR repeats is poor, and while the Coccomyxa ortholog branches close to PPR3/4/6, the Chlorella protein is far away in the protein tree (not shown).
What can be the function of a cyclin domain in the context of a PPR protein? Cyclins are regulators of cell cycle transitions, which they trigger by activating cyclin-dependent protein kinases. Their timely disappearance is controlled through their phosphorylation and ubiquitin-dependent degradation by the proteasome. The cyclin domains of PPR-cyclins (including PPR31) are quite remote from those of bona fide cyclins (Fig. 8B), so their involvement in cell cycle regulation is far from certain. The targeting predictions of PPR cyclins are ambiguous, with PPR3 and PPR4 being predicted as targeted to an organelle in Chlamydomonas and Volvox and to the cytosol in the Trebouxiophyceae, and the reverse for PPR6. Still, the ill-conserved N-terminal sequence preceding the well-conserved repeat domain does suggest the presence of a targeting peptide. Cyclin proteins have not been described before in organelles, nor has ubiquitinylation, and we have not been able to identify in the Chlamydomonas genome an organelle-targeted cyclin-dependent kinase that could be activated by PPR-cyclins. So we can only speculate as to the possible function of these proteins. If they are indeed organelle-located, and based on the presence of an SmR-like domain, we would like to propose that they function in replication or transcription of the plastid chromosomes in the nucleoid.
PPR-MT4 proteins probably act as RNA methyltransferases
The Mamiellophyceae-specific PPR19 and PPR26 not only are related via their PPR domains (see Fig. 2), they also share a C-terminal MethylTransferase_4 domain (Pfam PF02390, Interpro IPR003358). We thus propose the name PPR-MT4 for these and related proteins. In addition, PPR19 proteins contain right after the PPR domain a short CCCH Zinc-finger domain, which in some proteins has been shown to bind AU-rich RNAs.45 However, its sequence is atypical (CX7CX5CX3H instead of CX8CX5CX3H, and the first conserved aromatic residue at position 12 is missing), and this domain is not found in other PPR-MT4 proteins.
The methyltransferase_4 domain is found in tRNA (m(7)G46) methyltransferases, called TrmB in Bacteria or trm8p in yeast, which methylate tRNAs at position G46, in the variable loop.46 It usually occurs as a stand-alone protein, even though the yeast enzyme requires an accessory protein trm82p for stability and activity.47 In land plants, the methyltransferase_4 domain can also be found associated with PhosphoGlycerate Kinase or Glycosyl Hydrolase domains, but to our knowledge these proteins have not been functionally investigated. Within Viridiplantae, the combination of MT4 with a PPR domain is specific to Mamiellophyceae, as PPR19 and PPR26 are absent from other algae and land plants.
Are PPR-MT4 proteins really functional as tRNA (m(7)G46) methyltransferases? Sequence alignment of MT4 domains (Fig. S3) indicates that the three residues identified in the 3D-structure of the Bacillus enzyme48 as binding the S-adenosyl-l-methionine substrate, E44, E69 and D96, are conserved (except for D96 in PPR26). The catalytic residues (T/S) D at positions 153?154 linking to the guanine O6 and N2 atoms are also perfectly conserved, as is the aromatic character of residues presumed to stack with the guanine ring (Y/F at position 193, F/W at position 197). So there is little doubt that PPR-MT4 proteins can methylate a guanosine residue at position m7. However, the charged sequence 121-PKKRHEKR-128, which in the Bacillus enzyme is presumed to bind the D-arm of the tRNA and is well conserved in Trm8 and TrmB proteins, is completely missing in PPR-MT4. Instead, this region shows a large variability in sequence and length, casting doubt on the ability of the MT4 domain to bind a tRNA substrate. Based on the ability of PPR domains to bind RNA, we propose that the PPR moiety participates in substrate recognition and binding. Yet, the small number of repeats usually found for PPR-MT4 proteins and the fact that PPRs usually bind to extended, rather than folded, RNA structures, suggest that PPR-MT4 methylates guanosine residues in another RNA than tRNA, and probably with a limited specificity. The intracellular location of PPR-MT4 could be an important clue to guide us to its substrate(s). Amoebal PPR-MT4 proteins (see below) are all predicted cytosolic. But unfortunately, for the algal PPR19 and PPR26 that appear full-length at the N terminus, the predictions are ambiguous (see Table S1).
The evolutionary history of these PPRs is rather complex. In addition to Mamiellophyceae, PPR-MT4 proteins are also found in taxa that have undergone secondary plastid endosymbiosis, such as Diatoms (Phaedoactylum, Thalassiosira), Cryptophytes (Guillardia theta) and the Alveolate Perkinsus marinus. While these algae clearly have inherited most of their photosynthesis-related genes from a red algal endosymbiont, the Diatom genomes in particular show a clear phylogenetic signal derived from green algae.49 This has led to the conclusion that their evolutionary history includes another endosymbiosis event involving a Mamiellophyceae, which is clearly in line with the phylogeny of PPR-MT4 proteins (absent from red algae). Trees generated independently using either the PPR or the methyltransferase domains (Fig. 9A and B) show that the PPR-MT4 proteins of secondary endosymbionts are slightly more closely related to PPR19 than to PPR26, in both domains.
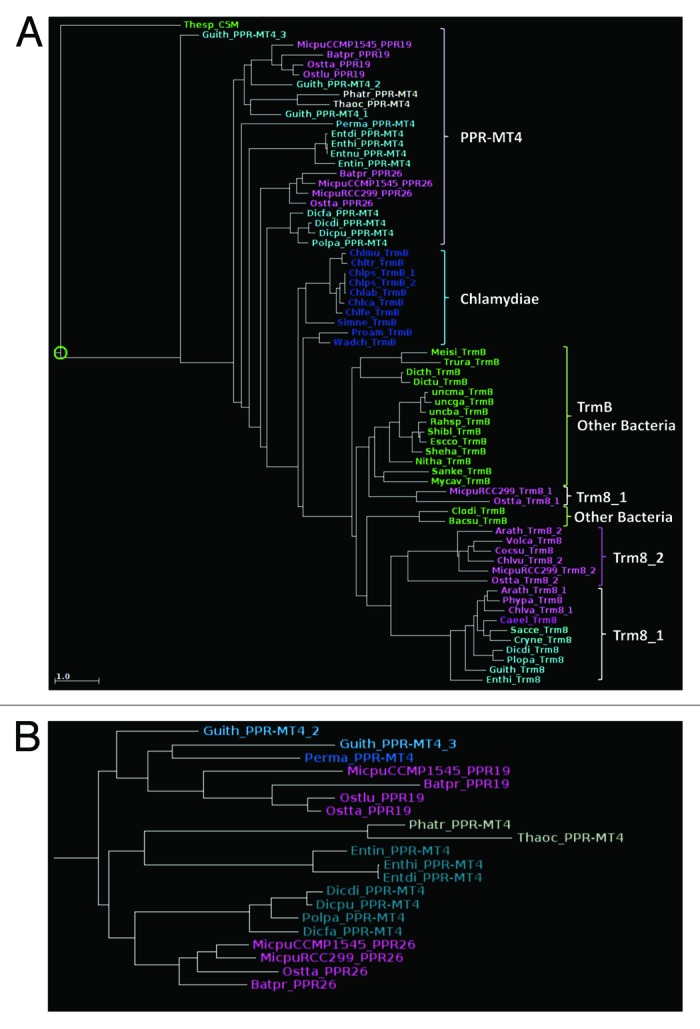
Figure 9. Phylogenetic trees (A) of the MT4 domains of PPR-MT4 along with representative TrmB and trm8 proteins; (B) of the PPR domains in PPR-MT4 proteins. Note that in both trees PPR19 tends to group with the PPR-MT4 of secondary endosymbionts, and PPR26 with the PPR-MT4 of Amoebas.
In contrast, PPR26 appears more closely related to yet another group of PPR-MT4 proteins, encountered in Amoebozoa (Dictyostelium, Entamoeba, etc., a single gene per genome). The presence of such a rare combination of domains in very distantly related taxa strongly suggests horizontal (not endosymbiotic) gene transfer, even if the phylogenetic trees are not sufficiently resolved to indicate in which direction. Still, arguments in favor of a transfer from the Amoeba to the alga can be found in the examination of the bacterial origin of the MT4 domain. Figure 9A and Figure S3 show that the MT4 domain of PPR-MT4 proteins is more closely related to TrmB proteins of Chlamydiae than to that of other bacteria. Chlamydiae live as intracellular parasites of eukaryotes, especially Amoebae,50 which can facilitate gene transfer. The rooting of PPR-MT4 in the phylogenetic tree is not within Chlamydiae, but examination of the alignments suggests that this is due to the presence in Chlamydiae TrmB of the charged region that binds the tRNA which, as mentioned above, is missing in PPR-MT4. Yet, other regions of the alignment show a high similarity between Chlamydiae and PPR-MT4, especially those of Amoebas (the replacement of the first G in the Rossman-fold motif by a C, the S(W/Y)F(E/D/N)xxW motif containing the guanine-stacking aromatic residues, in bold). This leaves little doubt as to the Chlamydial origin of the MT4 domain in PPR-MT4 proteins. Chlamydiae are believed to have played an important role as a source of plant genes, including essential starch metabolism genes, at the onset of plastid symbiosis.51 The story is different here, since PPR-MT4 is not found in other Archaeplastida. As a possible evolutionary scenario, we propose that Amoebas inherited a MT4 domain from their Chlamydiae parasites and hooked it to a PPR domain, before lateral gene transfer to an early Mamiellophyceae and duplication into PPR19 and PPR26 (and possibly into the related PPR20, which then lost its MT4 domain). Mamiellophyceae feed largely by phagotrophy, which can facilitate gene transfer according to the ?You are what you eat? model.52
Intra-protein duplication of repeats
How are the PPR repeats generated during evolution? The PPR genes themselves obviously duplicate during evolution and, for example, retrotransposition has been identified as a major route for the amplification of the family in land plants.7 But the variability in number of repeats within a group of orthologous PPR proteins suggests that loss and gain of repeats continues during the evolution of the gene, and our analysis of MCA1 is a good example (Fig. 5A). We tried to trace these events by identifying cases of high similarity between two repeats within a given protein, and verified them by comparing the different orthologs. For example, Figure 10A shows that the second and third repeats of PPR22 branch close to each other (and to repeat 4 in Micromonas), suggesting that they are derived from a tandem intra-protein repeat duplication. Other hints of intra-protein repeat duplication were observed in PPR20 (repeats 2 and 4, Fig. 10B), in PPR15 (repeats 2, 3, and 6), PPR23 (repeats 2 and 3), and in PPR25 (repeats 3 and 7). Note that most of these genes are specific to Mamiellophyceae, which thus appear as especially prone to intra-protein repeat duplication. In the list above, the duplicated repeats were often of low rank in the sequence order, and we asked whether this was a general trend in our entire set. Indeed, we found that within a protein, repeat 2 was overall the one with highest similarity to any other repeat in the same protein (data not shown), but the trend was not very strong, probably because duplications are the exception rather than the rule.

Figure 10. Excerpts from the PPR repeat tree, showing examples of probable intra-protein repeat duplication, in PPR22 (A) and in PPR20 (B).
Conclusion
PPR proteins of green alga present a fascinating array of properties, some of them shared with their homologs of land plants, others specific to algae. Sequence conservation across species can be very high, sometimes extending to the whole Chlorophyta or even to the entire Viridiplantae lineage. Our repeat-centered phylogenetic analysis appears capable of revealing some of the complex evolutionary scenarios that have led to today's PPR landscape, and we are also applying it to other repeat families like the OctotricoPeptide Repeat proteins. We find evidence for duplication of PPR genes (and of repeats within the genes), for gene loss and gene birth, for horizontal gene transfer of PPRs or of additional domains. While binding to the RNA may be the only relevant property (molecular function) of ?PPR-only? proteins, additional domains appear to be essential for conferring biological function to some PPRs. This can be RNA guanosine methylation (PPR-MT4), DNA binding (PPR-SmR and PPR-cyclins), ligand binding (PPR-CBS?), protein dimerization or partner-interaction (HCF152, MCA1), or other hitherto unrecognized functions. In these proteins, the PPR domain appears as a platform facilitating RNA binding, with some sequence specificity, while the additional domain represents the ?business end? of the protein. It is remarkable that these additional domains are always found C-terminal to the PPR domain. Because the order of repeats follows the 5??3? order of nucleotides in the target, this may indicate that the additional function concerns the RNA region donwnstream of the binding site, as is the case for editing in land plants. However, these additional domains are not necessarily devoted to RNA modification. For example, the possibility is intriguing that PPR-SmR and PPR-cyclins use their SmR domain to tether a short RNA molecule to the nucleoid upstream of a nick. DNA replication usually requires a short RNA serving as a primer.
Materials and Methods
Identification of PPR domains in green algal genomes
We retrieved from Phytozome 9.1 (http://www.phytozome.net/) the complete sets of protein sequences predicted from the genomes of Chlamydomonas reinhardtii (set 236), Coccomyxa subellipsoidea C169 (set 227), Ostreococcus lucimarinus (set 231), Micromonas pusilla CCMP1545 (set 228), M. pusilla RCC299 (set 229), and Volvox carteri (set 199). The protein sets of Chlorella variabilis NC64A (version 1), Ostreococcus sp.. RCC809 (version 2), and Ostreococcus tauri (version 2) were downloaded from the JGI Genome Portal (http://genome.jgi.doe.gov/). An alternative protein set for C. variabilis NC64A was also retrieved from the AUGUSTUS website of the University of Greifswald (http://augustus.gobics.de/predictions/Chlorella/). For red algae, the predicted protein sets of Cyanidioschyzon merolae 10D and Galdieria sulphuraria were obtained from GenBank, while those from Chondrus crispus was communicated by L. Meslet-Cladière (Station Biologique de Roscoff). For the glaucophyte Cyanophora paradoxa, protein sequences were downloaded from http://cyanophora.rutgers.edu/cyanophora/blast.php. Protein sequences from the plant Arabidopsis thaliana and the moss Physcomitrella patens were retrieved from TAIR 10 (http://www.arabidopsis.org/) and Phytozome 9.1, respectively. PPR domains were predicted by means of the FT-Rep program, using the PPR profile available in the PROSITE database,53 http://prosite.expasy.org/; profile accession number PS51375). The significance score cut-off values that were empirically determined for OctotricoPeptide (OPR) repeats (Cerrutti, Tourasse and Vallon, unpublished) were used in FT-Rep: -filter (filtering cut-off) 7.7; -C (psearch cut-off) 4.0; -AC (autocorrelation cut-off) 1.06. Low-complexity regions in protein sequences were hard-masked using segmasker (run with a window size of 25) from the NCBI BLAST+ toolkit (http://www.ncbi.nlm.nih.gov/books/NBK1763/) prior to FT-Rep search. The candidate PPR protein sequences were BLASTed against NCBI nr and against the other algal PPRs, and a few false positives (no significant repetitive hits to established PPRs) were eliminated. Sequence logos of the PPR multiple sequence alignments output by FT-Rep were generated using WebLogo54 (http://weblogo.berkeley.edu/).
Additional domains harbored by proteins containing PPR repeats were identified by scanning these proteins against the Pfam 27.0 database55 via the Pfam batch search facility (http://pfam.sanger.ac.uk/search).
Phylogenetic analyses
The ProtTest 2.4 software56 was used to find the amino-acid substitution model that best fits the PPR sequence data. To provide a reference tree to ProtTest, a phylogenetic tree was reconstructed using the Neighbor-Joining method57,58 applied to a matrix of observed pairwise distances between PPR domain sequences. Distances were calculated in the SEAVIEW 4 alignment editor59 as the percentage of amino-acid differences between sequences from the multiple alignment of all 1201 PPR domains produced by FT-Rep. The MtREV, MtMam, MtArt, RtREV, CpREV, HIVb, and HIVw substitution matrices were excluded from the set evolutionary models available in ProtTest. For the remaining models (WAG, Dayhoff, JTT, VT, Blosum62, LG, and DCMut), all versions including those with invariant sites, gamma distribution of substitution rates among sites, and empirical amino-acid frequencies were considered and the best-fit model was selected according to the Akaike Information Criterion (AIC) (ProtTest options: -sort C -S 0 -t1 F -t2 F -+I F -+I+G F). RAxML 7.2.660 was then used to compute pairwise maximum-likelihood (ML) evolutionary distances under the best-fit model (option -m PROTGAMMALGF, corresponding to the LG+G+F model61) and the reference tree (the LG+G+F model was in fact the best-fit model under the four selection criteria provided in ProtTest). The final phylogenetic tree of PPR domains was then built by the Neighbor-Joining method applied to the ML distance matrix. Pairwise patristic distances, corresponding to the sum of the branch lengths connecting any two sequences in the tree, were computed using the PATRISTIC program.62
To infer the phylogenetic relationships among proteins containing PPR domains, pairwise inter-protein evolutionary distances were calculated and used to reconstruct protein trees by Neighbor-Joining. The distance between two proteins was taken as either the minimum of all pairwise ML distances between the PPR domains of the two proteins or as the average of the minimum distances between each PPR domain of a protein and all PPR domains of the other protein.
Phylogenetic analyses of additional domains of PPR proteins (Methyltransferase, Cyclin, SmR) and selected homologs (identified by BLAST against GenBank) were also performed. Multiple sequence alignments were computed using CLUSTALW 2.163 and manually refined. Ambiguously aligned N- and C-terminal regions were excluded from further analyses. RAxML was then used to reconstruct maximum-likelihood phylogenetic trees from the alignments under the best-fit substitution model estimated by ProtTest, which was the LG+G+F model in all cases. The guide tree produced by CLUSTALW was provided as reference tree to ProtTest and as starting tree to RAxML, which was run with default parameters.
Phylogenetic trees were visualized in the Archaeopteryx tree viewer, the successor of the ATV viewer.64 Archaeopteryx allows to display annotations on the trees, such as phylogenetic origin of the species and protein domain structure, by making use of the recently developed PhyloXML file format.65 Trees were converted from the standard Newick/New Hampshire format to PhyloXML using the phyloxml_converter utility, and annotations were added to the PhyloXML files (Archaeopteryx and phyloxml_converter are available from http://www.phyloxml.org/). PPR-domain containing proteins were numbered according to orthology relationships inferred by examining the groupings in the phylogenetic trees, i.e., proteins that were judged as orthologous among different species were given the same ID number in all the species. The format for the repeats is taxid_PPRm_PPR_n, where taxid is an abbreviation of the taxon name (the first three letters of the genus name followed by the first two letters of the sp.ecies name), m is the PPR ID number based on orthology groups, and n is the positional number of a PPR repeat within a protein.
Supplementary Material
Acknowledgments
NJT was supported by the European contract GIAVAP (FP7-. KBBE.2010.3. GA No. 266401). Work in our laboratory is supported by the Centre National de la Recherche Scientifique (CNRS) and the Université Pierre et Marie Curie (UPMC).
Glossary
Abbreviations:
- PPR
pentatricopeptide repeat
- Mt
mitochondrion
- Cp
chloroplast
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/rnabiology/article/26127/
Footnotes
Previously published online: www.landesbioscience.com/journals/rnabiology/article/26127
References
- 1.Delannoy E, Stanley WA, Bond CS, Small ID. Pentatricopeptide repeat (PPR) proteins as sequence-specificity factors in post-transcriptional processes in organelles. Biochem Soc Trans. 2007;35:1643–7. doi: 10.1042/BST0351643. [DOI] [PubMed] [Google Scholar]
- 2.Small ID, Peeters N. The PPR motif - a TPR-related motif prevalent in plant organellar proteins. Trends Biochem Sci. 2000;25:46–7. doi: 10.1016/S0968-0004(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 3.Ringel R, Sologub M, Morozov YI, Litonin D, Cramer P, Temiakov D. Structure of human mitochondrial RNA polymerase. Nature. 2011;478:269–73. doi: 10.1038/nature10435. [DOI] [PubMed] [Google Scholar]
- 4.Barkan A, Rojas M, Fujii S, Yap A, Chong YS, Bond CS, Small I. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012;8:e1002910. doi: 10.1371/journal.pgen.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagi Y, Hayashi S, Kobayashi K, Hirayama T, Nakamura T. Elucidation of the RNA recognition code for pentatricopeptide repeat proteins involved in organelle RNA editing in plants. PLoS One. 2013;8:e57286. doi: 10.1371/journal.pone.0057286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipinski KA, Puchta O, Surendranath V, Kudla M, Golik P. Revisiting the yeast PPR proteins--application of an Iterative Hidden Markov Model algorithm reveals new members of the rapidly evolving family. Mol Biol Evol. 2011;28:2935–48. doi: 10.1093/molbev/msr120. [DOI] [PubMed] [Google Scholar]
- 7.O?Toole N, Hattori M, Andres C, Iida K, Lurin C, Schmitz-Linneweber C, Sugita M, Small I. On the expansion of the pentatricopeptide repeat gene family in plants. Mol Biol Evol. 2008;25:1120–8. doi: 10.1093/molbev/msn057. [DOI] [PubMed] [Google Scholar]
- 8.Hayes ML, Mulligan RM. Pentatricopeptide repeat proteins constrain genome evolution in chloroplasts. Mol Biol Evol. 2011;28:2029–39. doi: 10.1093/molbev/msr023. [DOI] [PubMed] [Google Scholar]
- 9.Johnson X, Wostrikoff K, Finazzi G, Kuras R, Schwarz C, Bujaldon S, Nickelsen J, Stern DB, Wollman FA, Vallon O. MRL1, a conserved Pentatricopeptide repeat protein, is required for stabilization of rbcL mRNA in Chlamydomonas and Arabidopsis. Plant Cell. 2010;22:234–48. doi: 10.1105/tpc.109.066266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lurin C, Andrés C, Aubourg S, Bellaoui M, Bitton F, Bruyère C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell. 2004;16:2089–103. doi: 10.1105/tpc.104.022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karpenahalli MR, Lupas AN, Söding J. TPRpred: a tool for prediction of TPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinformatics. 2007;8:2. doi: 10.1186/1471-2105-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugita M, Ichinose M, Ide M, Sugita C. Architecture of the PPR gene family in the moss Physcomitrella patens. RNA Biol. 2013;10 doi: 10.4161/rna.24772. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price DC, Chan CX, Yoon HS, Yang EC, Qiu H, Weber AP, Schwacke R, Gross J, Blouin NA, Lane C, et al. Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science. 2012;335:843–7. doi: 10.1126/science.1213561. [DOI] [PubMed] [Google Scholar]
- 14.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–16. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 15.Tardif M, Atteia A, Specht M, Cogne G, Rolland N, Brugière S, Hippler M, Ferro M, Bruley C, Peltier G, et al. PredAlgo: a new subcellular localization prediction tool dedicated to green algae. Mol Biol Evol. 2012;29:3625–39. doi: 10.1093/molbev/mss178. [DOI] [PubMed] [Google Scholar]
- 16.Loiselay C, Gumpel NJ, Girard-Bascou J, Watson AT, Purton S, Wollman FA, Choquet Y. Molecular identification and function of cis- and trans-acting determinants for petA transcript stability in Chlamydomonas reinhardtii chloroplasts. Mol Cell Biol. 2008;28:5529–42. doi: 10.1128/MCB.02056-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pazour GJ, Agrin N, Leszyk J, Witman GB. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 2005;170:103–13. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gumpel NJ, Ralley L, Girard-Bascou J, Wollman FA, Nugent JH, Purton S. Nuclear mutants of Chlamydomonas reinhardtii defective in the biogenesis of the cytochrome b6f complex. Plant Mol Biol. 1995;29:921–32. doi: 10.1007/BF00014966. [DOI] [PubMed] [Google Scholar]
- 19.Boulouis A, Raynaud C, Bujaldon S, Aznar A, Wollman FA, Choquet Y. The nucleus-encoded trans-acting factor MCA1 plays a critical role in the regulation of cytochrome f synthesis in Chlamydomonas chloroplasts. Plant Cell. 2011;23:333–49. doi: 10.1105/tpc.110.078170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raynaud C, Loiselay C, Wostrikoff K, Kuras R, Girard-Bascou J, Wollman FA, Choquet Y. Evidence for regulatory function of nucleus-encoded factors on mRNA stabilization and translation in the chloroplast. Proc Natl Acad Sci U S A. 2007;104:9093–8. doi: 10.1073/pnas.0703162104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choquet Y, Wostrikoff K, Rimbault B, Zito F, Girard-Bascou J, Drapier D, Wollman FA. Assembly-controlled regulation of chloroplast gene translation. Biochem Soc Trans. 2001;29:421–6. doi: 10.1042/BST0290421. [DOI] [PubMed] [Google Scholar]
- 22.Choquet Y, Zito F, Wostrikoff K, Wollman FA. Cytochrome f translation in Chlamydomonas chloroplast is autoregulated by its carboxyl-terminal domain. Plant Cell. 2003;15:1443–54. doi: 10.1105/tpc.011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 24.Johnson X. Manipulating RuBisCO accumulation in the green alga, Chlamydomonas reinhardtii. Plant Mol Biol. 2011;76:397–405. doi: 10.1007/s11103-011-9783-z. [DOI] [PubMed] [Google Scholar]
- 25.Zhelyazkova P, Hammani K, Rojas M, Voelker R, Vargas-Suárez M, Börner T, Barkan A. Protein-mediated protection as the predominant mechanism for defining processed mRNA termini in land plant chloroplasts. Nucleic Acids Res. 2012;40:3092–105. doi: 10.1093/nar/gkr1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wostrikoff K, Stern D. Rubisco large-subunit translation is autoregulated in response to its assembly state in tobacco chloroplasts. Proc Natl Acad Sci . 2007;104:6466–71. doi: 10.1073/pnas.0610586104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khrebtukova I, Spreitzer RJ. Elimination of the Chlamydomonas gene family that encodes the small subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase. Proc Natl Acad Sci U S A. 1996;93:13689–93. doi: 10.1073/pnas.93.24.13689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rensing SA, Ick J, Fawcett JA, Lang D, Zimmer A, Van de Peer Y, Reski R. An ancient genome duplication contributed to the abundance of metabolic genes in the moss Physcomitrella patens. BMC Evol Biol. 2007;7:130–9. doi: 10.1186/1471-2148-7-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meierhoff K, Felder S, Nakamura T, Bechtold N, Schuster G. HCF152, an Arabidopsis RNA binding pentatricopeptide repeat protein involved in the processing of chloroplast psbB-psbT-psbH-petB-petD RNAs. Plant Cell. 2003;15:1480–95. doi: 10.1105/tpc.010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baykov AA, Tuominen HK, Lahti R. The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem Biol. 2011;6:1156–63. doi: 10.1021/cb200231c. [DOI] [PubMed] [Google Scholar]
- 31.Kushwaha HR, Singh AK, Sopory SK, Singla-Pareek SL, Pareek A. Genome wide expression analysis of CBS domain containing proteins in Arabidopsis thaliana (L.) Heynh and Oryza sativa L. reveals their developmental and stress regulation. BMC Genomics. 2009;10:200. doi: 10.1186/1471-2164-10-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majeran W, Friso G, Asakura Y, Qu X, Huang M, Ponnala L, Watkins KP, Barkan A, van Wijk KJ. Nucleoid-enriched proteomes in developing plastids and chloroplasts from maize leaves: a new conceptual framework for nucleoid functions. Plant Physiol. 2012;158:156–89. doi: 10.1104/pp.111.188474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreira D, Philippe H. Smr: a bacterial and eukaryotic homologue of the C-terminal region of the MutS2 family. Trends Biochem Sci. 1999;24:298–300. doi: 10.1016/S0968-0004(99)01419-X. [DOI] [PubMed] [Google Scholar]
- 34.Fukui K, Kuramitsu S. Structure and Function of the Small MutS-Related Domain. Mol Biol Int. 2011;2011:691735. doi: 10.4061/2011/691735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zoschke R, Qu Y, Zubo YO, Börner T, Schmitz-Linneweber C. Mutation of the pentatricopeptide repeat-SMR protein SVR7 impairs accumulation and translation of chloroplast ATP synthase subunits in Arabidopsis thaliana. J Plant Res. 2013;126:403–14. doi: 10.1007/s10265-012-0527-1. [DOI] [PubMed] [Google Scholar]
- 36.Zoschke R, Kroeger T, Belcher S, Schöttler MA, Barkan A, Schmitz-Linneweber C. The pentatricopeptide repeat-SMR protein ATP4 promotes translation of the chloroplast atpB/E mRNA. Plant J. 2012;72:547–58. doi: 10.1111/j.1365-313X.2012.05081.x. [DOI] [PubMed] [Google Scholar]
- 37.Pfalz J, Liere K, Kandlbinder A, Dietz KJ, Oelmüller R. pTAC2, -6, and -12 are components of the transcriptionally active plastid chromosome that are required for plastid gene expression. Plant Cell. 2006;18:176–97. doi: 10.1105/tpc.105.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koussevitzky S, Nott A, Mockler TC, Hong F, Sachetto-Martins G, Surpin M, Lim J, Mittler R, Chory J. Signals from chloroplasts converge to regulate nuclear gene expression. Science. 2007;316:715–9. doi: 10.1126/science.1140516. [DOI] [PubMed] [Google Scholar]
- 39.Woodson JD, Perez-Ruiz JM, Schmitz RJ, Ecker JR, Chory J. Sigma factor-mediated plastid retrograde signals control nuclear gene expression. Plant J. 2012 doi: 10.1111/tpj.12011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terry MJ, Smith AG. A model for tetrapyrrole synthesis as the primary mechanism for plastid-to-nucleus signaling during chloroplast biogenesis. Front Plant Sci. 2013;4:14. doi: 10.3389/fpls.2013.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fukui K, Kosaka H, Kuramitsu S, Masui R. Nuclease activity of the MutS homologue MutS2 from Thermus thermophilus is confined to the Smr domain. Nucleic Acids Res. 2007;35:850–60. doi: 10.1093/nar/gkl735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diercks T, Ab E, Daniels MA, de Jong RN, Besseling R, Kaptein R, Folkers GE. Solution structure and characterization of the DNA-binding activity of the B3BP-Smr domain. J Mol Biol. 2008;383:1156–70. doi: 10.1016/j.jmb.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 43.Oldenburg DJ, Bendich AJ. Most chloroplast DNA of maize seedlings in linear molecules with defined ends and branched forms. J Mol Biol. 2004;335:953–70. doi: 10.1016/j.jmb.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 44.Eberhard S, Loiselay C, Drapier D, Bujaldon S, Girard-Bascou J, Kuras R, Choquet Y, Wollman FA. Dual functions of the nucleus-encoded factor TDA1 in trapping and translation activation of atpA transcripts in Chlamydomonas reinhardtii chloroplasts. Plant J. 2011;67:1055–66. doi: 10.1111/j.1365-313X.2011.04657.x. [DOI] [PubMed] [Google Scholar]
- 45.Hudson BP, Martinez-Yamout MA, Dyson HJ, Wright PE. Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat Struct Mol Biol. 2004;11:257–64. doi: 10.1038/nsmb738. [DOI] [PubMed] [Google Scholar]
- 46.De Bie LG, Roovers M, Oudjama Y, Wattiez R, Tricot C, Stalon V, Droogmans L, Bujnicki JM. The yggH gene of Escherichia coli encodes a tRNA (m7G46) methyltransferase. J Bacteriol. 2003;185:3238–43. doi: 10.1128/JB.185.10.3238-3243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexandrov A, Grayhack EJ, Phizicky EM. tRNA m7G methyltransferase Trm8p/Trm82p: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005;11:821–30. doi: 10.1261/rna.2030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zegers I, Gigot D, van Vliet F, Tricot C, Aymerich S, Bujnicki JM, Kosinski J, Droogmans L. Crystal structure of Bacillus subtilis TrmB, the tRNA (m7G46) methyltransferase. Nucleic Acids Res. 2006;34:1925–34. doi: 10.1093/nar/gkl116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moustafa A, Beszteri B, Maier UG, Bowler C, Valentin K, Bhattacharya D. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science. 2009;324:1724–6. doi: 10.1126/science.1172983. [DOI] [PubMed] [Google Scholar]
- 50.Horn M. Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol. 2008;62:113–31. doi: 10.1146/annurev.micro.62.081307.162818. [DOI] [PubMed] [Google Scholar]
- 51.Ball SG, Subtil A, Bhattacharya D, Moustafa A, Weber AP, Gehre L, Colleoni C, Arias MC, Cenci U, Dauvillée D. Metabolic effectors secreted by bacterial pathogens: essential facilitators of plastid endosymbiosis? Plant Cell. 2013;25:7–21. doi: 10.1105/tpc.112.101329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doolittle WF. You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998;14:307–11. doi: 10.1016/S0168-9525(98)01494-2. [DOI] [PubMed] [Google Scholar]
- 53.Sigrist CJ, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, Bougueleret L, Xenarios I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41(Database issue):D344–7. doi: 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–5. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 57.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 58.Studier JA, Keppler KJ. A note on the neighbor-joining algorithm of Saitou and Nei. Mol Biol Evol. 1988;5:729–31. doi: 10.1093/oxfordjournals.molbev.a040527. [DOI] [PubMed] [Google Scholar]
- 59.Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–4. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 60.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 61.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–20. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 62.Fourment M, Gibbs MJ. PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol Biol. 2006;6:1. doi: 10.1186/1471-2148-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 64.Zmasek CM, Eddy SR. ATV: display and manipulation of annotated phylogenetic trees. Bioinformatics. 2001;17:383–4. doi: 10.1093/bioinformatics/17.4.383. [DOI] [PubMed] [Google Scholar]
- 65.Han MV, Zmasek CM. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics. 2009;10:356. doi: 10.1186/1471-2105-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.