

Abstract
A major development in drug addiction research in recent years has been the discovery that immune signaling within the central nervous system contributes significantly to mesolimbic dopamine reward signaling induced by drugs of abuse, and hence is involved in the presentation of reward behaviors. Additionally, in the case of opioids, these hypotheses have advanced through to the discovery of the novel site of opioid action at the innate immune pattern recognition receptor Toll-like receptor 4 as the necessary triggering event that engages this reward facilitating central immune signaling. Thus, the hypothesis of major proinflammatory contributions to drug abuse was born. This review will examine these key discoveries, but also address several key lingering questions of how central immune signaling is able to contribute in this fashion to the pharmacodynamics of drugs of abuse. It is hoped that by combining the collective wisdom of neuroscience, immunology and pharmacology, into Neuroimmunopharmacology, we may more fully understanding the neuronal and immune complexities of how drugs of abuse, such as opioids, create their rewarding and addiction states. Such discoveries will point us in the direction such that one day soon we might successfully intervene to successfully treat drug addiction.
Keywords: Glia, cytokine, chemokine, innate immune, Toll-like Receptor
1. Drugs of abuse: where to begin?
The neurocircuitries that contribute to the adaptive and beneficial aspects of hedonia, learning, and memory are crucial for an organism's long-term survival. In contrast to activation of these multi-nuclei networked/patterned response systems by natural rewards (palatable food, salt, sex, etc.), several classes of foreign compounds (xenobiotics) are capable of directly “high-jacking” these systems, creating states of pharmacologically induced euphoria and reward. Repeated exposure to xenobiotics with these pharmacological properties can lead to neuronal and behavioral adaptations that produce states of addiction and dependence to the xenobiotic, which is self reinforcing leading to escalation of drug use, and in the absence of drug produces states of withdrawal; hence these agents are collectively termed drugs of abuse. These abused xenobiotics have diverse structures and pharmacologies of both biologically-derived and fully synthetic origins. And yet, the action of many drugs of abuse converge on the mesolimbic dopamine reward pathway, in which exposure to these xenobiotics results in activation of the dopamine neurons projecting from the ventral tegmental area to the nucleus accumbens shell and/or elevation of extracellular dopamine within the nucleus accumbens shell itself (Ikemoto, 2007). These xenobiotic effects occur via bypassing classical adaptive signaling pathways and directly manipulating neurotransporter function, altering activation and inhibition pathways and vesicular displacement of neurotransmitters. Additional neuronal-mediated complexities to this system have also been observed (Laviolette et al., 2002; Vargas-Perez et al., 2009).
These xenobiotics can be from both legal and illicit drug origins, but, irrespective of origin, when these drugs are administered purely for their rewarding properties their use causes a profound social and economic burden on the individual and the community around them. The state of addiction and dependence leads the individual to repeatedly administer these xenobiotics, resulting in extensive exposure of the central nervous system to the parent xenobiotic and/or its metabolites. This xenobiotic exposure has a variety of consequences for the central nervous system, including neuronal adaptation and toxicity (Büttner, 2011; Cappon et al., 1998; Fantegrossi et al., 2008; Salazar et al., 2008; Tilleux and Hermans, 2007; Weber et al., 2006). These xenobiotic-induced alterations in central nervous system homeostasis often lead to increased allosteric load. Thus, in the absence of the xenobiotic, or when its pharmacological action is blocked, behavioral signs of dependence and relapse are precipitated. However, these responses are varied across abused xenobiotics, thus demonstrating specificity of adaptation (Hyman et al., 2006).
Owing to the profound, abundant neuronal actions of these abused xenobiotics, much of the research focus over several decades has been on understanding the neurocircuitry, neuronal receptors, intra-neuronal signaling pathways and neuronal-sensitization events that lead to the physiological state of addiction and dependence. However, in the past two decades, a trickle of manuscripts examining the non-neuronal central nervous system immune consequences of drugs of abuse has now swollen to a significant body of work. Initially, these studies reported correlative evidence of central nervous system proinflammation resulting from exposure to the drugs of abuse demonstrating key implications for neurotoxicity and disease progression associated with, for example, HIV infection (Coller and Hutchinson, 2012). However, more recently, this drug-induced activation of central immune signaling is now understood to contribute substantially to the pharmacodynamic actions of drugs of abuse, by enhancing the engagement of classical mesolimbic dopamine reward pathways and withdrawal centers. Thus the hypothesis of major proinflammatory contributions to drug abuse was formed through the unification of the collective wisdoms of neuroscience, immunology and pharmacology; hence, Neuroimmunopharamcology.
Such discoveries of central nervous system immune involvement in the mesolimbic dopamine reward pathway have significant implications for how we understand reward to be modulated in beneficial adaptive situations versus maladaptive pathogen- and xenobiotic-induced reward conditions. However, whilst exciting in its implications, the hypothesis of major proinflammatory contributions to drug abuse also presents a series of quandaries which have rightly been raised by the addiction neuroscience establishment. None are less critical than the question: How can proinflammatory immune signaling be involved in drug reward and addiction when we don’t like being sick?
Thus, the aim of this review is to introduce and review the literature of the central immunology targets of drugs of abuse, highlighting the common mediators and mechanisms and the exciting opportunities these new targets have in identifying ‘at risk’ individuals and novel therapeutic opportunities. Additionally, some of the key conceptual and intellectual stumbling blocks that have impeded the proinflammatory hypothesis of drug abuse will be highlighted and explained in detail. Finally, the future of addiction research through “Neuroimmunopharmacology tinted glasses” will be surveyed to paint a picture of what the future of addiction research might hold.
Owing to the breadth of xenobiotics abused it will be difficult to cover all the developments in Neuroimmunopharmacology for each class. For a detailed review of these topics for abused xenobiotics where central immune signaling involvement has been established see our recent review (Coller and Hutchinson, 2012). Instead, here we will focus in this review on the evidence that we, and others, have generated over the past decade for opioid activation of central immune signaling and the impact this has on opioid reward and dependence.
2. An introduction to central immune signaling
Given that the hypothesis of major proinflammatory contributions to drug abuse requires both the knowledge of, and an appreciation for, neuroscience, immunology and pharmacology, a few key concepts need to be introduced in order to make this fascinating area optimally accessible. Firstly, the concept of immune-to-brain communication, which result in central immune signaling and subsequent altered behavior via neuronal-dependent adaptations will be examined.
It is very uncommon in an Immunology 101 course for any references to the central nervous system to be included, except perhaps when referring to immune involvement in neuroinflammatory diseases, such as Alzheimer’s and Multiple Sclerosis. The predominant focus of most basic immunology courses, and in fact the collective wisdom held by the general public, is that the immune system’s role is to defend the host organism from invading pathogens and to fight off infections. Whilst this host defense dogma is correct, the immune system has a far more nuanced role than we in western medicine and medical research currently give it credit for. The potential impact of peripheral immune cells, or immune signaling factors on brain function is commonly not discussed. However, this limited view of immune function is rapidly changing owing to a wealth of literature over more than 50 years.
Few of us will be unfamiliar with feeling sick at some point in our lives. But how do we feel sick and why don’t we like it? A standard systemic immune response to an insult such as endotoxin (lipopolysaccharide from gram negative bacteria) causes a profound alteration in behavior, termed sickness behavior or the illness response. The anhedonic qualities associated with the illness response have been well established in multiple domains such as animal husbandry as well as the clinic (Yirmiya et al., 2000). Clearly anhedonia is only one facet of the complex sickness response, which also includes lethargy, depression, anxiety, anorexia, heightened pain states (hyperalgesia and allodynia), and cognitive impairment (Dantzer et al., 1999). Many of these behaviors require significant central nervous system engagement, demonstrating that this peripheral immune response is capable of profoundly modifying behavior and thus must have the capacity to alter central nervous system function. These discoveries of immune-to-brain communication are a cornerstone of Psychoneuroimmunology (Besedovsky and Rey, 2007).
The cause of the altered behavior induced by illness had been postulated to be due to changes in metabolic reserves resulting from the full activation of the immense power, but energy hungry, immune system. But when it was discovered that a blood borne factor resulting from endotoxin exposure was capable of altering behavioral function without the need for endotoxin to cross the blood brain barrier (Holmes and Miller, 1963), the age of immune-to-brain signaling was born. These immune derived cytokines have been characterized to act by various humoral and neuronal routes to alter central nervous system neuronal function (Capuron and Miller, 2011; Miller et al., 2009). But this is not the only alteration in the brain during an illness response. The non-neuronal cells of the central nervous system, glia, also respond following a similar peripheral immune challenge (Laflamme and Rivest, 1999), causing the generation of proinflammatory cytokines and a myriad of other neuronal adaptations and sensitizations within the brain and spinal cord. Hence, illness-induced immune responses are capable of profoundly altering central nervous system function via multiple parallel routes.
3. But is this too non-specific? Are all brain nuclei altered to the same extent? How can this peripheral immune response cause a specific behavioral phenotype such as the illness response?
The key concepts of bioavailability, neuroanatomy and immune heterogeneity come to the forefront. Firstly, much of the central nervous system is protected from the primary factors of an invading organism, owing to the blood brain barrier, thus rendering factors, such as endotoxin, with poor central nervous system bioavailability and immediately restricting the triggers of central immune signaling. Secondly, discrete central nervous system nuclei are capable of responding to the immune challenge owing to their neural networks and structural characteristics. The circumventricular organs are sensitive to peripherally restricted factors like endotoxin and circulating cytokines, but likely choose this leaky blood brain barrier phenotype in order to sense peripheral challenges. The ascending sensory neural projects that sense peripheral immune responses via the sensory vagus nerve project to the nucleus of the tractus solitarius and area postrema and from there can secondarily recruit additional centers through specific, targeted pathways the activation of which leads to the generation of each element of the sickness response: fever, increased sleep, adipsia/aphagia, anhedonia, etc. Therefore, the majority of the central nervous system is not influenced by peripheral immune responses. Finally, even within these peripheral immune-influenced nuclei there is significant immune heterogeneity. Just as a dopamine neuron is as distinct in its neuroanatomical location, transcriptome and proteome from a serotonin neuron, it is apparent that the immune cells and immune signaling molecules of the central nervous system have similar regional specificity (Adler and Rogers, 2005). Thus, a peripheral immune response does not simply cause pan proinflammatory glial reactivity within the central nervous system. Rather discrete nuclei are sensitive to such immune stimuli and become activated (Laflamme and Rivest, 1999; Park et al., 2008). Moreover, the immune signals within the central nervous system may not be “inflammatory” per se, and rather can act at sub-inflammation levels functioning in a fashion akin to neurotransmitters (Adler and Rogers, 2005). Interestingly, the range of factors that have been identified as capable of triggering central immune signals has expanded to include endogenous danger signals (DAMPs; danger associated molecular patterns) that elicit responses from the innate immune system, such as via pattern recognition receptors (Buchanan et al., 2010).
By examining a combination of these neuronal and non-neuronal adaptations following peripheral illness, a series of hypotheses have been put forward to explain the altered behavior caused by the initial immune response. These include, but are not limited to, active transport of cytokines into CNS (Erickson et al., 2012),, induction of neuroinflammatory mediators in brain as a consequence of blood-borne cytokines (Dantzer et al., 2008), acute depletion of serotonin owing to microglial upregulation of indolamine-2,3-oxygenase which causes a direction of tryptophan away from serotonin synthesis and instead into the kynurenic acid pathway; and alterations in glutamate homeostasis due to proinflammatory signaling induced down regulation of astrocytic glutamate transporters which cause acute loss of extracellular glutamate control (Capuron and Miller, 2011; Miller et al., 2009; Yirmiya and Goshen, 2011a).
4. So where do drugs of abuse fit in and how is proinflammatory immune signaling within the central nervous system involved? How can proinflammatory immune signaling be involved in drug reward and addiction when we don’t like being sick? If illness is something we don’t enjoy shouldn’t illness counteract the rewarding pharmacodynamic actions of drugs of abuse?
Examples of alterations of central nervous system immunology by drugs of abuse began surfacing in the early 1990s with profound opioid-induced changes in glial cellular morphology and phenotypic receptor/immunohistological marker expression. For example, after long-term systemic morphine administration, a significant increase in astrocytic GFAP (glial fibrillary acidic protein; a cell surface marker of astrocyte reactivity) expression was observed in the ventral tegmental area but not in the substantia nigra, locus ceruleus, cerebral cortex, or spinal cord, thereby displaying significant regional heterogeneity (Beitner-Johnson et al., 1993), perhaps in part reflective of known heterogeneities in astrocytes (Chaboub and Deneen, 2012). Moreover, these opioid-induced differences in GFAP expression were strain specific (Beitner-Johnson et al., 1993). It is noteworthy that Beitner-Johnson et al., (1993) hypothesized a possible role for these opioid-induced GFAP adaptations in response to drugs of abuse, because they occurred primarily in the mesolimbic dopamine system and strain differences were correlated with preference for drugs of abuse. However, this line of research fell silent until the turn of the millennium.
Immune involvement, in general, was first demonstrated by Dafny and Dougherty et al. (Dafny et al., 1990; Dougherty et al., 1990) who found that several non-selective immunosuppressive treatments ameliorated morphine withdrawal behaviors in rats. However, these studies were conducted prior to an appreciation of the importance of glial cells in the behaviorally relevant functioning of the central nervous system. Additionally, concerns about the animals’ ability to display withdrawal behaviors under such generalized immunosuppressant regimens led to the end of the work (Berthold et al., 1989; Dantzer et al., 1987). However, one intriguing aspect to Dafny’s research was that his use of systemically administered immunosuppressants would likely have drastically altered immune-to-brain and central immune signaling capacities.
The first implication of central immune signaling modulation of morphine reward behaviors was accomplished by direct injection of astrocyte-conditioned medium (that likely contained important soluble co-factors such as cytokines and chemokines, but not endotoxin) into the nucleus accumbens (Narita et al., 2006). This caused heightened morphine conditioned place preference, a preclinical behavioral index of reward that relies upon Pavlovian conditioning of neutral environmental stimuli and pairing with the administration of a drug (Narita et al., 2006). Through the use of pharmacological proinflammatory glial attenuating drugs, this study (Narita et al., 2006), and others to follow, demonstrated that blockade of proinflammatory glial activation could: block morphine and oxycodone conditioned place preference (Hutchinson et al., 2009; Hutchinson et al., 2008b; Hutchinson et al., 2012; Narita et al., 2006); block drug-induced reinstatement of morphine conditioned place preference (Schwarz et al., 2011); attenuate remifentanil self administration (Hutchinson et al., 2012); block morphine and oxycodone spontaneous and precipitated withdrawal (Hutchinson et al., 2009; Hutchinson et al., 2010b), and simultaneously prevent brain increases or caused brain decreases of cytokines and chemokines in various brain regions shown to mediate withdrawal (e.g. in the ventral tegmental area and in the nucleus accumbens)(Hutchinson et al., 2009); and block morphine-induced elevations of dopamine in the nucleus accumbens (Bland et al., 2009; Hutchinson et al., 2012). Collectively, this body of work from multiple laboratories has entwined central immune signaling in the complexities of the mesolimbic dopamine reward neurocircuitry.
Despite opioid-induced central immune signaling being found to modify opioid-induced dopamine release in the nucleus accumbens, much of the prior and perhaps parallel events that lead to the elevated behavioral reward and dependence endpoints remain poorly characterized. Clearly, opioid-induced central immune signaling must have neuronal consequences in order to precipitate these changes in behavior. The neuronal consequence of opioid-induced proinflammatory immune signaling that have been characterized to date suggest that the response is dependent on proinflammatory-induced down-regulation of glutamate transporter expression, which can lead to a dysregulation of extracellular glutamate (Nakagawa et al., 2005; Nakagawa and Satoh, 2004; Ozawa et al., 2004; Ozawa et al., 2001; Wang et al., 2003), potentially contributing at minimum to opioid withdrawal (Nakagawa and Satoh, 2004). Additionally, proinflammatory cytokines themselves have been proposed as potential contributors to opioid reward (Coller and Hutchinson, 2012) via their neuroexcitatory effects (Watkins et al., 2009), GABA receptor downregulation (Stellwagen et al., 2005), upregulation of AMPA/NMDA expression and function (De et al., 2003), and enhancement of neurotransmitter release (Youn et al., 2008). The opioid-induced modulation of glial derived neurotrophic factors such as GDNF are also of significant interest owing to the established behavioral significance for GDNF on nucleus accumbens dopamine levels and the expression of reward behaviors (Airavaara et al., 2006; Airavaara et al., 2004). However, the significance and consequences of the regional heterogeneity of opioid-induced central immune signaling in the very early Beitner-Johnson et al. (1993) studies are at present unclear.
Whilst evidence for these major proinflammatory contributions to drug abuse has been presented here for morphine and similar opioids, similar central proinflammatory responses have been observed for a diverse range of abuse xenobiotics, such as alcohol, cocaine, methamphetamine, and 3,4-methylenedioxymethamphetamine (see Coller et al., (2012) for review). Interestingly, as time passes and the proinflammatory hypothesis of drug abuse becomes more widely known and accepted, increasing numbers of manuscripts have been published that transition this associative relationship between xenobiotic exposure and proinflammatory central immune responses, to a causative pharmacodynamic relationship. To date, evidence has now been published demonstrating central immune signaling involvement in behavioral responses to alcohol, cocaine, methamphetamine, and 3,4-methylenedioxymethamphetamine (Coller and Hutchinson, 2012). However, the mechanistic link between the xenobiotic-induced central immune signaling and altered mesolimbic dopamine reward neurocircuitry remains to be characterized and contrasted for the different classes of abused substances.
5. How do structurally diverse xenobiotics have a similar central immune proinflammatory phenotype but such diverse neuropharmacologies?
The apparent non-specificity of the activation of the central immune response has raised concerns that this event is a byproduct of direct neuronal actions of the drugs of abuse rather than a specific triggering of immune signaling and their activation pathways. Whilst this is a very valid hypothesis supported by generic neurotoxicity events triggered for example by xenobiotic-induced glutamate excitotoxicity, recent evidence suggests an entirely new site of xenobiotic action. Whilst first characterized for opioids, this new non-opioid receptor site of opioid action likely has implications for a very diverse range of xenobiotics, both of abused and non-abused pharmacologies (Coller and Hutchinson, 2012; Hutchinson et al., 2012; Hutchinson et al., 2011).
Opioid research has long focused on opioid ligand action at classical neuronal opioid receptors and their role in disinhibition of the mesolimbic dopamine reward pathway and modification of several other nuclei in the presentation of acute reward, development of dependence and presentation of withdrawal. Classical neuronal opioid receptors and their associated pharmacodynamic actions are well characterized as stereoselective, that is the naturally occurring (−)-isomer of opioid ligands has far greater activity than the unnatural, fully synthetic, (+)-isomer (Takagi et al., (1960). However, the early work of Takagi et al. (1960) demonstrated the pharmacodynamic relevance of non-classical non-stereoselective opioid actions. Since this early work, only a handful of papers have examined the pharmacological and behavioral significance of opioid actions at these non-stereoselective sites (Hutchinson et al., 2010a; Hutchinson et al., 2011; Hutchinson et al., 2008c). Several sites have been proposed, such as Filamin A (Burns and Wang, 2010; Wang et al., 2008) and NADPH oxidase (Wang et al., 2012a). However, our interest has focused on a fascinating class of innate immune receptors called Toll-like receptors (TLR), owing to their unique and diverse detection capacity and downstream signaling events, some of which had already been implicated in opioid pharmacodynamics. Interestingly, we have generated evidence that some of the other sites of potential action of opioids may lie downstream of TLR engagement increasing the importance of TLRs to opioid pharmacodynamics (Hutchinson et al., 2012).
As introduced previously, structurally diverse drugs of abuse, including opioids can be viewed as xenobiotics; that is, chemicals which are foreign to the organism. The body has a range of well-characterized systems to detect these foreign chemicals such as the liver’s pregnane X receptor (Matic et al., 2007). The question we posed was whether there were other receptors that would recognize chemicals as substances “foreign” to the central nervous system and trigger a central immune signaling response, and thus be the entity upstream of the proinflammatory central immune signals contributing to opioid reward and dependence behaviors.
Within the central nervous system, pattern recognition receptors, such as TLRs, can serve this sentinel role identifying “molecular patterns” as “non-self” or “danger” signals (Buchanan et al., 2010). These receptors are expressed by the innate immune system cells of the central nervous system, including endothelial cells, microglia, and some astrocytes, but rarely by adult neurons under nonpathological conditions (Buchanan et al., 2010; Chen and Nunez, 2010; Rivest, 2009). However, in some cases neuronal TLR expression has been reported (Li et al., 2009; Wadachi and Hargreaves, 2006), although the behavioral role of such expression is yet to be clarified. Unlike neuronal receptors for neurotransmitters, which display ligand selectivity and specificity, pattern recognition receptors have explicitly evolved to recognize multiple diverse conserved pathogen-associated molecular patterns (PAMPs) that are associated with microbial pathogens or cellular signals of danger or stress (danger-associated molecular patterns; DAMPs). Harking back to the original psychoneuroimmunology work in the 1960s, TLR4 has been characterized as the innate immune receptor responsible for detecting the presence of endotoxin (Hoshino et al., 1999). Binding of agonist ligands to TLR4 and its accessory molecules such as MD2 and CD14 activates downstream intracellular signaling pathways similar to those activated by interleukin-1β (O’Neill, 2008), and in fact belongs to the interleukin-1 receptor/TLR superfamily, inducing the production and release of proinflammatory, neuroexcitatory mediators via MyD88-dependent intracellular pathways (Yirmiya and Goshen, 2011b). Therefore, TLRs may play a pivotal sensing role for the innate immune system within the central nervous system by detecting the presence of xenobiotics, such as opioids, and translating their presence into a central immune signal.
Over recent years, supported by National Institute on Drug Abuse funding, we have examined the xenobiotic-mediated TLR4 actions of opioids using a collection of in vivo, in vitro, molecular and in silico strategies, demonstrating that various opioids, including morphine, activate TLR4 signaling (Hutchinson et al., 2012; Hutchinson et al., 2010b; Wang et al., 2012b) through binding to an accessory protein of TLR4, MD2, thereby inducing TLR4 oligomerization and triggering immune signaling within the central nervous system (Hutchinson et al., 2012; Wang et al., 2012b). With specific relevance to opioid reward we have demonstrated that pharmacological blockade of TLR4 or genetic deletion of MyD88-TLR4-dependent signaling suppresses opioid-induced conditioned place preference; reduces remifentanil self-administration and reduces morphine-induced elevations of extracellular dopamine in the nucleus accumbens (Hutchinson et al., 2012). Importantly, these actions of opioids at TLR4 do not produce a unidirectional influence on opioid pharmacodynamics, as blockade of opioid action at TLR4 or reduction of opioid-induced proinflammatory central immune signaling enhances acute and chronic opioid analgesia (Hutchinson et al., 2007; Hutchinson et al., 2008a; Hutchinson et al., 2010a; Hutchinson et al., 2009; Hutchinson et al., 2008b; Hutchinson et al., 2012; Hutchinson et al., 2011; Hutchinson et al., 2010b; Thomas and Hutchinson, 2012; Wang et al., 2012b). Thus we have established at minimum opioid-induced TLR4 as an initiating site of proinflammatory central immune signaling which occurs parallel to actions at opioid receptors, collectively creating the behavioral opioid phenotypic responses.
6. Whose fault is it, the parent or the “child”? The effect of drug metabolites on central immune signaling
In order for the body to clear xenobiotics from the system, they can undergo a myriad of biotransformations, often catalyzed by specific enzymatic reactions, such as those carried out by the families of Cytochrome P450s and glucuronyltransferases, just to name two. Traditionally, these reactions are considered bioinactivation steps that serve the dual purpose to also facilitate the hydrophilic clearance of the xenobiotic. However, in some cases the opposite is true and bioactivation can occur through the unmasking of an active functional group, thus metabolism of the parent xenobiotic gives birth to an active metabolite “child”, through which the pharmacological activity of the parent xenobiotic continues. This pharmacological activity of the metabolite can be equal to, or greater than the parent, and critically, can share the same, lose or gain pharmacological characteristics.
The main metabolites of morphine, morphine-3- and morphine-6-glucuronide are intriguing beasts. Owing to the established structure activity relationship of the morphine 4,5-epoxymorphian structure established with simpler structures such as codeine/morphine and hydrocodone/hydromorphone, one can immediately hypothesize that morphine-6-glucuronide will have maintained opioid activity, since the critical 3′ hydroxyl group is maintained, whilst morphine-3-glucuronide will have profound loss of opioid function (Chen et al., 1991). What is not anticipated is the newfound neuroexcitatory activity of morphine-3-glucuronide (Labella et al., 1979), which does not involve direct morphine-3-glucuronide activity at NDMA receptors (Hemstapat et al., 2009). Thus, despite extensive searchers for direct neuronal targets, the mechanism of morphine-3-glucuronide action remained unclear.
Until recently the acknowledgment of the immune modulatory capacity of opioids had been limited to their parent molecules, with little to no attention paid to the much longer lasting and abundant metabolites. We identified that morphine-3-glucuronide displayed profound TLR4 activity, greater than that of its parent morphine, and was capable of inducing in vitro microglial proinflammatory activation (isolated from the spinal cord) and release of Interleukin-1β, with similar in vivo effects also observed and noted to produce profound nociceptive TLR4-dependent behavioral consequences (Lewis et al., 2010). This discovery of immune-biasing of morphine via 3-glucuronidation has profound ramifications as it opens the door for other glucuronidated xenobiotics to follow a similar path to become recognized as xenobiotic associated molecular patterns (XAMPs) by pattern recognition receptors such as TLR4. The first extension of this hypothesis is born out in our recent work that demonstrates that a major ethanol metabolite, ethyl-glucuronide, possesses TLR4 activity and associated in vitro molecular and in vivo behavioral consequences (Lewis et al., 2013; Schäfer, 2013). How many other xenobiotics of either licit or illicit use, naturally occurring or in the Pharmacopoeia undergo similar immunobioactivation steps? Only time and further research will tell.
The reason why xenobiotic metabolism has such an immunobioactivation action may be found in early immunogenicity research of haptenation (Landsteiner, 1936). A hapten is a small chemical functional group, which alone possesses minimal immune activity, but when combined with a carrier molecule, such as a protein, now gains immunogenicity to which the immune system can respond. This strategy has been commercialized to create, for example, many of the antibodies used for immunohistochemistry or other research purposes today. Haptenization can be employed by the host organism to unmask the presence of an otherwise immunologically invisible parasite (Palm and Medzhitov, 2009). Interestingly, many of the haptens which were discovered early last century have been forgotten, instead only the most active agents are still employed to engender strong acquired immune antibody responses (Palm and Medzhitov, 2009). Of note is that glucuronidation is among the haptenation that Landsteiner (1936) characterized to possess immunogenicity. Recently, additional advancements in understanding the immune response to haptens was clarified by Palm and Medzhitov (2009), which has further implicated the innate immune system and TLRs in the complexities of hapten detection and subsequent innate and acquire immune responses. Therefore, could it be that biotransformation enzymes and the innate immune system are joining forces to detect the presence of an evolutionarily ancient foe, similar to TLR4 detection of endotoxin? This is an area with wider implications than the drug abuse field and disserves significant attention.
7. But hang on, we have just said endotoxin and hence TLR4 activation produces sickness behaviors and anhedonia. How can xenobiotic-induced TLR4 activation contribute to drug reward, but pathogen-induced TLR4 signaling causes sickness behaviors? Aren’t these contradictory?
Rewarding behaviors, such as the ingestion of palatable foods, are altered by sickness induced TLR4 signaling. This signaling is initiated out in the body as a consequence of pathogen/endotoxin recognition by TLR4-expressing peripheral immune cells. Alteration in palatable food intake is the behavioral culmination of the complex balance of both anhedonic and hedonic neurocircuitries (Baldo and Kelley, 2007; Berridge and Kringelbach, 2013). Importantly, a distinction here needs to be made when comparing the central immune signaling following administration of a xenobiotic of abuse and exposure to a peripheral immune stimulant like endotoxin or gram negative bacteria. Xenobiotics of abuse, unlike endotoxin or gram negative bacteria from which endotoxin is derived, can broadly access brain nuclei including those associated with drug reward owing to their penetrance across the blood brain barrier. Xenobiotics of abuse directly engage TLR4-dependent central immune signaling, rather than relying on indirect humoral and neuronal pathways activated as a consequence of strictly peripheral activation of TLR4. Thus a distinct pattern of activation should occur following CNS-penetrant xenobiotic exposure versus following peripherally-restricted endotoxin/gram negative bacteria, and hence each sums to produce substantially different behavioral responses.
Whilst, behaviorally, illness associated TLR4 signaling gives rise to reduction in palatable food intake, this effect is not solely caused by a reduction in the hedonic value of the food but is also due to combined influences of illness-induced aphagia, fatigue, anxiety and other illness associated outcomes as well (Borowski et al., 1998; Park et al., 2008). Therefore, even during peripherally triggered TLR4-dependent illness responses, were an ill organism to encounter a rewarding stimulus, activation of the reward neurocircuitries by such stimuli can still occur, despite the behavioral response appearing as anhedonia (Borowski et al., 1998; Park et al., 2008). A key example of this is the activation of the caudal nucleus accumbens by palatable food consumption under both control and peripheral endotoxin-induced sickness conditions (Park et al., 2008). The caudal nucleus accumbens is a key nuclei contributing to the positive hedonic value of numerous pharmacological and non-pharmacological stimuli (McBride et al., 1999). Importantly, the behavioral anhedonia reported by Park et al., (2008) likely resulted from endotoxin-induced activation of the rostral nucleus accumbens, an area linked with anhedonia (Berridge and Kringelbach, 2013; McBride et al., 1999).
But can a peripheral TLR4-dependent illness response or central immune signaling event alone engage any part of the reward neurocircuitry? This has been examined by Borowski et al., (1998), who demonstrated that systemic endotoxin (lipopolysaccharide; LPS) administration caused significant elevations in nucleus accumbens dopamine release, a neurochemical response classically associated with hedonic rewarding experiences, despite this being associated with decreased intracranial self stimulation on ascending but not descending rate-intensity functions. As nucleus accumbens dopamine release can also be induced by non-rewarding stimuli, like shock or other stressors, the interpretation of dopamine release in response to sickness remains to be defined.
However, given the fact that either central immune signaling alone (caused by centrally administered astrocyte conditioned media (Narita et al., 2006)) or TLR4 activation alone (produced by central endotoxin administration (Holmes and Miller, 1963)) fail to present behaviorally as rewarding experiences indicates, additional engagement of other reward pathways is require. And yet, as outlined previously, opioid reward in TLR4 null mutant mice, or when TLR4 is pharmacologically blocked by an antagonist, fail to produce behavioral reward (Hutchinson et al., 2012). Therefore, the two systems are interdependent and activation of both appears to be necessary to produce behavioral reward. However, the precise mechanism of this fascinating relationship requires further examination.
This does raise a quizzical point of methodological contention: does decreased intracranial self stimulation, palatable food consumption or self administration of a xenobiotic of abuse mirror discounting of their rewarding qualities thus rendering responses inherently less valued, or alternatively, does this reflect an increased rewarding value of the stimuli and hence reduced responses are required to reach hedonic satisfaction? Sure enough, examples of both interpretations can be found in the literature (Park et al., 2008; Ranaldi and Beninger, 1994), emphasizing the value of testing multiple preclinical models. However, future methodological complications presented by examining the role of central immune signaling in reward behavior and neurochemistry need to be examined carefully using multiple compatible methodologies. Collectively, these data open a fascinating window into the potential, and yet extremely complex relationship between illness responses and drug reward, and thus the likely involvement of TLR4 and central immune signaling in both.
Thus, opioids together with their newly identified TLR4 actions, affect the mesolimbic dopamine system to amplify opioid-induced elevations in extracellular dopamine levels, with especial attention being drawn to the caudal nucleus accumbens. This means that one could expect that activation of TLR4 or pharmacologically mimicking its downstream proinflammatory central immune signals would potentiate drug reward. This has already been demonstrated for alcohol (Blednov et al., 2011), and as discussed and reviewed previously (Coller and Hutchinson, 2012), blockade of TLR4 and related immune signaling pathways drastically alters the behavioral response to multiple drugs of abuse. However, why behaviorally alcohol consumption under these TLR4-burdened conditions presents as an increase in response rate, whilst palatable food consumption does not, needs greater examination acknowledging a role of the central immune signaling and reward neurochemistry events.
Thus, to reiterate, it is hypothesized that if proinflammatory central immune signaling is triggered in key central nervous system nuclei combined with activation of mesolimbic dopamine reward pathways the “perfect storm” of neuronal and immune signals are created to produce the full range of reinforcing signals that present behaviorally as drug reward (see schematic in Figure 1). However, it must be emphasized that a great deal of additional work is required to consolidate these hypotheses with established neural pathways and temporal signaling events.
Figure 1. Hypothesized central immune signaling contributions to opioid reward.
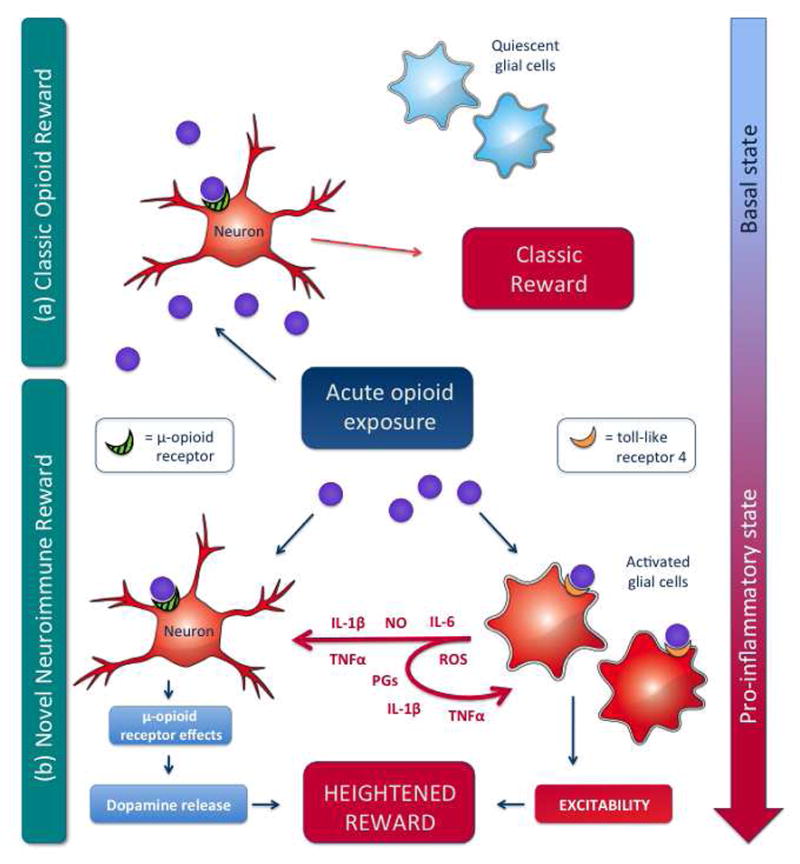
a) The classic view of opioid reward suggests that opioids bind to neuronal opioid receptors resulting in the eventual activation of the cortico-mesolimbic-dopamine reward pathway and the presentation of associated reward behaviors. In this model, glia, such as astrocytes and microglia, are not accounted for, beyond their homeostatic roles.
b) The recent appreciation of non-neuronal actions of opioids and opioid-induced proinflammatory glial activation necessitates additions to the solely neuronal hypotheses of opioid reward. The wealth of information of neuronal signaling and adaptations that occurs following opioid exposure and neuronal opioid receptor activation can now be complimented by the parallel TLR4-dependent activation of central immune signaling, and alteration of neuronal excitability due to a collection of opioid-TLR4 dependent glial adaptations. Collectively, the neuronal opioid receptor-dependent and complementary opioid-TLR4 dependent mechanisms produce a heightened opioid reward signal and ensuing presentation of altered behavior.
8. Do we have any human data to support the proinflammatory hypothesis of drug abuse?
Currently there are no clinically applicable pharmacological therapies that target the central nervous system immune signaling pathways. Even the range of immunomodulatory agents that are clinically prescribed have been designed to alter peripheral not central immune function and as such often have very poor central nervous system bioavailability. As such, pharmacological testing of this proinflammatory hypothesis is currently difficult, although promising data are on the horizon (www.clinicaltrials.gov; search ibudilast; studies NCT01740414 and NCT00723177). Nevertheless, given that up to 60% of drug dependence is heritable (Tsuang et al., 1998), if central immune signaling were a major contributor to drug abuse one would expect that genetic variability in immune pathways would be associated with drug dependent populations. Moreover, if the TLR4 immunobioactivation hypothesis above is correct, one would hypothesize that similar immunogenetic related traits would be associated with multiple drugs of abuse. Additionally, such a hypothesis would also aid in the scaling of the ladder of drugs of abuse from the perceived less harmful drugs of abuse to those whose consumption is profoundly detrimental.
To date we have examined functionally important single nucleotide polymorphisms in the Interleukin-1β gene and their association with dependence to opioid or alcohol in two separate populations. Here we positively linked −511C/T and −31T/C in both populations (Liu et al., 2009). Mechanistically how these genes agree with the central hypothesis of proinflammation linked to risk of dependence, the Interleukin-1B wild-type sequence at −511 and −31 is associated with increased Interleukin-1β release, hence it is hypothesized that carriers of the wild-type allele have greater central immune signaling and proinflammatory responses following opioid exposure (Liu et al., 2009) and hence greater risk of dependence which is supported by preclinical data (Liu et al., 2011). Examination of a wider range of genetic polymorphisms in immune genes is near completion with exciting implications for the identification of at risk individuals and the personalization of pharmacological dependence treatment to a central immune targeted therapy when immunogenetic screening indicates probable proinflammatory involvement in a case of drug abuse.
9. Can we pharmacologically or non-pharmacologically intervene to reduce drug abuse by targeting central immune signaling pathways?
Abstinence programs and substitution based maintenance therapies are commonly employed, but unfortunately are ineffective in the long run, and in some countries face stiff political opposition. Non-pharmacological therapies such as cognitive based therapy are widely used with some success. Thus the newly discovered role of central immune signaling in drug reward and dependence provides an exciting novel target to achieve a drug addiction treatment and supplement existing treatment regimens.
However, as mentioned previously, there are no specifically designed or developed pharmacotherapies for the targeted indication of intervening in a proinflammatory signaling event within the central nervous system, either one associated with drug abuse or any other neuroimmune pathology. However, several agents are in various stages of development. The relative success of cognitive based therapy in treating drug addiction is intriguing. Has the “talk and thought ” based therapy altered the hedonic and anhedonic neurocircuitry thereby breaking the maladaptive rewarding association with drugs of abuse? Or has the therapy strengthened other life skill coping mechanisms providing an alterative outlet other than drug abuse? Whilst not the focus of our research, a significant psychoneuroimmunology based literature of therapy modulation of immune function has developed. Could it be that “talk and thought ” based therapies have activity, at least in part due to their influence on brain-to-immune and immune-to-brain communication? This is a fascinating as yet unexplored hypothesis.
As we are still at the infancy of drug development pipelines, especially when examining the treatment of drug addiction it is opportune to raise several key questions. What should a new drug target and pharmacodynamically achieve if it were to intervene in this central immune signaling process associated with drug reward? Pan blockade of glial activation? Create an anti-inflammatory environment within the central nervous system, thus reducing active or limiting subsequent proinflammatory signals? Specifically target a key secondary signaling kinase(s)? Block a proinflammatory protein receptor or neutralize the molecule before it can act? Block the initial detection step the immune response? Should it be an antagonist, agonist, partial agonist or inverse agonist? Will it be able to reverse an established central immune signaling event and hence an established drug addiction? If multiple drugs of abuse are shown to rely on central immune signaling to produce their pharmacodynamic response should a pharmacological therapy be specific for a particular drug of abuse or capable of blocking multiple drugs of abuse?
Our preclinical investigations, and those of our peers, have focused on several pharmacological agents that have been opportunistically identified historically to have the right pharmacokinetic and pharmacodynamic profiles to attenuate, or block, various forms of central immune signaling. These include the exploitation of the anti-inflammatory properties of minocycline, propentofylline and more recently, ibudilast. In the cases where bioavailability issues, owing to hindrances such as the impermeability of the blood brain barrier, direct central administration has been employed. However, when looking to the future clinical application of pharmacotherapies and imagining the perfect drug to counter addiction a different profile immerges.
Oral bioavailability would be beneficial, with the option of sufficient functional potency to allow for the application of indwelling long-term delivery methods, such as subcutaneous rods or pellets to aid compliance in the traditionally non-compliant or treatment mandated populations. The drug’s pharmacokinetics would need to be sufficient to allow for once or at most twice daily administration, again to aid in the compliance of treatment. The best precise pharmacological target requires gazing into an as yet still cloudy crystal ball. Pan blockade of all glial functions, as occurs with glial metabolic inhibitors, is not plausible as this would produce catastrophic seizures owing to loss of homeostatic glial functions (Watkins and Maier, 2003). Pan blockade of glial activation, per se is not specific enough, as such activation may be beneficial in an anti-inflammatory or pro-inflammatory direction. Pro-inflammatory blockade by either neutralization of proinflammatory cytokine mechanisms or intracellular cascades may have unwanted off target consequences, as central immune signaling in all its diversity plays key roles in a myriad of normal health central nervous system functions. Creation of an anti-inflammatory environment within the central nervous system may be beneficial, but how would one direct this to the nuclei most in need for altering drug reward behaviors? No, in the case of opioids, we believe the best target to pharmacologically block lies at the first step of the central immune response, TLR4. The long-term safety consequences of such a treatment are unclear, but seem less harmful than the likely immunosuppression implications of pan blockade of immune responses generally or inhibition of pan monocyte-like cell function. In this case it is fortuitous that TLRs are evolutionarily and old system, thus much redundancy has been built around them. In fact, a significant portion of the population has a genetic deficiencies in TLR4 rendering them with diminished responsiveness, and yet these individuals thrive (eg Koch et al., (2011)). However, the association of similar genetic polymorphisms in drug abuse has yet to be examined.
To this end we have capitalized on our discovery that the (+)-isomers of opioid receptor antagonists, such as (+)-naloxone and (+)-naltrexone, which we have characterized as functional antagonists of the MD2/TLR4 activity of opioid ligands. Critically, (+)-isomers have little to no opioid receptor activity owing to the stereoselectivity of neuronal opioid receptors. To date we have established that (+)-naloxone and (+)-naltrexone are capable of the range of anti-opioid reward and dependence characteristics outlined previously, and yet maintain and in some cases potentiate acute beneficial opioid analgesia. These characteristics of reducing abuse liability of opioids whilst potentiating their indicated clinical utility suggest formulations of existing opioids with such drugs could beneficially impact the clinical safety and efficacy of the existing repertoire of opioid agonists.
An alternative future approach would be to design an opioid that either lacks TLR4 activity or actively blocks it in the one molecule. Interestingly, a novel opioid agonist, PTI-609, developed by Pain Therapeutics (Burns and Wang, 2010), that combines filamin A inhibitor functions and is also capable of reduced TLR4 signaling has been found in animal models to engender limited conditioned place preference, whilst maintaining opioid analgesia. Collectively, these are key data demonstrating that the beneficial analgesic actions of opioids can be separated from the unwanted rewarding properties, by either co-administration of glial-targeted activation inhibitors or rationale drug design to avoid xenobiotic-induced activation of central immune signaling. Further refinement of these approach promises great things in the efforts for improved drug abuse treatment and avoiding drug abuse liability of clinically diverted agents.
10. What does “glial activation” actually mean?
At this juncture, it is appropriate to note that the phrase “glial activation”, one which we have used on numerous occasions, is no longer sufficiently specific enough to render it useful in describing the responses of the immunocompetent cells of the central nervous system. Nor is it sufficient to class glially targeted drugs as glial activation inhibitors or attenuators. Rather we propose the use of the phrase “central immune signaling” in the future. The reason for this change is terminology is that it is quite apparent that whilst glia are the predominant and classically held immune-like cells of the central nervous system, who are the probable sources of many central immune signals, they are by no means the only origins of these signals, nor may they be the key population that affects a pharmacodynamic response. There is evidence that nearly every cell of the central nervous system, either resident like neurons, oligodendrocytes and blood brain barrier endothelial cells, or transient populations such as migrating peripheral immune cells are all capable of contributing and modifying this central immune signal. Also residing at the heart of this change in terminology is the acknowledgment that whilst glia are the new kids on the block and disserve scrutiny, the established neuronal networks must be tested for their non-classical immune signaling contributions to the proinflammatory hypothesis of drug addiction and other neuroimmune related pathologies.
11. Viewing addiction through Neuroimmunopharmacology tinted glasses: what does the future hold?
“Very lame and imperfect theories are sufficient to suggest useful experiments which serve to correct those theories and give birth to others more perfect. These, then occasion farther experiments which bring us still nearer to the truth; and in this method of approximation we must be content to proceed, and we ought to think ourselves happy if, in this slow method, we make any real progress”.
Joseph Priestley (1733–1804)
The discoveries and their associated hypotheses outlined here for the proinflammatory hypothesis of addiction have been built on the established wealth of addiction neuroscience, with a touch of re-examination and re-experimentation. However, as beautifully put by Priestley, even the hypotheses here are imperfect and require further refinement and sharpening. Nevertheless, the groundswell of supporting evidence for further Neuroimmunopharmacological investigations into drug addiction mechanisms is immense.
In order to capitalize on these exciting developments, predominantly in the preclinical domain, every effort should be made to translate them to the clinical setting. The development, testing and approval of new central immune signaling targeted pharmacotherapies is a key step in causally implicating these hypothesized mechanisms in drug reward and abuse, and therefore moving forward to more efficacious treatments. Less expensive and mechanistically critical studies to characterize immunological predispositions to drug abuse should also be examined, such as the immunogenetic differences between drug abuse and healthy populations.
Examination of the full clinical landscape of drug dependent populations and those at high risk of drug abuse should be fully assessed in the future for factors that may be associated with enhanced central immune signaling. For example, exciting developments in examining early life stressors that trigger later life drug abuse propensity have begun to mechanistically implicate central immune signaling (Schwarz and Bilbo, 2013; Schwarz et al., 2011), and can be reversed by immune targeted pharmacological interventions. Another example would be to minimize or eliminate chronic peripheral inflammation that may facilitate detrimental drug-reward enhancing central immune signaling. Liver damage, associated with a life of substance abuse, left untreated during an addiction treatment regimen may maintain central immune signaling patterns in their drug abuse primed states. Our recent clinical work developing a novel clinical neuroimmune model of pain by combining intravenous endotoxin and intradermal capsaicin (Hutchinson et al., 2013) demonstrates that modeling of these complex neuroimmune interactions is possible in healthy volunteer patients. Many other such systems level changes may prove cumulatively beneficial in normalizing peripheral immunology thus minimizing the detrimental impact of central immune signals and their action in drug abuse.
On the flip side of our efforts to understand the mechanisms of drug reward and addiction that lead to drug abuse, we need to be mindful of the efforts by some to increase the rewarding actions of existing and novel drugs of abuse through targeting these central immune system signaling pathways. As we have yet to fully appreciate the complexity of this multi system response this concern is not on our horizon, but legislative efforts should be at the ready to intervene and to provide the disincentives to parties who may wish to exploit this route.
In closing, appreciating the existence and impact of xenobiotic-induced central immune signaling combined with the established knowledge of neuronal actions of drug of abuse is key to fully appreciate the complete pharmacodynamic actions of multiple drugs of abuse. Whilst our research to date has focused extensively on opioids, glimpses of data suggest this area could be a hot bed of activity for multiple drugs of abuse. We hope in the near future that such an appreciation has a meaningful impact on avoiding drug addiction the treatment of dependent populations.
Highlights.
Central immune signaling contributes significantly to reward created by multiple drugs of abuse.
Toll-like receptor 4 is capable of triggering proinflammatory reward facilitating signals that are necessary for reward behaviors.
Pharmacological interventions targeting central immune signaling may prove to be crucial in future drug addiction interventions.
Acknowledgments
This work was supported in part by NIH Grants DA023132, DA024044, and DE017782 and an Australian Research Council Research Fellow (DP110100297)
Abbreviations
- TLR
Toll-like receptor
- LPS
Lipopolysaccharide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dr Mark R Hutchinson, Discipline of Physiology, School of Medical Sciences, University of Adelaide, Adelaide, South Australia, Australia, 5005.
Prof Linda R Watkins, Department of Psychology & Neuroscience, University of Colorado at Boulder, Boulder, Colorado, USA 80309.
References
- Adler MW, Rogers TJ. Are chemokines the third major system in the brain? J Leukoc Biol. 2005;78:1204–1209. doi: 10.1189/jlb.0405222. [DOI] [PubMed] [Google Scholar]
- Airavaara M, Mijatovic J, Vihavainen T, Piepponen TP, Saarma M, Ahtee L. In heterozygous GDNF knockout mice the response of striatal dopaminergic system to acute morphine is altered. Synapse. 2006;59:321–329. doi: 10.1002/syn.20245. [DOI] [PubMed] [Google Scholar]
- Airavaara M, Planken A, Gäddnäs H, Piepponen TP, Saarma M, Ahtee L. Increased extracellular dopamine concentrations and FosB/DeltaFosB expression in striatal brain areas of heterozygous GDNF knockout mice. Eur J Neurosci. 2004;20:2336–2344. doi: 10.1111/j.1460-9568.2004.03700.x. [DOI] [PubMed] [Google Scholar]
- Baldo BA, Kelley AE. Discrete neurochemical coding of distinguishable motivational processes: insights from nucleus accumbens control of feeding. Psychopharmacology (Berl) 2007;191:439–459. doi: 10.1007/s00213-007-0741-z. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Guitart X, Nestler EJ. Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fischer strain differences in the rat ventral tegmental area. J Neurochem. 1993;61:1766–1773. doi: 10.1111/j.1471-4159.1993.tb09814.x. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Kringelbach ML. Neuroscience of affect: brain mechanisms of pleasure and displeasure. Curr Opin Neurobiol Epub. 2013 doi: 10.1016/j.conb.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold H, Borel JF, Flückiger E. Enigmatic action of ciclosporine A on the naloxone-precipitated morphine withdrawal syndrome in mice. Neuroscience. 1989;31:97–103. doi: 10.1016/0306-4522(89)90032-8. [DOI] [PubMed] [Google Scholar]
- Besedovsky HO, Rey AD. Physiology of psychoneuroimmunology: a personal view. Brain Behav Immun. 2007;21:34–44. doi: 10.1016/j.bbi.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Bland ST, Hutchinson MR, Maier SF, Watkins LR, Johnson KW. The glial activation inhibitor AV411 reduces morphine-induced nucleus accumbens dopamine release. Brain Behav Immun. 2009;23:492–497. doi: 10.1016/j.bbi.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011;25:S92–S105. doi: 10.1016/j.bbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski T, Kokkinidis L, Merali Z, Anisman H. Lipopolysaccharide, central in vivo biogenic amine variations, and anhedonia. Neuroreport. 1998;9:3797–3802. doi: 10.1097/00001756-199812010-00006. [DOI] [PubMed] [Google Scholar]
- Buchanan MM, Hutchinson MR, Watkins LR, Yin H. Toll-like receptor 4 in CNS pathologies. J Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns LH, Wang HY. PTI-609: A Novel Analgesic that Binds Filamin A to Control Opioid Signaling. Recent Patents CNS Drug Discov. 2010;5:210–220. doi: 10.2174/157488910793362386. [DOI] [PubMed] [Google Scholar]
- Büttner A. Review: The neuropathology of drug abuse. Neuropathol Appl Neurobiol. 2011;37:118–134. doi: 10.1111/j.1365-2990.2010.01131.x. [DOI] [PubMed] [Google Scholar]
- Cappon GD, Morford LL, Vorhees CV. Enhancement of cocaine-induced hyperthermia fails to elicit neurotoxicity. Neurotoxicol Teratol. 1998;20:531–535. doi: 10.1016/s0892-0362(98)00004-x. [DOI] [PubMed] [Google Scholar]
- Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–238. doi: 10.1016/j.pharmthera.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaboub LS, Deneen B. Developmental origins of astrocyte heterogeneity: the final frontier of CNS development. Dev Neurosci. 2012;34:379–388. doi: 10.1159/000343723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZR, Irvine RJ, Somogyi AA, Bochner F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991;48:2165–2171. doi: 10.1016/0024-3205(91)90150-a. [DOI] [PubMed] [Google Scholar]
- Coller JK, Hutchinson MR. Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther. 2012;134:219–245. doi: 10.1016/j.pharmthera.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Dafny N, Dougherty PM, Drath D. Immunosuppressive agent modulates the severity of opiate withdrawal. NIDA Res Monogr. 1990;105:553–555. [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Satinoff E, Kelley KW. Cyclosporine and alpha-interferon do not attenuate morphine withdrawal in rats but do impair thermoregulation. Physiol Behav. 1987;39:593–598. doi: 10.1016/0031-9384(87)90158-2. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Wollman E, Vitkovic L, Yirmiya R. Cytokines and depression: fortuitous or causative association? Mol Psychiatry. 1999;4:328–332. doi: 10.1038/sj.mp.4000572. [DOI] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–142. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- Dougherty PM, Pellis NR, Dafny N. The brain and the immune system: an intact immune system is essential for the manifestation of withdrawal in opiate addicted rats. Neuroscience. 1990;36:285–289. doi: 10.1016/0306-4522(90)90293-d. [DOI] [PubMed] [Google Scholar]
- Erickson MA, Dohi K, Banks WA. Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation. 2012;19:121–130. doi: 10.1159/000330247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantegrossi WE, Ciullo JR, Wakabayashi KT, de La Garza R, Traynor JR, Woods JH. A comparison of the physiological, behavioral, neurochemical and microglial effects of methamphetamine and 3,4-methylenedioxymethamphetamine in the mouse. Neuroscience. 2008;151:533–543. doi: 10.1016/j.neuroscience.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemstapat K, Le L, Edwards SR, Smith MT. Comparative studies of the neuro-excitatory behavioural effects of morphine-3-glucuronide and dynorphin A(2–17) following spinal and supraspinal routes of administration. Pharmacol Biochem Behav. 2009;93:498–505. doi: 10.1016/j.pbb.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Holmes JE, Miller NE. Effects of Bacterial Endotoxin on Water Intake, Food Intake, and Body Temperature in the Albino Rat. J Exp Med. 1963;118:649–658. doi: 10.1084/jem.118.4.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. ScientificWorldJournal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Buijs M, Tuke J, Kwok YH, Gentgall M, Williams D, Rolan P. Low-dose endotoxin potentiates capsaicin-induced pain in man: Evidence for a pain neuroimmune connection. Brain Behav Immun. 2013;30:3–11. doi: 10.1016/j.bbi.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Coats BD, Lewis SS, Zhang Y, Sprunger DB, Rezvani N, Baker EM, Jekich BM, Wieseler JL, Somogyi AA, Martin D, Poole S, Judd CM, Maier SF, Watkins LR. Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain Behav Immun. 2008a;22:1178–1189. doi: 10.1016/j.bbi.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, Watkins LR. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010a;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, Brzeski A, Northcutt A, Vietz CM, Judd CM, Maier SF, Watkins LR, Johnson KW. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23:240–250. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Chao LW, Kearney JJ, Zhang Y, Berkelhammer DL, Loram LC, Rozeske RR, Bland ST, Maier SF, Gleeson TT, Watkins LR. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain Behav Immun. 2008b;22:1248–1256. doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, Watkins LR. Opioid Activation of Toll-Like Receptor 4 Contributes to Drug Reinforcement. J Neurosci. 2012;32:11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008c;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler JL, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010b;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch A, Hamann L, Schott M, Boehm O, Grotemeyer D, Kurt M, Schwenke C, Schumann RR, Bornstein SR, Zacharowski K. Genetic variation of TLR4 influences immunoendocrine stress response: an observational study in cardiac surgical patients. Crit Care. 2011;15:R109. doi: 10.1186/cc10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labella FS, Pinsky C, Havlicek V. Morphine derivatives with diminished opiate receptor potency show enhanced central excitatory activity. Brain Res. 1979;174:263–271. doi: 10.1016/0006-8993(79)90849-7. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor kappaB alpha within specific cellular populations of the rat brain. J Neurochem. 1999;73:309–321. doi: 10.1046/j.1471-4159.1999.0730309.x. [DOI] [PubMed] [Google Scholar]
- Landsteiner K. The Specificity of Serological Reactions. C.C. Thomas; Springfield, IL; Baltimore, MD: 1936. [Google Scholar]
- Laviolette SR, Nader K, van der Kooy D. Motivational state determines the functional role of the mesolimbic dopamine system in the mediation of opiate reward processes. Behav Brain Res. 2002;129:17–29. doi: 10.1016/s0166-4328(01)00327-8. [DOI] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, Rice KC, Watkins LR. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience. 2010;165:569–583. doi: 10.1016/j.neuroscience.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Zhang Y, Hund DK, Maier SF, Rice KC, Watkins LR. Glucuronic acid and the ethanol metabolite ethyl-glucuronide cause toll-like receptor 4 activation and enhanced pain. Brain Behav Immun. 2013;30:24–32. doi: 10.1016/j.bbi.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun X, Zhang Y, Huang J, Hanley G, Ferslew KE, Peng Y, Yin D. Morphine promotes apoptosis via TLR2, and this is negatively regulated by beta-arrestin 2. Biochem Biophys Res Commun. 2009;378:857–861. doi: 10.1016/j.bbrc.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Liu L, Coller JK, Watkins LR, Somogyi AA, Hutchinson MR. Naloxone-precipitated morphine withdrawal behavior and brain IL-1β expression: comparison of different mouse strains. Brain Behav Immun. 2011;25:1223–1232. doi: 10.1016/j.bbi.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hutchinson MR, White JM, Somogyi AA, Coller JK. Association of IL-1B genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenet Genomics. 2009;19:869–876. doi: 10.1097/FPC.0b013e328331e68f. [DOI] [PubMed] [Google Scholar]
- Matic M, Mahns A, Tsoli M, Corradin A, Polly P, Robertson GR. Pregnane X receptor: promiscuous regulator of detoxification pathways. Int J Bioch Cell Biol. 2007;39:478–483. doi: 10.1016/j.biocel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Ikemoto S. Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res. 1999;101:129–152. doi: 10.1016/s0166-4328(99)00022-4. [DOI] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Fujio M, Ozawa T, Minami M, Satoh M. Effect of MS-153, a glutamate transporter activator, on the conditioned rewarding effects of morphine, methamphetamine and cocaine in mice. Behav Brain Res. 2005;156:233–239. doi: 10.1016/j.bbr.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Satoh M. Involvement of glial glutamate transporters in morphine dependence. Ann N Y Acad Sci. 2004;1025:383–388. doi: 10.1196/annals.1307.047. [DOI] [PubMed] [Google Scholar]
- Narita M, Miyatake M, Narita M, Shibasaki M, Shindo K, Nakamura A, Kuzumaki N, Nagumo Y, Suzuki T. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2006;31:2476–2488. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Sekiya Y, Minami M, Satoh M. Effect of gene transfer of GLT-1, a glutamate transporter, into the locus coeruleus by recombinant adenoviruses on morphine physical dependence in rats. Eur J Neurosci. 2004;19:221–226. doi: 10.1111/j.1460-9568.2004.03101.x. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Nakagawa T, Shige K, Minami M, Satoh M. Changes in the expression of glial glutamate transporters in the rat brain accompanied with morphine dependence and naloxone-precipitated withdrawal. Brain Res. 2001;905:254–258. doi: 10.1016/s0006-8993(01)02536-7. [DOI] [PubMed] [Google Scholar]
- Palm NW, Medzhitov R. Immunostimulatory activity of haptenated proteins. Proc Natl Acad Sci U S A. 2009;106:4782–4787. doi: 10.1073/pnas.0809403105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SM, Gaykema RPA, Goehler LE. How does immune challenge inhibit ingestion of palatable food? Evidence that systemic lipopolysaccharide treatment modulates key nodal points of feeding neurocircuitry. Brain Behav Immun. 2008;22:1160–1172. doi: 10.1016/j.bbi.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranaldi R, Beninger RJ. Rostral-caudal differences in effects of nucleus accumbens amphetamine on VTA ICSS. Brain Res. 1994;642:251–258. doi: 10.1016/0006-8993(94)90929-6. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nature. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Salazar M, Pariente JA, Salido GM, González A. Ethanol induces glutamate secretion by Ca2+ mobilization and ROS generation in rat hippocampal astrocytes. Neurochem Int. 2008;52:1061–1067. doi: 10.1016/j.neuint.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Schäfer M. The painful Toll of ethanol and its metabolites: a new molecular pattern of recognition by Toll like receptors? Brain Behav Immun. 2013;30:22–23. doi: 10.1016/j.bbi.2013.01.085. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci. 2013;33:961–971. doi: 10.1523/JNEUROSCI.2516-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Hutchinson MR, Bilbo SD. Early-Life Experience Decreases Drug-Induced Reinstatement of Morphine CPP in Adulthood via Microglial-Specific Epigenetic Programming of Anti-Inflammatory IL-10 Expression. J Neurosci. 2011;31:17835–17847. doi: 10.1523/JNEUROSCI.3297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi K, Fukuda H, Watanabe M, Sato M. Studies on antitussives. III.(+)-Morphine and its derivatives. J Pharm Soc Jap. 1960;80:1506–1509. [Google Scholar]
- Thomas J, Hutchinson MR. Exploring neuroinflammation as a potential avenue to improve the clinical efficacy of opioids. Expert Rev Neurother. 2012;12:1311–1324. doi: 10.1586/ern.12.125. [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85:2059–2070. doi: 10.1002/jnr.21325. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, True W, Lin N, Toomey R, Eaves L. Co-occurrence of abuse of different drugs in men: the role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry. 1998;55:967–972. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- Vargas-Perez H, Ting-A-Kee R, Walton CH, Hansen DM, Razavi R, Clarke L, Bufalino MR, Allison DW, Steffensen SC, van der Kooy D. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009;324:1732–1734. doi: 10.1126/science.1168501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res. 2006;85:49–53. doi: 10.1177/154405910608500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Frankfurt M, Burns LH. High-affinity naloxone binding to filamin a prevents mu opioid receptor-Gs coupling underlying opioid tolerance and dependence. PLoS ONE. 2008;3:e1554. doi: 10.1371/journal.pone.0001554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang Q, Zhou H, Gao H, Chen SH, Chu CH, Wilson B, Hong JS. Naloxone inhibits immune cell function by suppressing superoxide production through a direct interaction with gp91phox subunit of NADPH oxidase. J Neuroinflammation. 2012a;9:32. doi: 10.1186/1742-2094-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A. 2012b;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312:60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Weber M, Modemann S, Schipper P, Trauer H, Franke H, Illes P, Geiger KD, Hengstler JG, Kleemann WJ. Increased polysialic acid neural cell adhesion molecule expression in human hippocampus of heroin addicts. Neuroscience. 2006;138:1215–1223. doi: 10.1016/j.neuroscience.2005.11.059. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011a;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011b;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Pollak Y, Morag M, Reichenberg A, Barak O, Avitsur R, Shavit Y, Ovadia H, Weidenfeld J, Morag A, Newman ME, Pollmächer T. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917:478–487. doi: 10.1111/j.1749-6632.2000.tb05412.x. [DOI] [PubMed] [Google Scholar]
- Youn DH, Wang H, Jeong SJ. Exogenous tumor necrosis factor-alpha rapidly alters synaptic and sensory transmission in the adult rat spinal cord dorsal horn. J Neurosci Res. 2008;86:2867–2875. doi: 10.1002/jnr.21726. [DOI] [PubMed] [Google Scholar]