

Abstract
Mounting evidence suggests that the pathological hallmarks of Alzheimer’s disease (AD), neurofibrillary tangles and parenchymal amyloid plaques, are downstream reflections of neurodegeneration caused by the intraneuronal accumulation of amyloid-β proteins (Aβ), particularly Aβ42 and Aβ40. While the neurotoxicity of more amyloidogenic but less abundant Aβ42 is well documented, the effect of Aβ40 on neurons has been understudied. The Aβ40 expression in the presymptomatic AD brain is ten times greater than that of Aβ42. However, the Aβ40:42 ratio decreases with AD progression and coincides with increased amyloid plaque deposition in the brain. Hence, it is thought that Aβ40 protects neurons from the deleterious effects of Aβ42. The pathophysiological pathways involved in the neuronal uptake of Aβ40 or Aβ42 have not been clearly elucidated. Lack of such critical information obscures therapeutic targets and thwarts rational drug development strategies aimed at preventing neurodegeneration in AD. The current study has shown that fluorescein labeled Aβ42 (F-Aβ42) is internalized by neurons via dynamin dependent endocytosis and is sensitive to membrane cholesterol, whereas the neuronal uptake of F-Aβ40 is energy independent and nonendocytotic. Following their uptake, both F-Aβ40 and F-Aβ42 did not accumulate in early/recycling endosomes; F-Aβ42 but not F-Aβ40 accumulated in late endosomes and in the vesicles harboring caveolin-1. Furthermore, F-Aβ42 demonstrated robust accumulation in the lysosomes and damaged their integrity, whereas F-Aβ40 showed only a sparse lysosomal accumulation. Such regulated trafficking along distinct pathways suggests that Aβ40 and Aβ42 exercise differential effects on neurons. These differences must be carefully considered in the design of a pharmacological agent intended to block the neurodegeneration triggered by Aβ proteins.
Keywords: Alzheimer’s disease, cellular trafficking, cholesterol, endocytosis, lysosomes, neurodegeneration
INTRODUCTION
Deposition of Aβ proteins as parenchymal plaques is the conventional pathological hallmark of AD. However, extensive neuropathological and biochemical observations made in AD transgenic mouse models and in human AD patients suggest that intraneuronal accumulation of Aβ proteins is the pivotal event that triggers neurodegeneration in AD.1–3 The appearance of intraneuronal Aβ leads to profound deficits in hippocampal long-term potentiation, facilitates tau hyperphosphorylation, and disrupts proteosomal as well as mitochondrial functions.4 Removal of intraneuronal Aβ with passive immunotherapy was shown to halt neurodegeneration and reverse behavioral deficits in AD mouse models.2,5 However, immunotherapy with antiamyloid antibodies immobilizes extraneuronal amyloid deposits, jams the cerebral vasculature with amyloid, and causes life-threatening conditions such as meningoencephalitis.6 Hence, targeted therapies that can specifically clear neurons of toxic Aβ proteins are warranted; development of such therapies requires a thorough understanding of pathophysiological mechanisms that promote neuronal uptake of toxic Aβ isoforms.
In the early stages of AD, reuptake of extracellular Aβ proteins, rather than the accumulation of Aβ generated within the neurons through ER/Golgi processing of the amyloid precursor protein (APP), is believed to contribute significantly to the intraneuronal Aβ pool.7 Published reports have claimed several receptors and even nonendocytotic mechanisms to mediate intraneuronal accumulation of Aβ proteins.8 Previously, we reported nonsaturable, energy independent, and nonendocytotic uptake of fluorescein labeled Aβ40 in neuronal cells.9 In this study, we have shown that Aβ42 is internalized via endocytosis, which is energy dependent.
It is becoming increasingly evident that Aβ40 protects neurons from the deleterious effects of Aβ42.10–14 Moreover, Aβ40/Aβ42 ratios are associated with the severity and location of AD pathology;15,16 a lower Aβ40/Aβ42 ratio was shown to enhance amyloid deposition in the parenchyma,17 whereas higher Aβ40/Aβ42 ratio drives cerebrovascular amyloid accumulation.18 It is obvious from these reports that Aβ40 and Aβ42 play distinct roles in AD, and their relative distributions in various physiological compartments may modulate AD pathology. To exercise such differential impact, Aβ40 and Aβ42 must be trafficked and regulated via distinct pathways, which need to be carefully considered in the design of a pharmacological agent that can block their accumulation in neurons.
MATERIALS AND METHODS
Synthesis of Aβ40 Proteins
Aβ40 and Aβ42 are synthesized on an ABI 433 peptide synthesizer (Foster City, CA) with Val-NovaSyn TGA resin (Calbiochem-Novabiochem, San Diego, CA) employing HBTU activation and synthesis protocols as described in our earlier publication.9 Fluorescein was tagged to Aβ40 or Aβ42 on the Fmoc column, which was thoroughly washed with dimethylformamide and dichloromethane to remove excess NHS-fluorescein. The fluorescein labeled Aβ40 or Aβ42 (F-Aβ40 or F-Aβ42) thus obtained was purified by high performance liquid chromatography (HPLC) methods. Hence it is unlikely to have any free fluorescein in the F-Aβ40 or F-Aβ42 solutions. F-Aβ40 and F-Aβ42 monomers were prepared as per the procedure described by Klein et al.19 Various microscopy markers such as Alexa Fluor 633 labeled transferrin (AF633-Trf), Dil labeled low density lipoprotein (Dil-LDL), Alexa Fluor 647 labeled cholera toxin (AF647-CT) and Lysotracker Red DND 99 (LR) were obtained from Invitrogen (Carlsbad, CA).
Cell Cultures
Pheochromocytoma (PC12) cells were purchased from ATCC and grown in a 50:50 mixture of DMEM and Hams F12 (Mediatech Inc., Herndon, VA) enriched with 10% cosmic calf serum (Hyclone Hudson, NH). For the live cell imaging experiments, the PC12 cells were plated on sterile Delta-T culture dishes (Bioptechs, Butler, PA) at a density of 5000 cells/dish and cultured in the growth medium supplemented with 100 ng/mL of nerve growth factor (NGF) (Harlan Biosciences, Indianapolis, IN) and 1% serum for at least 4 days until the neurite growth was prominent. The differentiated PC12 cells were transfected using effectene transfection kits (Qiagen Valencia, CA) with m-cherry fluorescent protein fused to various cellular targets. In addition, the differentiated PC12 cells were cultured on 6-well culture plates, and on glass coverslip bottomed dishes for flow cytometry and laser confocal microscopy, respectively.
Live Cell Imaging
Following the incubation with various fluorophores the cells were washed twice, maintained under an atmosphere humidified with 5% CO2 in air, and imaged live using a TE-2000-S inverted microscope (Chiyoda-ku, Tokyo 100-8331, Japan) equipped with Nikon FITC HQ and m-Cherry-A-zero filters. The images were captured using Nikon’s NIS elements AR 3.0 software and processed using Adobe Photoshop CS4 software (Adobe Systems Inc., San Jose, CA).
Flow Cytometry
The PC12 cells incubated with various fluorophores were washed twice with ice-cold PBS, removed from the substrate by gentle trypsinization, suspended in ice cold PBS, and immediately scanned using FACScan (Becton Dickinson FACS canto, San Jose, CA). The fluorescence signal from fluorescein labeled protein was detected using a 488 nm laser and 530/30 band-pass filter whereas the signal for protein labeled with AF633-Trf was analyzed using a 633 nm laser and 660/20 band-pass filter.
Confocal Microscopy
The PC12 cells treated with various fluorophores were imaged using an Axiovert 100 M microscope/LSM 510 system (Carl Zeiss Micro Imaging, Inc., Thornwood, NY) equipped with 200 mW argon ion and 15 mW helium–neon ion (HeNe) lasers. F-Aβ40 or F-Aβ42 was imaged by the 488 nm line of the 200 mW argon ion laser and a 505–550 nm band-pass filter. The LR was visualized with the 543 nm line of the HeNe laser and a 560–615 BP filter. The Dil-LDL was imaged using the 568 nm line of the HeNe laser and a long pass 584 nm filter. The images were acquired with a C-Apochromat 63 X/1.2 NA water-immersion lens. The pinhole was set at 1.25 μM for all image acquisitions; photomultiplier, gain, and offset settings were maintained the same for all images.
Temperature Dependent Uptake of F-Aβ42 in PC12 Cells
The differentiated PC12 cells were preincubated in DMEM containing 10 mM HEPES buffer (DMEM/HEPES) at 4 °C for 30 min. The cells were subsequently incubated with ice-cold DMEM/HEPES containing 3.5 μM F-Aβ42 and 75 nM LR at 4 °C for 30 min, gently washed with ice-cold DMEM/HEPES twice, and imaged using confocal microscopy.
Influence of Cellular ATP on F-Aβ42 Internalization by PC12 Cells
The differentiated PC12 cells were preincubated with glucose free DMEM containing 0.1% sodium azide and 50 mM 2-deoxy-D-glucose for 30 min. Then F-Aβ42 (3.5 μM) was added to the cells and incubated for 30 min. AF633-Trf (15 μg/mL) was added to the cells 20 min before terminating the experiment. At the end of the experiment, the cells were washed with PBS and imaged. The control experiments were conducted similarly, but the cells were preincubated with DMEM.
Role of Endocytosis on the Internalization of F-Aβ40 or F-Aβ42 by PC12 Cells
The PC12 cells grown on delta T dishes were preincubated with 80 μM Dynasore (Tocris Bioscience, Ellisville, MO) in DMEM for 30 min. In the control experiments, the cells were incubated with DMEM alone. Following the preincubation, F-Aβ40 or F-Aβ42 solutions (3.5 μM) were added to the cells and incubated for 60 min. Then the cells were washed with DMEM without phenol red and imaged by live cell microscopy.
1. Accumulation of F-Aβ40 and F-Aβ42 in the Vesicles Harboring Caveolin 1
PC12 cells that stably express m-CFP/CAV-1(m-Cherry Fluorescent protein fused to Caveolin-1) grown on delta T dishes were incubated with 3.5 μM F-Aβ40 or F-Aβ42 for 60, 120, 180, and 240 min. The cells were washed with DMEM without phenol red and imaged using live cell microscopy.
2. Effect of Methyl-β-cyclodextrin (MβCD) on the Uptake of F-Aβ40 or F-Aβ42
The PC12 cells were preincubated with either DMEM or 10 mM methyl-β-cyclodextrin (MβCD) for 60 min. Then 3.5 μM F-Aβ42 or F-Aβ40 and AF633-Trf were added to the cells and incubated for 15, 30, 60, or 180 min. Then the cells were thoroughly washed, and the intracellular Aβ was quantified by flow cytometry or by Western blots.
3. Effect of AF647-CT on the Uptake of F-Aβ40 and FAβ42
PC12 cells grown on the delta T dishes were coincubated with 3.5 μM F-Aβ40 or F-Aβ42 and 5 μg/mL AF647-CT for 60 min. The cells were washed and imaged using live cell microscopy. Similar treatments were performed on PC12 cells grown on 6-well culture plates, and cellular fluorescence was quantified by flow cytometry.
Intracellular Itinerary of F-Aβ40 and F-Aβ42
1. Accumulation in Early Endosomes
Differentiated PC12 cells grown on delta T dishes were incubated with 3.5 μM F-Aβ40 or F-Aβ42 for 60 min. An early endosomal marker AF633-Trf (20 μg/mL) was added 20 min prior to the termination of the experiment. The cells were washed thoroughly and imaged by live cell microscopy.
2. Accumulation in Secondary Endosomes
The cells were treated with 3.5 μM F-Aβ40 or F-Aβ42 and Dil-LDL (Dilcomplexed low density lipoprotein) (15 μg/mL) for 30 or 60 min. Then the cells were thoroughly washed and imaged live with confocal microscopy.
3. Lysosomal Accumulation
Differentiated PC12 cells that stably express m-CFP/LAMP-1 (m-Cherry fluorescent protein fused to lysosomal-associated membrane protein-1) were incubated with either F-Aβ40 or F-Aβ42 for 60, 120, and 180 min respectively. Then the cells were washed with DMEM and imaged by live cell microscopy. The fluorescein and m-CFP signals in the images were superimposed, and the extent of colocalization was estimated by Pearson’s correlation coefficients. Approximately 50–100 cells were evaluated for each time point.
4. Influence of F-Aβ40 or F-Aβ42 Accumulation on the Lysosomal Integrity
PC12 cells that stably express m-CFP/LAMP-1 were incubated with 3.5 μM F-Aβ40 or F-Aβ42 for 15, 30, 60, 90, 120, and 180 min. The cells were washed with DMEM without phenol red and imaged using live cell microscopy. The areas of individual lysosomes in each cell were measured using Image J software. At least 50 cells were assayed for each time point. Change in lysosomal area was plotted against time.
Live Cell Imaging and Quantification of Digital Images
Each reported image is a composite of three images obtained at low, medium, and high exposure times, which ensure complete definition of the intracellular fluorescence signal. Caution was exercised to set the maximum exposure times below the signal saturation limits. Replicates in each study were run simultaneously and imaged at the same exposure setting. For inhibition studies, cells treated with inhibitors were imaged first with autoexposure settings and controls were imaged later with the same settings.
The cellular fluorescence in micrographs was quantified using Image J software (National Institute of Mental Health, Bethesda, Maryland). A minimum of 25 cells was quantified for each group in an experiment. From the gray images obtained directly from the microscope, individual cells were selected using polygon selection tool, and parameters such as minimum and maximum intensities, area, integrated density, and mean gray value were obtained. The minimum and maximum intensities of a typical image varied between 0 and 255 RU. The mean gray values ranged between 20 and 75, and the mean background values were between 5 and 15. The background fluorescence was subtracted from the mean gray values obtained for various control and treatment groups in a study, and the differences among them were statistically analyzed using Graphpad Prism software (La Jolla, CA).
For the calculation of lysosomal areas, the threshold of gray images was adjusted to reduce the background. The lysosomal areas were measured using the analyze particle tool. The vesicles <300 (pixel)2 were considered as small vesicles and the vesicles >300 (pixel)2 were considered as large vesicles which were assumed to be formed from the aggregation of smaller vesicles.
Immunoprecipitation and Western Blot
Following the uptake experiments the cell pellet was treated with 200 μL of cell lysis buffer consisting of RIPA buffer (Sigma-Aldrich, St. Louis, MO) and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Then the cell pellet was subjected to several quick freeze thaw cycles followed by probe sonication (Fisher Scientific, Pittsburgh, PA). To a 50 μL aliquot of the cell lysate was added 250 μg of IgG 4.1 antibody raised against human fibrillar Aβ42, and this was incubated for 2 h.20 Then the IgG 4.1–Aβ complex was separated from the cell lysate with 100 μL of immobilized protein A bead slurry (Thermo Fisher Scientific Inc., Rockford IL). Aβ protein was separated from the beads by boiling for 5 min at 95 °C in tris tricine sample buffer (Bio-Rad Hercules, CA) containing 2% v/v 6-mercaptoethanol. A 30 μL aliquot of this supernatant was loaded onto 10–20% gradient tris tricine peptide precast gels (Bio-Rad, Hercules, CA). The separated protein bands were transferred onto a 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA). Subsequently, the membrane was blocked with 5% nonfat milk and incubated with 6E10 monoclonal antibody (Coavance, Dedham, MA) overnight and further incubated with goat anti-mouse IgG secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The membrane was briefly incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific Rockford, IL) and imaged.
RESULTS
Energy Dependent Internalization of F-Aβ42 in Differentiated PC12 Cells
The Z-stack image of the PC12 cells incubated with F-Aβ42 for 30 min at 37 °C showed clear intracellular accumulation (Figure 1B), which reduced significantly when the incubation temperature was changed to 4 °C (Figure 1A). Upon incubation with F-Aβ42 and AF633-Trf (a clathrin-mediated endocytosis marker) at 37 °C, PC12 cells accumulated green (F-Aβ42) and red fluorescence (AF633-Trf) in the perinuclear region (Figure 1C). However, the accumulation of either F-Aβ42 or AF633-Trf (Figure 1D) decreased in the PC12 cells depleted of cellular ATP. The intracellular fluorescence intensities quantified using ImageJ confirmed these observations and demonstrated that the differences in the uptake of F-Aβ42 and AF633-Trf between normal and ATP depleted PC12 cells are statistically significant (Figure 1I).
Figure 1.

A, B: Effect of temperature on the uptake of F-Aβ42 by PC12 cells. (A) Z-Stack images of differentiated PC12 cells incubated with 3.5 μM F-Aβ42 and 75 nM Lysotracker Red (LR) for 30 min at 4 °C. (B) Z-Stack images of differentiated PC12 cells incubated with 3.5 μM F-Aβ42 and 75 nM Lysotracker Red (LR) for 30 min at 37 °C. C, D, J: Effect of ATP depletion on the uptake of F-Aβ42 by PC12 cells. Differentiated PC12 cells were incubated with 3.5 μM of F-Aβ42 and 15 μg/mL of Alexa Fluor labeled transferrin (AF633-Trf) for 60 min. (C) Intracellular accumulation of F-Aβ42; and intracellular AF633-Trf, overlay of images F-Aβ42 and AF633-Trf. (D) Uptake of F-Aβ42 and AF633-Trf in differentiated PC12 cells depleted of cellular ATP, overlay of F-Aβ42 and AF633-Trf images. (I) Quantification of intracellular fluorescence intensities using Image J software. Data obtained from 25 to 30 cells was presented as mean ± SEM. One-way ANOVA followed by Tukey post-test showed the following: ***p < 0.001, F-Aβ42 uptake in normal versus ATP depleted cells; ††p < 0.01, AF633 Trf uptake in normal versus ATP depleted cells. E–H, J: Inhibition of F-Aβ40 uptake by Dynasore. (E, F) Differentiated PC12 cells were treated with 3.5 μM F-Aβ40 for 60 min. (E) Intracellular F-Aβ40 fluorescence and overlay on differential interference contrast image (DIC). (F) Uptake of F-Aβ40 by differentiated PC12 cells following pretreatment with 80 μM Dynasore, a dynamin inhibitor and image G overlaid on the DIC image. Inhibition of F-Aβ42 uptake by Dynasore (G, H): intracellular F-Aβ42 fluorescence in differentiated PC12 cells. (G) Intracellular F-Aβ42 fluorescence and overlay on DIC image. (H) F-Aβ42 uptake following a 60 min pretreatment with 80 μM Dynasore overlay on DIC image. (J) Intracellular fluorescence intensities were quantified using Image J software. Data obtained from 30 to 40 cells was presented as mean ± SEM. One-way ANOVA followed by Tukey post-test showed the following: ***p < 0.01, FAβ42 uptake in control PC12 cells versus F-Aβ42 uptake in PC12 cells treated with Dynasore. Scale bars = 25 μm.
Dynasore Inhibits the Uptake of F-Aβ42 but Not FAβ40
Dynasore is a potent dynamin inhibitor and interferes with the endocytotic processes that involve dynamin in clathin-coated pits. Uptake of F-Aβ40 by the PC12 cells was not affected by Dynasore treatment (Figure 1E,F,J), further proving fact that the uptake is nonendocytotic as demonstrated in our earlier study.9 However, F-Aβ42 uptake in the PC12 cells pretreated with 80 μM Dynasore decreased significantly compared to that in the normal cells (Figure 1G,H,J).
Role of Caveolae Mediated Endocytosis in the Intracellular Uptake of F-Aβ Proteins
PC12 cells stably expressing m-CFP fused caveolin-1 (m-CFP/CAV1) were employed to examine the endosomal pathway involved in the internalization of F-Aβ42. In these cells, F-Aβ42 (Figure 2E–H) but not F-Aβ40 (Figure 2A–D) colocalized with m-CFP/CAV1 at various incubation times ranging between 60 and 240 min.
Figure 2.

A–D: Colocalization of F-Aβ42 and F-Aβ40 with caveolin-1 in PC12 cells. Differentiated PC12 cells stably expressing m-cherry fluorescent protein fused to caveolin-1 (m-CFP/CAV1) incubated with 3.5 μM F-Aβ40 for up to 4 h. Accumulation following incubations at (A) 60 min; (B) 120 min; (C) 180 min; (D) 240 min. E–H: Differentiated PC12 cells stably expressing m-CFP/CAV1 incubated with F-Aβ42 for up to 4 h. Intracellular accumulation of F-Aβ42 after (E) 60 min; (F) 120 min; (G) 180 min; and (H) 240 min incubation times. Scale bars = 25 μm.
In PC12 cells treated with MβCD (depletes membrane cholesterol required for maintaining the integrity of lipid rafts and also for caveolae formation) for 60 (Figure 3D) or 180 min (Figure 3E), F-Aβ42 remained mostly at the cell periphery but the untreated cells showed punctate deposition of F-Aβ42 in the perinuclear region (Figure 3C). Flow cytometry analysis showed lower accumulation of F-Aβ42 (Figure 3F) as well as AF633-Trf (Figure 3G) in MβCD treated PC12 cells than in the normal cells. Moreover, the Western blots of the immunoprecipitated lysates obtained from the PC12 cells incubated with F-Aβ42 for 15 or 30 min following a 1 h pretreatment with MβCD showed substantially lower F-Aβ42 uptake than the normal cells (Figure 3H).
Figure 3.

A–H: Uptake of F-Aβ proteins in methyl-β-cyclodextrin (10 mM) treated PC12 cells. (A, B) Confocal micrographs showing the uptake of F-Aβ40 (green) and AF633-Trf (red) in (A) normal PC12 cells and in (B) PC12 cells pretreated with 10 mM methyl-β-cyclodextrin (mβCD) for 60 min. (C–E) Confocal micrographs of F-Aβ42 (green) and AF633-Trf (red) treated PC12 cells. (C) Normal PC12 cells; (D) PC12 cells pretreated with 10 mM mβCD for 60 min; (E) PC12 cells pretreated with 10 mM mβCD for 180 min. (F, G) Flow cytometry analysis of (F) F-Aβ42 and (G) AF633-Trf in control and 10 mM mβCD treated cells. (H) Western blots of internalized Aβ42 in normal and mβCD treated PC12 cells. Aβ42 uptake following 15 min incubation in (i) normal PC12 cells and (ii) the PC12 cells pretreated with mβCD for 60 min. Aβ42 uptake following 30 min incubation in (iii) normal and (iv) mβCD treated PC12 cells. Scale bars = 25 μm.
GM1 Receptor and the Endocytosis F-Aβ42
Interestingly, the uptake of F-Aβ42 decreased in the presence of AF647-CT (Figure 4E–H), a GM1 receptor ligand, but is not affected by the AF633-Trf (Figure 4A–D). In addition to the microscopy images, the flow cytometry data also confirmed the inhibitory effect of AF647-CT on the F-Aβ42 uptake by PC12 cells (Figure 4II). In the presence of AF647-CT, the geometric mean (±coefficient of variance) of F-Aβ42 fluorescence intensity reduced from 487.4 ± 22.2 to 289.6 ± 18.3. As reported in our earlier publication, F-Aβ40 did not colocalize with either AF633-Trf or AF647-CT and neither did the markers impact F-Aβ40 uptake (Figure 4I).
Figure 4.

A–H: Inhibition of F-Aβ40 and F-Aβ42 uptake by Alexa Fluor labeled cholera toxin. (A–D) Uptake of F-Aβ42 (green) and AF633-Trf (red) in differentiated PC12 cells after 60 min of incubation. (A) F-Aβ42; (B) AF633-Trf; (C) overlay of images A and B; (D) differential interface contrast (DIC) image overlaid on image C. (E-H) Uptake of F-Aβ42 (green) and AF647-CT (red) in differentiated PC12 cells after 60 min of incubation. (E) F-Aβ42; (F) AF647-CT; (G) merge image of E and F; (H) DIC image is overlaid on the image G. (I, II) Flow cytometry analysis of differentiated PC12 cells incubated with either F-Aβ40 or F-Aβ42 along with AF647-CT. (I) Histograms of PC12 cells treated with F-Aβ40 and AF647-CT; (II) histograms of PC12 cells treated with F-Aβ42 and AF647-CT along with the comparison of geometric means of F-Aβ40 and F-Aβ42 fluorescence in control and AF647-CT treated cells. Scale bars = 25 μm.
Intracellular Itinerary of Aβ42 versus Aβ40
Early Endosomes
Like F-Aβ40 that has been described in our earlier studies,9 F-Aβ42 did not show any appreciable localization in the early endosomes labeled with AF633-Trf (Figure 3C).
Late Endosomes
When coincubated with Dil-LDL, a marker for late endosomes, F-Aβ42 showed remarkable colocalization as evidenced by the punctate yellow stain on the X–Z and Y–Z images of the PC12 cells incubated with the fluorophores for 60 min (Figure 5A). F-Aβ42 exhibited a similar trend following 30 min incubation with the Dil-LDL (Figure 5B), while F-Aβ40 did not show colocalization with the marker under similar incubation conditions (Figure 5C).
Figure 5.

A–C: Confocal micrographs depicting the extent of colocalization of F-Aβ proteins with Dil-LDL in differentiated PC12 cells. (A) Z-Series image of F-Aβ42 colocalization with Dil-LDL, a late endosome marker, following 60 min incubation. (B) F-Aβ42 and Dil-LDL colocalization following 60 min incubation. (C) Absence of F-Aβ40 and Dil-LDL colocalization after 60 min incubation. Scale bars = 25 μm.
Lysosomes
Differentiated PC12 cells stably expressing m-CFP/LAMP1 were incubated with F-Aβ42 or F-Aβ40 to examine their lysosomal accumulation patterns for up to 180 min. F-Aβ42 showed distinct lysosomal accumulation (Figure 6A–E) whereas F-Aβ40 showed only a partial localization in the lysosomes (Figure 6F–J). Pearson’s correlation coefficients of F-Aβ42 colocalization with m-CFP/LAMP1 were greater than 0.6 even at incubation times as short as 30 min (Figure 6K). Pearson’s correlation coefficient 1 is considered as 100% colocalization. In contrast, F-Aβ40 was unable to colocalize with lysosomes as much as F-Aβ42 did during the initial time points. The Pearson’s correlation coefficients describing the colocalization of F-Aβ40 with lysosomal m-CFP/LAMP1 varied between 0.3 and 0.6, but increased above 0.6 only at later time points (Figure 6K).
Figure 6.

A–K: Time dependent lysosomal accumulation of F-Aβ proteins and subsequent changes in lysosomal integrity (scale bars = 25 μm). (A–E) Accumulation of F-Aβ42 in the lysosomes expressing m-cherry fluorescent protein fused LAMP-1 (m-CFP/LAMP1). (A) Lysosomal accumulation of F-Aβ42 after 60 min incubation. The inset of the cell body shows colocalization of F-Aβ42 with lysosomes; (B, C) lysosomal accumulation of F-Aβ42 after 120 min of incubation; (D, E) F-Aβ42 colocalized with lysosomes following 180 min of incubation. (F–J) Accumulation of F-Aβ40 in the lysosomes expressing m-CFP/LAMP1. (F) Intracellular accumulation of FAβ40 colocalized with lysosomes after 60 min of incubation; (G, H) after 120 min incubation; (I, J) after 180 min incubation. (K) Pearson’s correlation coefficients describing the colocalization of fluorescent proteins expressed in lysosomes plotted against time.
The damage caused to lysosomal integrity due to the accumulation of F-Aβ proteins was assessed by the following: tracking disrupted lysososmes that show up as red blotches due the leakage of lysosomal contents into cytoplasm; monitoring changes in the mean lysosomal areas; and evaluating vesicles larger than 300 (pixel)2 that are indicative of clumped lysosomes. More red blotches were found in the cells incubated with F-Aβ42 (Figure 7A–C) than those incubated with F-Aβ40 (Figure 7D–F). The areas of smaller lysosomal vesicles in FAβ42 or F-Aβ40 treated PC12 cells showed a gradual increase up to 60 min and reached a plateau thereafter (Figure 7G). But the appearance of clumped lysosomes increased linearly with time in F-Aβ42 treated cells but not in the cells treated with FAβ40 (Figure 7H).
Figure 7.

A–H: Detrimental effects of F-Aβ42 on the lysosomal integrity. (A–C) PC12 cells stably expressing m-CFP/LAMP1 incubated with 3.5 μM F-Aβ42 up to 180 min (scale bars = 25 μm). (A) Intravesicular accumulation of F-Aβ42 followed by 180 min of incubation; (B) lysosomes after 180 min of treatment with F-Aβ42; (C) merge image of A and B with yellow color indicating the colocalization, punctate regions with blue arrows indicating the lysosomes without F-Aβ42 and orange arrows indicating severely damaged lysosomes due to accumulation of F-Aβ42. (D–F) Effects of F-Aβ40 on the integrity of lysosomes (scale bars = 25 μm). PC12 cells stably expressing m-CFP/LAMP1 was incubated with F-Aβ40 for 180 min. (D) Intravesicular accumulation of F-Aβ40 followed by 180 min of incubation; (E) lysosomes after 180 min of incubation with F-Aβ40; (F) merge images of E and F. (G, H) changes in the areas of small lysosomal vesicles and large lysosomal vesicles most likely formed by the aggregation of smaller vesicles in PC12 cells treated with F-Aβ42 or F-Aβ40 for various lengths of time. (G) Small lysosomal vesicles change in area with time; (H) large lysosomal vesicles change in area with time.
DISCUSSION
While parenchymal amyloid plaques and intraneuronal tangles, the most visible pathological hall marks of AD, are considered as the mere downstream reflections of AD pathology,21 the neurodegeneration in AD is believed to be actually triggered by the intraneuronal accumulation of Aβ proteins. Understanding Aβ accumulation in the neurons is important so that treatments to prevent neurodegeneration could be identified. Endocytotic pathways involving α7 nicotinic acetylcholine (α7-NAch) receptor,22 N-methyl D-aspartate (NMDA) receptor,23 low density lipoprotein receptor-related protein 1 (LRP1)/apolipoprotein E (APOE),24–26 or receptor for advanced glycosylated end products (RAGE) were implicated in the neuronal uptake of soluble Aβ. In addition, nonsaturable and nonendocytotic uptake of both Aβ40 and Aβ42 in PC12 cells8 and in human neuroblastoma cells27 was also proposed. Such a confusing array of targets renders the discovery of therapeutic strategies to prevent intraneuronal Aβ accumulation untenable.
The reduction in the Aβ40:Aβ42 ratio in the AD brain, which correlates with the severity of neurodegeneration, makes it imperative to understand the relative interactions of each isoform with neurons.28 Many of the aforementioned studies were focused on the neuronal uptake of more amyloidogenic but less abundant Aβ42 and mostly ignored the interactions of Aβ40 with neurons. The concentration of Aβ40 in the brain at the onset of AD pathology is about 10 times greater than that of Aβ42. If the neuronal uptake of both isoforms is mediated by the same receptor, then Aβ40 can inhibit Aβ42 uptake substantially and alleviate the neurons of its toxicity. Alternatively, if Aβ40 and Aβ42 are internalized by neurons via different routes, then their uptake mechanisms must be carefully investigated before devising strategies to reduce their neuronal uptake.
We have been conducting extensive studies to resolve this dilemma and showed that Aβ40 selectively accumulates in a subpopulation of cortical or hippocampal neurons primarily by nonsaturable, energy independent, and nonendocytotic pathways but only to a minor extent via endocytosis.9 Specifically, these studies have demonstrated that the uptake of F-Aβ40 by rat primary hippocampal neurons or differentiated PC12 cells was not inhibited at 4 °C, which is indicative of energy independent uptake.9 In contrast, we now demonstrate that the uptake of F-Aβ42 by PC12 cells is inhibited at 4 °C, and hence is mediated by energy dependent endocytosis (Figure 1A,B). Like the rodent embryonic neurons, neuronal crest-derived PC12 cells are well studied and are widely employed in the investigation of various neurophysiological processes, including intraneuronal protein trafficking.29,30 It was also shown that the differentiation of PC12 cells with the nerve growth factor increases the expression of various receptors implicated in Aβ42 endocytosis to the levels observed in central nervous system derived neuronal cell lines.31,32 Our studies have clearly demonstrated that Aβ40 uptake mechanisms observed in PC12 cells are verifiable in primary rat embryonic cortical neurons.9
F-Aβ42 uptake in differentiated PC12 cells but not of F-Aβ40 occurs by a dynamin dependent endocytosis that is inhibited by dynasore (Figure 1), a potent inhibitor of dynamin involved in the scission of clathrin- and caveolin-coated vesicles as well as phagosomes.33 Energy-dependent endocytosis of F-Aβ42 could be mediated by clathrin, which is involved in the endocytosis of α7-NAch,22 LRP1/APOE,24–26 and NMDA receptors;23 caveolae; or independently of clathrin and caveolin. Saavedra et al.34 have questioned the involvement of clathrin coated pits and the role of α7NAch or LRP1/APOE in the neuronal uptake of Aβ42. In addition, they also concluded against the involvement of caveolae-mediated enodcytosis, because Aβ42 did not colocalize with caveolin 1. In our study, we have shown that Aβ42 and to a minor extent Aβ40 accumulated in the vesicles harboring mcherry/caveolin1 following 60–180 min incubation (Figure 2). This discrepancy in observations could be due to differences in the experimental techniques employed in both studies. Saavedra et al. used immunofluorescence methods to locate Aβ42 in the cell. The extensive processing steps involved in this method may not only extract small proteins such as Aβ42 from the cell but also facilitate their translocation within the cellular compartments. To eliminate these fixation artifacts, we employed live cell imaging coupled with genetic methods to establish the colocalization of Aβ42 with caveolin-1.
Furthermore, in the PC12 cells treated with mβCD, which extracts cholesterol from the cell membranes so that the lipid rafts are disrupted, we found a significant reduction in Aβ42 uptake while F-Aβ40 uptake was unaffected (Figure 3). Both Aβ40 and Aβ42 were shown to increase the annular and bulk fluidity of cholesterol rich cortical and hippocampal synaptic membranes, but not of the synaptic membranes obtained from the cerebellum that are low in cholesterol.35,36 It could be inferred from these reports that cholesterol depletion may restrict F-Aβ40 and F-Aβ42 interactions with the cell membranes. However, reduction of only F-Aβ42 uptake in mβCD treated cells suggests that the uptake differences are most likely caused by the disruption of endocytotic processes that are exclusively accessed by F-Aβ42, rather than the biophysical changes in the cell membrane, which could affect both F-Aβ40 and F-Aβ42 uptake.
Additionally, the uptake of F-Aβ42 but not of F-Aβ40 was reduced in the presence of cholera toxin, a GM1 receptor ligand localized in the lipid rafts (Figure 4). The caveolin-1 is closely associated with lipid rafts in the neurons where it forms scaffolds to coordinate membrane proteins and regulate cell signaling and exists as caveolar vesicles that mediate endocytosis. Hence, the colocalization of F-Aβ42 with caveolin-1 and the critical role played by the lipid rafts in FAβ42 endocytosis suggest that F-Aβ42 is internalized by PC12 cells via caveolae mediated endocytosis after binding to the GM1 receptor localized in the lipid rafts (Figure 4).
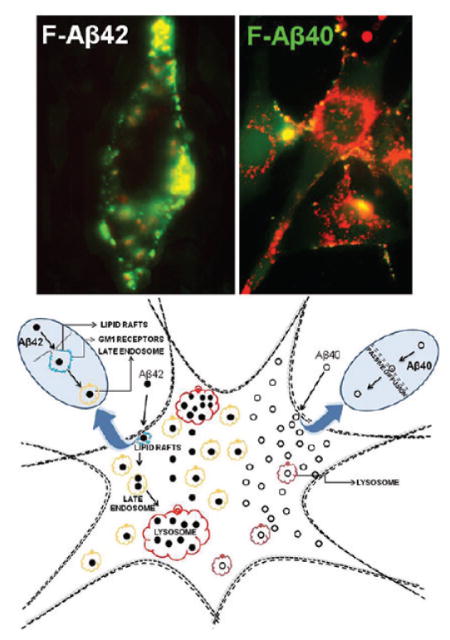
These observations suggest that neurons internalize Aβ40 and Aβ42 via distinct mechanisms. After internalization, a major portion of F-Aβ40 was shown to spread diffusely throughout the cytoplasm, whereas F-Aβ42 was found to accumulate mostly in multivesicular bodies and lysosomes.37–39 At low lysosomal pH, Aβ42 reportedly forms insoluble aggregates and leads to the lysosomal disruption. It was even hypothesized that the insoluble fibrils released into the brain extracellular space after the death of the distressed neurons act as nidus for amyloid plaque formation in the AD brain. Some investigators opined that Aβ40 does not have a similar impact on the lysosomal integrity because of its lower propensity to aggregate and greater susceptibility to lysosomal proteases. Nevertheless, Aβ40 was found to aggregate as quickly as Aβ42 at the late endosomal/lysosomal pH ~5.40 Moreover, neurons are exposed to several fold higher Aβ40 concentrations present in the brain extracellular space than that of Aβ42. If Aβ40 could reach lysosomal compartment to the same extent as that of Aβ42, the proteosomal susceptibility of Aβ40 could be offset by the magnitude of its accumulation in the lysosomes. Therefore, it is important to investigate the intracellular pharmacokinetics of Aβ40 and Aβ42 following their uptake at the plasma membrane and evaluate their tendency to accumulate in various cellular compartments that are susceptible to Aβ toxicity.
In the PC12 cells incubated with F-Aβ40 or F-Aβ42 for 60 min, neither isoforms showed appreciable localization in the early/recycling endosomes. Following 30 or 60 min incubation, F-Aβ42 but not F-Aβ40 accumulated in the late endosomes labeled with Dil-LDL (Figure 5), a late endosomal marker. From the late endosomes, F-Aβ42 emptied into lysosomes stably expressing mcherry/LAMP1 and the colocalization with the mcherry signal increased substantially between 30 and 180 min. On the other hand, F- Aβ40 exhibited only a partial localization in the lysosomes (Figure 6). Upon accumulation, F-Aβ42 caused more damage to lysosomes than F-Aβ40. However, it is hard to ignore the correlation between the extent of lysosomal damage and the magnitude of F-Aβ accumulation in the lysosomes (Figure 7). In F-Aβ42 or FAβ40 treated PC12 cells, only those lysosomes that harbored the proteins were disrupted and lysosomes devoid of any green fluorescence that signifies the absence of F-Aβ accumulation remained intact. Notably, intact lysosomes are more prevalent in F-Aβ40 treated cells than in F-Aβ42 treated cells, which is most likely due to the inability of F-Aβ40 to accumulate in the lysosomes as much as F-Aβ42.
In summary, this study outlines differences between the mechanisms of uptake and intracellular itinerary of Aβ42 and Aβ40 following their acute exposure in differentiated PC12 cells, whereas the cortical and hippocampal neurons in AD patients are subjected to chronic exposure of Aβ proteins at only 0.1–2% of the concentrations used in this study.41 Even under the chronic exposure, the endosomal–lysosmal system in the neurons is disrupted,42 thereby suggesting that the primary mechanisms through which Aβ proteins are internalized by the neurons and exhibit their toxicity are most likely to be the same with acute or chronic exposures. Hence, the observations made in the current in vitro study could be extrapolated to understand pathophysiological processes driving neurodegeneration in AD patients and transgenic animals in vivo.
By maintaining a mechanism of uptake and cellular pharmacokinetics different from Aβ42, the question arises as to whether Aβ40 plays a protective role in counteracting the toxic effects of Aβ42 on the neurons. Or is Aβ40 just a less toxic version of Aβ42? If the former is true, an ideal drug intended to treat neurodegeneration in AD is expected to specifically target neuronal uptake of Aβ42 without interfering with Aβ40. If the latter scenario were to be true, then the potential drug would be expected to inhibit the neuronal uptake of both Aβ42 and Aβ40. More studies are being conducted in our lab to clarify these questions.
Acknowledgments
The authors acknowledge the financial assistance provided by Alzheimer’s Association Grant NIRG-09-133017 (K.K.K.) and NIH/NCRR/RCMI Grant G12RR03020 (K.K.K.). The sponsors had no role in study design; data collection, analysis, and interpretation; and played no role in the decision to submit this paper for publication.
Footnotes
The authors declare no competing financial interest.
References
- 1.Christensen D, Kraus S, Flohr A, Cotel M-C, Wirths O, Bayer T. Transient intraneuronal Aβ rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol. 2008;116(6):647–655. doi: 10.1007/s00401-008-0451-6. [DOI] [PubMed] [Google Scholar]
- 2.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-[beta] in Alzheimer’s disease. Nat Rev Neurosci. 2007;8(7):499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 3.Wirths O, Multhaup G, Bayer TA. A modified β-amyloid hypothesis: intraneuronal accumulation of the β-amyloid peptide—the first step of a fatal cascade. J Neurochem. 2004;91(3):513–520. doi: 10.1111/j.1471-4159.2004.02737.x. [DOI] [PubMed] [Google Scholar]
- 4.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156(1):15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. A[beta] Immunotherapy Leads to Clearance of Early, but Not Late, Hyperphosphorylated Tau Aggregates via the Proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Jicha GA. Is passive immunization for Alzheimer’s disease ‘alive and well’ or ‘dead and buried’? Expert Opin Biol Ther. 2009;9(4):481–491. doi: 10.1517/14712590902828285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poduslo JF, Gilles EJ, Ramakrishnan M, Howell KG, Wengenack TM, Curran GL, Kandimalla KK. HH domain of Alzheimer’s disease Abeta provides structural basis for neuronal binding in PC12 and mouse cortical/hippocampal neurons. PLoS One. 2010;5(1):e8813. doi: 10.1371/journal.pone.0008813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdick D, Kosmoski J, Knauer MF, Glabe CG. Preferential adsorption, internalization and resistance to degradation of the major isoform of the Alzheimer’s amyloid peptide, A beta 1–42, in differentiated PC12 cells. Brain Res. 1997;746(1–2):275–284. doi: 10.1016/s0006-8993(96)01262-0. [DOI] [PubMed] [Google Scholar]
- 9.Kandimalla KK, Scott OG, Fulzele S, Davidson MW, Poduslo JF. Mechanism of neuronal versus endothelial cell uptake of Alzheimer’s disease amyloid beta protein. PLoS One. 2009;4(2):e4627. doi: 10.1371/journal.pone.0004627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan Y, Wang C. A[beta]40 Protects Non-toxic A[beta]42 Monomer from Aggregation. J Mol Biol. 2007;369(4):909–916. doi: 10.1016/j.jmb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. A{beta}40 Inhibits Amyloid Deposition In Vivo. J Neurosci. 2007;27(3):627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens Kl, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum Mutat. 2006;27(7):686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- 13.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, DeLucia M, Lin W-L, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. A[beta]42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron. 2005;47(2):191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zou K, Kim D, Kakio A, Byun K, Gong J-S, Kim J, Kim M, Sawamura N, Nishimoto S-i, Matsuzaki K, Lee B, Yanagisawa K, Michikawa M. Amyloid β-protein (Aβ)1–40 protects neurons from damage induced by Aβ1–42 in culture and in rat brain. J Neurochem. 2003;87(3):609–619. doi: 10.1046/j.1471-4159.2003.02018.x. [DOI] [PubMed] [Google Scholar]
- 15.Shoji M. Cerebrospinal fluid Abeta40 and Abeta42: natural course and clinical usefulness. Front Biosci. 2002;7:d997–1006. doi: 10.2741/A826. [DOI] [PubMed] [Google Scholar]
- 16.Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, Blennow K. Prediction of Alzheimer’s Disease Using the CSF Aβ42/Aβ40 Ratio in Patients with Mild Cognitive Impairment. Dementia Geriatr Cognit Disord. 2007;23(5):316–320. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- 17.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman MLC, Staufenbiel M, Mathews PM, Jucker M. A[beta] is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7(9):954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- 18.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human Apolipoprotein E4 Alters the Amyloid-β 40:42 Ratio and Promotes the Formation of Cerebral Amyloid Angiopathy in an Amyloid Precursor Protein Transgenic Model. J Neurosci. 2005;25(11):2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein WL. A[beta] toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41(5):345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 20.Poduslo JF, Ramakrishnan M, Holasek SS, Ramirez-Alvarado M, Kandimalla KK, Gilles EJ, Curran GL, Wengenack TM. In vivo targeting of antibody fragments to the nervous system for Alzheimer’s disease immunotherapy and molecular imaging of amyloid plaques. J Neurochem. 2007;102(2):420–33. doi: 10.1111/j.1471-4159.2007.04591.x. [DOI] [PubMed] [Google Scholar]
- 21.Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol. 2008;172(6):1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110(2):199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 23.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109(5):813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 24.Fuentealba RA, Liu Q, Zhang J, Kanekiyo T, Hu X, Lee JM, LaDu MJ, Bu G. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Abeta42 uptake and lysosomal trafficking. PLoS One. 2010;5(7):e11884. doi: 10.1371/journal.pone.0011884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G. Neuronal cell death in Alzheimer’s disease correlates with apoE uptake and intracellular Abeta stabilization. J Clin Invest. 1997;100(2):310–320. doi: 10.1172/JCI119536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281(47):36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 27.Morelli L, Prat MI, Castano EM. Differential accumulation of soluble amyloid beta peptides 1–40 and 1–42 in human monocytic and neuroblastoma cell lines. Implications for cerebral amyloidogenesis. Cell Tissue Res. 1999;298(2):225–232. doi: 10.1007/s004419900109. [DOI] [PubMed] [Google Scholar]
- 28.Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, Vandersteen A, Segers-Nolten I, Van Der Werf K, Subramaniam V, Braeken D, Callewaert G, Bartic C, D’Hooge R, Martins IC, Rousseau F, Schymkowitz J, De Strooper B. Neurotoxicity of Alzheimer’s disease A[beta] peptides is induced by small changes in the A[beta]42 to A[beta]40 ratio. EMBO J. 2010;29(19):3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Sweeney D, Greengard P, Gandy S. Metabolism of Alzheimer beta-amyloid precursor protein: regulation by protein kinase A in intact cells and in a cell-free system. Proc Natl Acad Sci USA. 1996;93(9):4081–4084. doi: 10.1073/pnas.93.9.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuda M, Yamamoto A. Effect of forskolin on synaptotagmin IV protein trafficking in PC12 cells. J Biochem. 2004;136(2):245–253. doi: 10.1093/jb/mvh116. [DOI] [PubMed] [Google Scholar]
- 31.Nery AA, Resende RR, Martins AH, Trujillo CA, Eterovic VA, Ulrich H. Alpha 7 nicotinic acetylcholine receptor expression and activity during neuronal differentiation of PC12 pheochromocytoma cells. J Mol Neurosci. 2010;41(3):329–339. doi: 10.1007/s12031-010-9369-2. [DOI] [PubMed] [Google Scholar]
- 32.Bu G, Sun Y, Schwartz AL, Holtzman DM. Nerve Growth Factor Induces Rapid Increases in Functional Cell Surface Low Density Lipoprotein Receptor-related Protein. J Biol Chem. 1998;273(21):13359–13365. doi: 10.1074/jbc.273.21.13359. [DOI] [PubMed] [Google Scholar]
- 33.Abban CY, Bradbury NA, Meneses PI. HPV16 and BPV1 infection can be blocked by the dynamin inhibitor dynasore. Am J Ther. 2008;15(4):304–311. doi: 10.1097/MJT.0b013e3181754134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saavedra L, Mohamed A, Ma V, Kar S, de Chaves EP. Internalization of beta-amyloid peptide by primary neurons in the absence of apolipoprotein E. J Biol Chem. 2007;282(49):35722–35732. doi: 10.1074/jbc.M701823200. [DOI] [PubMed] [Google Scholar]
- 35.Chochina SV, Avdulov NA, Igbavboa U, Cleary JP, O’Hare EO, Wood WG. Amyloid β-peptide1–40 increases neuronal membrane fluidity: role of cholesterol and brain region. J Lipid Res. 2001;42(8):1292–1297. [PubMed] [Google Scholar]
- 36.Avdulov NA, Chochina SV, Igbavboa U, O’Hare EO, Schroeder F, Cleary JP, Wood WG. Amyloid β-Peptides Increase Annular and Bulk Fluidity and Induce Lipid Peroxidation in Brain Synaptic Plasma Membranes. J Neurochem. 1997;68(5):2086–2091. doi: 10.1046/j.1471-4159.1997.68052086.x. [DOI] [PubMed] [Google Scholar]
- 37.Shirwany NA, Payette D, Xie J, Guo Q. The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr Dis Treat. 2007;3(5):597–612. [PMC free article] [PubMed] [Google Scholar]
- 38.Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA. 2009;106(47):20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almeida CG, Takahashi RH, Gouras GK. beta-Amyloid Accumulation Impairs Multivesicular Body Sorting by Inhibiting the Ubiquitin-Proteasome System. J Neurosci. 2006;26(16):4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overly CC, Lee KD, Berthiaume E, Hollenbeck PJ. Quantitative measurement of intraorganelle pH in the endosomal-lysosomal pathway in neurons by using ratiometric imaging with pyranine. Proc Natl Acad Sci USA. 1995;92(8):3156–3160. doi: 10.1073/pnas.92.8.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Aβ40 Inhibits Amyloid Deposition In Vivo. J Neurosci. 2007;27(3):627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belinson H, Lev D, Masliah E, Michaelson DM. Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J Neurosci. 2008;28(18):4690–4701. doi: 10.1523/JNEUROSCI.5633-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]