

Abstract
Basic fibroblast growth factor (bFGF) has been implicated in tumor growth via interactions with its receptors (FGFRs) on the cell surface and therefore, bFGF/FGFRs are considered essential targets for cancer therapy. Herein, a consensus heptapeptide (LSPPRYP) was identified for the first time from a phage display heptapeptide library after three sequential rounds of biopanning against FGFR-expressing cells with competitive displacement of phage by bFGF, followed by subtraction of non-specific binding by FGFR-deficient cells. Phage bearing LSPPRYP showed high levels of binding to Balb/c 3T3 cells expressing high-affinity bFGF-binding FGFR (bFGFR), but not to the cells that do not express bFGFR (Cos-7), or express a very low affinity bFGFR (HaCat). The selected-phage-derived peptide synthesized by solid phase method using a rapid and practical Fmoc strategy was found to specifically compete with bFGF for binding to its receptors, inhibit bFGF-stimulated cell proliferation by inducing cell cycle arrest, and block bFGF-induced activation of Erk1 and Erk2 kinase in B16-F10 melanoma cells. Importantly, treatment of melanoma-bearing mice with the synthetic peptide significantly suppressed tumor growth. The results demonstrate a strong anticancer activity of the isolated bFGFR-binding peptide (and its future derivatives), which may have great potential for cancer therapy.
Keywords: bFGF, phage display, proliferation, cancer therapy
INTRODUCTION
Basic fibroblast growth factor (bFGF) plays essential roles in both physiological and pathological processes by interacting with specific receptors on the cell surface [1, 2]. Because overexpression of bFGF and/or its receptors is commonly detected in tumors, the development of antagonists to bFGF and its receptors has been considered as a potential strategy for cancer therapy [3, 4].
Phage display technology has been used to identify desirable peptides and is an important tool for drug discovery. We have successfully applied the technique to identify a high-affinity bFGF-binding peptide with strong inhibitory activity against bFGF-induced cell proliferation and angiogenesis [5]. Herein, the goal was to isolate a FGF receptor (FGFR)-binding peptide with this technique. The phage display library used consisted of phages bearing random heptapeptides fused to the N-terminus of the coat protein pIII of the M13 Phage. Using FGFR-deficient Cos-7 cells as the subtractive cells, three consecutive rounds of panning with the library against Balb/c 3T3 cells expressing exuberant FGF receptors were performed to identify a binding peptide sequence (LSPPRYP), by extrapolating from the corresponding DNA sequences of 5 positive phage clones eluted from the cells by bFGF. The phage bearing LSPPRYP was evaluated for its cell-binding specificity. Moreover, because bFGF greatly contributes to melanoma growth and progress [6, 7], the synthetic peptide consisting of this sequence and a spacer sequence GGGS was further tested for antitumor activity using B16-F10 murine melanoma cells for in vitro experiments, and introduced into C57BL/6 mice for in vivo experiments. The results demonstrated that the identified synthetic peptide could reverse the effects of bFGF on cell proliferation, cell cycle progression, Erk1/Erk2 activation of melanoma cells, and significantly inhibit tumor growth in mice.
RESULTS
Isolation of phages binding to bFGF receptors
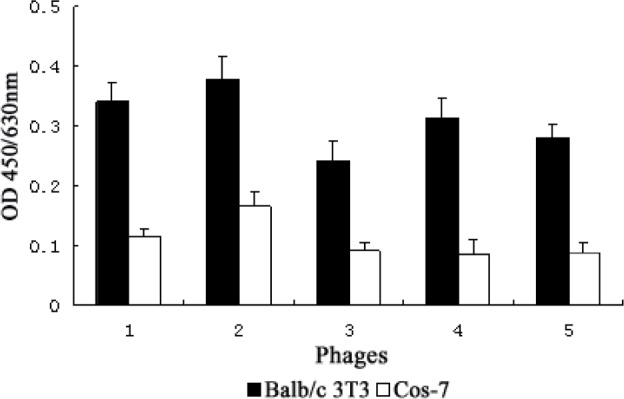
Specific phages capable of binding to bFGF receptors were selected by three rounds of biopanning against positive cells expressing high-affinity bFGF receptors on the cell surface. In order to diminish the background of screening, bound phages were specifically eluted with bFGF and subtractive panning was carried out against cells that were deficient in bFGF receptors. In the first round, a lower concentration of PBST (0.05%) was applied to wash for higher eluate titers. In order to enrich highly specific and affinity phages, nonspecifically binding phages were absorbed by subtractive cells before screening, and the concentration of PBST was then increased to 0.1% from the second round. In the last round of panning, low affinity phages eluted within 1 h were discarded, and the phages further eluted with bFGF for an additional 1 h were analyzed by ELISA to identify high-affinity bFGF receptor-binding clones. Phage clones that exhibited a binding affinity (i.e, OD value) to Balb/c 3T3 2-fold greater than observed for Cos-7 cells were considered positive. As shown in Fig. 1, we identified 5 positive clones from a total of 13 phage clones.
Figure 1. Specific binding of the positive phage clones to bFGF receptors.
The binding affinities of 5 positive phage clones to Balb/c 3T3 cells and Cos-7 cells were detected by ELISA assay. Data presented are the mean OD values (±SDs) of triplicate samples.
Sequence analysis and property prediction of positive phages
Total DNA of the positive phages was isolated and sequenced using −96gⅢ primers. The amino acid sequences of the peptides displayed on the corresponding phages were deduced from the DNA sequences and Bioedit and ProtParam programs were applied to analyze the sequences and predict the peptide properties. As shown in Table 1, 5 clones shared consensus sequences (LSPPRYP). Comparison of the amino acid sequences of the heptapeptide (P9) with that of bFGF revealed that the P9 contained 6 amino acids identical to the adjacent amino acids (L3, S9, P13, P14, R120, Y124) of the 3D structure of bFGF, which are located within the motifs (P13~K18 and R120~K125), which are involved in receptor binding and mitogenic activity of bFGF. Furthermore, similar to bFGF, P9 also carried positive charges under physiological conditions, suggesting that electrostatic interaction might also be involved in their binding to FGF receptors.
Table 1. Properties of peptides displayed by positive phages.
pI, Isoelectric Point.
GRAVY, Grand Average of Hydropathicity.
Specificity of selected phage clone for binding cells
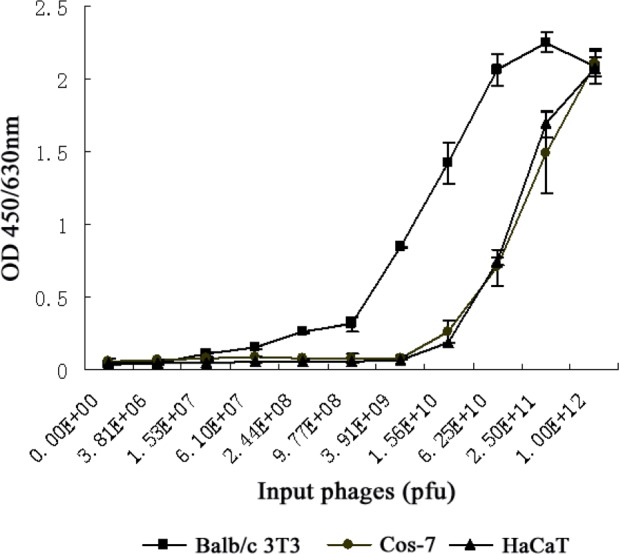
It has been shown that Balb/c 3T3 cells express high-affinity bFGF receptors (e.g., FGFR1c and FGFR2c) on the cell surface, while HaCat cells exclusively express a specific isoform of FGFR2 (also known as FGFR2b or KGFR) with a very low affinity to bFGF [8, 9]. To assess the binding specificity of the selected phage clone, we compared the ability of the phages to bind Balb/c 3T3, HaCat and FGFR-deficient Cos-7 cells [10, 11]. As shown in Figure 2, the affinity of the phage clone LSPPRYP to Balb/c 3T3 cells was markedly stronger than to HaCaT and Cos-7 cells. The Kd value for Balb/c 3T3 cells was between 3.91×109 pfu and 1.56×1010 pfu, which is approximately 16 times less than the Kd value (between 6.25×1010 pfu and 2.50×1011 pfu) for HaCaT and Cos-7 cells (Fig. 2). The results revealed that the LSPPRYP phage exhibits greater binding to the cells expressing high-affinity bFGF receptors than to the cells with low affinity bFGF receptors or without bFGF receptors.
Figure 2. Comparison of binding affinity of LSPPRYP phage for different cell lines.
Phages were first incubated with Balb/c 3T3, HaCat, and Cos-7 cells respectively, and then washed with PBST. The quantity of the phages binding to the cells was detected with anti-M13 mAb. Data presented are the mean OD value (±SDs) of triplicates.
Specificity of the synthetic peptide for bFGF binding to its receptors
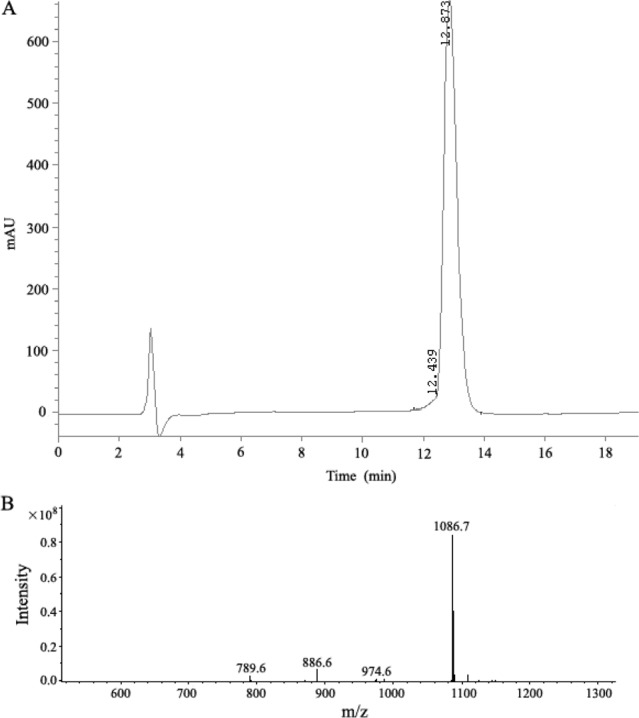
In a phage display heptapeptide library, the C-terminus of the random peptide is fused to the phage via a short spacer (GGGS), having no free negatively charged carboxylate. Therefore, a spacer sequence (GGGS) was added to the C-terminus and the C-terminal carboxylate was amidated to block the negative charge when synthesizing the peptide corresponding to the selected sequence (LSPPRYP). The selected-phage-derived peptide was synthesized on solid phase using a rapid and practical Fmoc strategy. The crude compound was subjected to RP-HPLC purification. As shown in Fig. 3, high purity grade (98%) was obtained after purification and the synthetic peptide was characterized for its identity by mass spectrometry analysis.
Figure 3. Analysis of the synthetic P9 peptides.
(a) HPLC chromatogram at 214 nm. (b) electrospray ionization mass spectrum of the synthetic peptide.
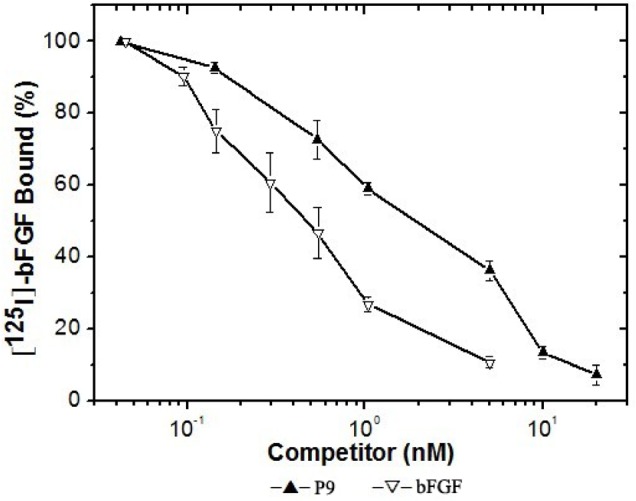
To characterize the effect of the synthesized P9 peptides (LSPPRYPGGGS) on bFGF binding to its receptors, P9 peptides were compared to bFGF for their ability to compete directly for 125I -bFGF binding on Balb/c 3T3 cells expressing exuberant bFGF receptors. As shown in Fig. 4, P9 is highly efficient in specific competing for 125I-bFGF binding to its receptors, in which about 2.5 nM of P9 was able to displace 50% of 125I-bFGF. Scatchard analysis of 125I-bFGF binding on Balb/c 3T3 revealed a receptor population consisting of approximately 9,600 sites/cell with a dissociation constant (KD) of 100 pM. These affinity and capacity values are within the range of values previously published for the high affinity bFGF receptors (KD = 10 - 200 pM, 0.2-10 ×104 sites/cell). In the presence of P9 peptides, specific high affinity binding of 125I-bFGF to Balb/c 3T3 cells decreased significantly with a KD of 500 pM and a receptor population of about 27,000 sites/cell. These results indicate that P9 peptides are able to block the binding of bFGF to its receptors, and may have the potential to antagonize bFGF bioactivity.
Figure 4. Specific binding of 125I -bFGF to Balb/c 3T3 cells competed by increasing concentrations of bFGF or the synthetic P9 peptides.
Binding assays were performed as described in Materials and Methods. Values shown are the mean value (±SDs) of triplicates.
Effect of the synthetic peptide on bFGF-stimulated proliferation of melanoma cells
We next determined the efficacy of the synthetic P9 peptides in blocking bFGF-stimulated tumor cell proliferation. B16-F10 murine melanoma cells with high levels of FGFR expression [12, 13] were incubated with 30 ng/mL bFGF alone or along with various concentrations of the P9 peptides. As shown in Fig. 5, the synthetic peptides inhibited B16-F10 cell proliferation in a dose-dependent manner and nearly 90% of inhibition was achieved at 10 nM.
Figure 5. Inhibition of bFGF-stimulated proliferation of B16-F10 cells by the synthetic P9 peptides.
Starved cells were treated with 30 ng/ml bFGF alone, or 30 ng/ml bFGF plus P9 at the indicated concentrations. Cell proliferation was determined 48 h after treatment. Data are presented as the mean (±SDs) of 3 independent experiments performed in triplicate.
Effect of the synthetic peptides on bFGF-induced cell cycle progression
The effect of the synthetic peptides on cell cycle progression induced by bFGF was determined by flow cytometry analysis. As shown in Fig. 6, treatment with bFGF significantly increased the percentage of S phase cells, and decreased the ratio of G0/G1 phase cells. In contrast, cells treated with bFGF plus the synthetic peptides had a higher G0/G1-phase population and lower S-phase population than those treated with bFGF alone, suggesting that the synthetic peptides reversed the S-phase increase induced by bFGF, and arrested the cells at the G0/G1 phase.
Figure 6. Flow cytometry analysis of the effect of the synthetic P9 peptides on cell cycle distribution of bFGF-stimulated cells.
Panel A, (a) Control cells without treatment of bFGF or P9. (c-f) B16-F10 cells were starved for 24 h and then treated with (b) 30 ng/ml bFGF, (c) 30 ng/ml bFGF plus 1 nM P9, (d) 30 ng/ml bFGF plus 10 nM P9, (e) 30 ng/ml bFGF plus 100 nM P9, or (f) 30 ng/ml bFGF plus 1000 nM P9 for 48 h. Cells were then fixed in 70% ice-cold ethanol, washed with PBS, stained with PI, and subjected to flow cytometry analysis. Data shown are the representative pictures of 3 independent experiments. Panel B, cell cycle distribution of the control and treated cells. Data are presented as the mean (±SDs) of 3 independent experiments. * P< 0.01 vs control group; # P< 0.01 vs bFGF group.

Effect of the synthetic peptides on bFGF-induced MAP kinase activation
The effect of the synthetic P9 peptide on bFGF signal transduction was determined by measuring its capacity to inhibit the activation of two MAPKs (Erk1 and Erk2) in B16-Fl0 cells after stimulation by bFGF. Pretreatment of B16-Fl0 cells with the synthetic peptides at a concentration of 10 nM or higher for 5 min prior to bFGF stimulation completely suppressed bFGF-induced Erk1 and Erk2 phosphorylation (Fig. 7, lanes 3-6). The levels of Erk1/Erk2 activation in the cells treated with bFGF and P9 peptides at a concentration of 10 nM or higher were significantly lower than those in the cells treated with bGFG alone, and comparable to the untreated control B16-Fl0 cells.
Figure 7. Synthetic P9 peptides inhibit bFGF-induced MAP kinase activation.
Panel A. Before stimulation with 30 ng/ml bFGF for 20 min, starved B16-F10 cells were pretreated with P9 at the indicated concentrations for 5 min. Cell lysates were resolved with 10% SDS-PAGE, transferred to PVDF membrane, and blotted with anti-pErk1/pErk2 mAb. Data shown are the representative pictures of 3 independent experiments. Panel B. Density ratios of pErk1/pErk2 to Erk1/Erk2. Data are presented as the mean (±SDs) of 3 independent experiments.
The synthetic peptides suppressed tumor growth in vivo
We further investigated the in vivo antitumor efficacy of the synthetic peptides in a mouse model of melanoma. Tumor was established by subcutaneously implanting the C57BL/6-derived B16-Fl0 cells into C57BL/6 mice. The synthetic peptides were administrated at dose of 0.5 mg/kg or 2.5 mg/kg on alternate days starting at day 7, and the growth of the tumors was assessed for 9 days. Treatment with the synthetic peptides resulted in a significant reduction in tumor volume compared to that observed as a result of PBS treatment (P<0.01 vs PBS group). A dose-dependent inhibition of tumor growth of 38% and 82% was observed on day 9 for animals treated with the synthetic peptides at a dose of 0.5 mg/kg and 2.5 mg/kg, respectively (Fig. 8A). All mice were sacrificed at day 9, one day after the last injection of the P9 peptides (16 days after tumor injection, and 9 days after first administration of the synthetic peptides). Immunostaining with anti-phospho-Erk1/Erk2 (pErk1/pErk2) antibody showed a dose-dependent inhibition of Erk1/Erk2 activation. The numbers of pErk1/pErk2-positive cells were markedly reduced in tumors from mice treated with P9 peptides compared to tumors in control animals (Fig. 8B). The data showed a potent inhibitory effect of the P9 peptides on tumor growth in vivo, which was associated with significant inhibition of Erk1/Erk2 activation in tumor cells.
Figure 8. Synthetic P9 peptides inhibit the growth of the murine melanoma B16-F10 cells in mice.
(A) B16-F10 cells were injected subcutaneously into female C57BL/6 mice. When tumors were visible on all mice on day 7, treated mice were intraperitoneally injected with P9 on alternate days at 0.5, or 2.5 mg/kg, whereas control mice were injected with PBS (n=10 mice for each group). (B) P9 inhibits Erk1/Erk2 activation in tumors in vivo. On the next day after last injection, the B16-F10 tumors were extracted, fixed, and embedded in paraffin. Tissue sections were immunostaining for phospho-Erk1/2 as described in the Material and Methods. Meanwhile, negative control was incubated with PBS instead of monoclonal anti-phospho-Erk1/2. Negative control (Panel a), PBS (Panel b), 0.5 mg/kg (Panel c), and 2.5 mg/kg (Panel d). Data are representative of 2 independent experiments.
DISCUSSION
Phage display technology provides an efficient tool to identify the desirable sequences binding specifically to targets, and is widely used in diagnostic and therapeutic protein developments in recent years [14-16]. FGFRs have been shown to act as driving oncogene in certain cancers to maintain the malignant properties of tumor cells via a cell autonomous manner [17]. In this study herein, we developed a biopanning strategy using phage display technology for identifying bFGF receptor-binding peptides. The selected phage clone exhibits a remarkable binding affinity to Balb/c 3T3 cells that express high-affinity bFGF receptors, while binding poorly to cells that do not express bFGF receptors (Cos-7), or express low affinity bFGF receptors. Furthermore, the identified peptide specifically competed for bFGF binding to its receptors, and was able to inhibit bFGF-stimulated cancer proliferation in vitro and suppress tumor growth in vivo.
There is a growing body of evidence that bFGF promotes cancer cell proliferation through the activation of the Ras/Raf/MEK/ERK pathway [18-21]. Cell proliferation is regulated during the G0/G1 phase in the cell cycle. CDK4 and CDK6 interacting with the cyclin D family of proteins drive G0/G1 phase into the DNA synthesizing S-phase [22]. ERK activity plays a pivotal role in cell proliferation via regulation of cyclin D1 expression during mid-G1 and driving cells past the G1-restriction point [23, 24]. In order to explore the potential of the isolated bFGF receptor-binding peptide P9 in cancer treatment, we investigated the effects of P9 on bFGF-induced proliferation, cell cycle distribution, and MAKP signal transduction in B16-F10 cells. Our results showed that P9 inhibited the proliferation of B16-F10 cells stimulated by bFGF in a dose-dependent manner. In accordance with the inhibitory effect of P9 on cell proliferation, cell cycle analysis showed that P9 arrested bFGF-stimulated cells at the G0/G1 phase, and the MAP kinase activation assay revealed that P9 reduced the levels of Erk1 and Erk2 phosphorylation in B16-F10 cells induced by bFGF. Downregulation of Erk1 and Erk2 activation may contribute to more G0/G1 cells in P9-treated cells than in the untreated control cells via restriction of cyclin D1 expression during mid-G1 and inhibition of cells passing the G1-restriction point. In a small animal model system, P9 significantly suppressed tumor growth and blocked Erk1/Erk2 activation in the tumors. The synthetic peptide attenuates the mitogenic activity of bFGF possibly via interfering with the interaction between bFGF and its receptors.
As bFGF/FGFRs have been considered as a target for cancer therapy, efforts have been made to develop the antagonists targeting bFGF/FGFRs including antibodies and small molecule tyrosine kinase inhibitors. When compared to antibodies, small peptides may have several advantages for therapy. First, the peptides are easy to synthesize and modify according to their pharmacokinetic requirements [25]. Second, they are small and easily penetrate through the tissue [26]. Third and most importantly, they have low immunogenicity [25, 27]. There are also disadvantages for utilizing small peptides [27]. One disadvantage is their rapid proteolysis by endogenous peptidases and proteases. There are several ways to overcome this obstacle including the usage of D-amino acids, and substitution of the peptide bonds. Another disadvantage for small peptides is the loss of binding affinity when conjugated to other therapeutic molecules, which could be addressed by keeping the conjugated molecules apart from the receptor-binding sequence. Compared to chemical agents such as tyrosine kinase inhibitors which are not tumor specific and bear the risk of severe side effects [28], P9 has potential for high selectivity because it targets interactions between bFGF and its receptors, and may be safer to use from a clinical point of view.
In summary, our results demonstrate that the isolated bFGF receptor-binding peptide P9 provides an effective bFGF/FGFR antagonist, and may have potential application for the treatment of proliferative disorders, including a variety of cancers with upregulation of bFGF/FGFRs.
MATERIALS AND METHODS
Reagents and cell lines
Ph.D.-7™ Phage Display Peptide Library Kit and Escherichia coli ER2738 were purchased from New England Biolabs Inc. (Beverly, MA, USA). Horseradish peroxidase (HRP)-anti-M13 mAb was purchased from Amersham Pharmacia Biotech (Uppsala, Sweden). Phospho-Erk1/2 (pErk1/pErk2) rabbit mAb, Erk1/2 (Erk1/Erk2) rabbit mAb and goat anti-rabbit IgG conjugated with horseradish peroxidase antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-rat immunoglobulin sensitive S-P detection kit (UltraSensitive™ S-P) was purchased from Maixin Limited Corporation (Fuzhou, Fujian, China). Balb/c 3T3, HaCat, Cos-7, and B16-F10 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen Corporation, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS).
Biopanning of a heptapeptide phage display library
Balb/c 3T3 cells grown to 90 % confluence in 25 cm2 tissue culture flask were washed 2 times and blocked with DMEM-2% dry milk for 2 h at 37°C, 5% CO2. After the cells were washed 5 times with 0.05% Tween-20 in PBS (0.05% PBST), the heptapeptide phage library containing 2 × 1011 pfu diluted in 2 ml DMEM was added and agitated gently at room temperature for 1 h. The cell-bound phages were washed 10 times with 0.05 % PBST, then eluted with 555 nM bFGF with gentle agitation at room temperature for 2 h. The eluate was then amplified, purified and titered for the second round of panning. The phages containing 2 × 1011 pfu obtained from the first round of panning were diluted in 2 ml DMEM-2% dry milk, added to 25 cm2 flask with a 90 % confluency of Cos-7 cells, and agitated gently at room temperature for 1 h. The supernatant was collected and added to the flask cultured with Balb/c 3T3 cells. Steps were repeated similar to the first round of panning except that the concentration of Tween-20 in PBS used for washing was increased from 0.05% to 0.1%. For the last round of selection, low affinity cell-bound phages were first eluted with bFGF for 1 h, discarded, and high-affinity cell-bound phages were then eluted with bFGF for an additional 1 h. The phage clones obtained from the eluate were subsequently subjected to ELISA analysis.
ELISA assay for positive phages
Balb/c 3T3 cells and Cos-7 cells (negative control) were plated at a density of 1 × 104 cells per well in a 96-well plate and incubated at 37°C, 5% CO2 for 24 h. After the plates were blocked with PBS-2% dry milk (PBSM) at 37°C for 1 h and washed 3 times with 0.05% PBST, phage clones (~1010 pfu/well) serially diluted 4 times were added and incubated with gentle agitation at room temperature for 1 h. PBS was used as the blank control. The plates were then washed 3 times with 0.05% PBST before 200 μl of HRP-anti-M13 mAb (1:5000) was added to each well, and incubated with gentle agitation at room temperature for 1 h. After 3 washes with 0.05 % PBST, 50 μl of freshly-made 3,3',5,5'-tetramethylbenzidine (TMB) solution was added to each well and kept at room temperature in the dark for 20 min. The reaction was terminated by adding 50 μl of 2 M H2SO4 to each well. Absorbance was measured at 450 nm with reference at 630 nm.
Evaluation of cell-binding specificity of selected phage clone
Balb/c 3T3, HaCaT and Cos-7 were grown to confluence in 96-well plates. After the plates were blocked with PBSM at 37°C, 5% CO2 for 1 h and washed 3 times with 0.05% PBST, phage clones (~1012 pfu/well) serially diluted 4 times were added and incubated at room temperature for 1 h with gentle agitation. PBS was used as the blank control. The cells were washed briefly with PBST before incubation with HRP-anti-M13 mAb (1: 5000) at room temperature for 1 h. After 3 washes with PBST, freshly-made TMB solution was added to react at room temperature in the dark for 20 min. The reaction was terminated and the absorbance was measured in the manner as described earlier.
DNA sequencing and peptide synthesis
DNA sequences of the positive phage clones were determined using an automated DNA sequencer at Invitrogen Company (Shanghai, China), and analyzed with Bioedit and ProtParam programs (http://au.expasy.org/tools/protparam.html).
In order to mimic the structure of peptide displayed on the phage, a spacer sequence Gly-Gly-Gly-Ser was added to the C-terminus, and the C- terminal carboxylate was amidated for blocking the negative charge when the peptide corresponding to the selected sequence was synthesized on a Liberty Microwave Peptide Synthesizer (CEM Corp., Matthews, NC) by solid phase method following the Fmoc chemistry protocols. Briefly, the first amino acid derivative Fmoc-Ser(tBu)-OH was loaded on the amide resin, and the peptide chain was elongated by sequential coupling and Fmoc deprotection of Fmoc-Gly-OH, Fmoc-Gly-OH, Fmoc-Gly-OH, Fmoc-Pro-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Arg(Mtr)-OH, Fmoc-Pro-OH, Fmoc-Pro-OH, Fmoc-Ser(tBu)-OH, and Fmoc-Leu-OH. The peptide was then cleaved from the solid support and simultaneously removed of all protecting groups. The crude product was isolated by precipitation, dried in vacuo, and purified by RP-HPLC with a C18 HPLC column. A gradient of 30–100% acetonitrile containing 0.1% trifluoroacetic acid was applied to elute the column with continuous measurement of the absorbance at 214 nm. The synthetic product was further confirmed by mass spectrometry analysis.
Receptor binding assay
The recombinant bFGF was iodinated by the modified chloramine-T method. Briefly, bFGF (5 μg/50 μ1 in 0.5 M phosphate buffer, pH 7.4) was incubated with chloramine-T (20 μg/10 μ1 of phosphate buffer) and 125I-labeled sodium (1.12 mCi/10 μ1) at room temperature for 55 sec. The reaction was immediately terminated by the addition of sodium metabisulflte (250 μg/200 μl) and 1% potassium iodide (100 μl), and the mixture was applied to a Sephadex G-50 column (1×20 cm) preequilibrated in 50 mM phosphate buffer containing 0.1% BSA to separate 125I-labeled bFGF from free 125I.
Confluent Balb/c 3T3 cells (0.5×106/well) were incubated with binding buffer (DMEM, 20mM HEPES, pH7.4) containing 125I-bFGF (50000 cpm) and different concentrations of unlabeled ligand (bFGF or the synthetic peptide) for 2.5 h at 4°C. For nonspecific binding, cells were incubated with 125I-bFGF (50000 cpm) and an excess of unlabeled bFGF (200 nM). The cells were washed with (3 × 1 ml) cold PBS (50 mM, PH7.4), and lysed with 1% Triton X-100 (500 μl). The radioactivity of the extracts was measured in a γ counter. Specific binding was determined as the difference between total and nonspecific binding, and data were analyzed by Scatchard analysis.
Cell proliferation assay
Cells were seeded in 96-well plates (5 × 103 cells/well) in DMEM containing 10% FBS. After incubation at 37°C for 24 h, the culture medium was replaced by DMEM containing 0.4% FBS to starve the cells for 24 h. The medium was then replaced by DMEM containing 30 ng/ml bFGF, or 30 ng/mL bFGF plus peptide at various concentrations. After culturing for 48 h, the number of viable cells was determined by the methylthiazoletetrazolium (MTT) method as previously described [5].
Flow cytometric analysis of cell cycle
Cells were starved for 24 h, and treated with 30 ng/ml bFGF or 30 ng/mL bFGF plus serially-diluted peptides for 48 h. After fixing in 70% ice-cold ethanol for 30 min at 4 °C and washing with PBS for 3 times, cells were stained with propidium iodide (PI) in the dark at room temperature for 30 min. The ModFit DNA analysis program was applied to analyze the percentages of cells at various phases of the cell cycle.
MAP kinase activation assay
Starved cells were pretreated with serially-diluted peptides for 5 min before 20 min of stimulation with bFGF (30 ng/ml). Cells were lysed in SDS-PAGE loading buffer after being washed with cold PBS. Cell lysates were separated by 10% SDS–PAGE, then transferred to PVDF membranes, and immunoblotted, respectively, with anti-pErk1/pErk2 mAb and anti-Erk1/Erk2 mAb followed by a goat anti-rabbit IgG, HRP-linked antibody. The blots were visualized with the ECL detection system.
In vivo antitumor studies
B16-F10 murine melanoma cells were harvested, and injected subcutaneously into the flanks (5 × 105 cells) of 7-week old female C57BL/6 mice. When tumors were visible on all mice on day 7, treated mice were intraperitoneally injected with the synthetic peptides on alternate days for a total of 5 injections at a dosage of 0.5, or 2.5 mg/kg, whereas control mice were injected with PBS. The tumor volumes were determined by measuring length (l) and width (w) and calculating volume (V = lw2/2) as described elsewhere [29]. On the next day after the last injection, mice were sacrificed and the tumors were extracted and fixed in 10% neutral buffered formalin and embedded in paraffin. Tissue sections cut from the paraffin blocks were deparaffinized and processed to microwave antigen retrieval before being subjected to stain with a monoclonal rabbit anti-phospho-Erk1/2 at a dilution of 1: 200 overnight at 4°C. Meanwhile, tissue sections incubated with PBS instead of monoclonal anti-phospho-Erk1/2 were used as the negative control. Visualization of the antigen-antibody reaction was performed using an UltraSensitive™ S-P (Rabbit) kit according to the manufacturer's suggestions.
Statistical analysis
Statistical analysis was carried out with SPSS version 13.0. One-way ANOVA analysis was applied to determine the statistical differences between groups. Data were presented as mean ± SDs. Statistical significance was accepted at P<0.05.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30973671, 81071800), the Natural Science Foundation of Guangdong Province of China (9151064001000031), the Science and Technology Planning Project of Wenzhou (Y20090244), the Natural Science Foundation of Zhejiang Province of China (Y4090379), the Guangdong Provincial Science and Technology Program (2010B060900040), the Fundamental Research Funds for the Central Universities (X. Wu), Guangdong Provincial “Thousand-Hundred-Ten Talent Project” (X. Wu), and Key Laboratory of Functional Protein Research of Guangdong Higher Education Institutes, Jinan University.
REFERENCES
- 1.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug. Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets. 2009;9:639–651. doi: 10.2174/156800909789057006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rusnati M, Presta M. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr. Pharm. Des. 2007;13:2025–2044. doi: 10.2174/138161207781039689. [DOI] [PubMed] [Google Scholar]
- 4.Cronauer MV, Schulz WA, Seifert HH, Ackermann R, Burchardt M. Fibroblast growth factors and their receptors in urological cancers: basic research and clinical implications. Eur. Urol. 2003;43:309–319. doi: 10.1016/s0302-2838(03)00005-8. [DOI] [PubMed] [Google Scholar]
- 5.Wu X, Yan Q, Huang Y, Huang H, Su Z, Xiao J, Zeng Y, Wang Y, Nie C, Yang Y, Li X. Isolation of a novel basic FGF-binding peptide with potent antiangiogenetic activity. J. Cell. Mol. Med. 2010;14:351–356. doi: 10.1111/j.1582-4934.2008.00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsunoda S, Nakamura T, Sakurai H, Saiki I. Fibroblast growth factor-2-induced host stroma reaction during initial tumor growth promotes progression of mouse melanoma via vascular endothelial growth factor A-dependent neovascularization. Cancer Sci. 2007;98:541–548. doi: 10.1111/j.1349-7006.2007.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribatti D, Vacca A, Ria R, Marzullo A, Nico B, Filotico R, Roncali L, Dammacco F. Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. Eur J Cancer. 2003;39:666–674. doi: 10.1016/s0959-8049(02)00150-8. [DOI] [PubMed] [Google Scholar]
- 8.Miki T, Bottaro DP, Fleming TP, Smith CL, Burgess WH, Chan AM, Aaronson SA. Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. U. S. A. 1992;89:246–250. doi: 10.1073/pnas.89.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Igarashi M, Finch PW, Aaronson SA. Characterization of recombinant human fibroblast growth factor (FGF)-10 reveals functional similarities with keratinocyte growth factor (FGF-7) J. Biol. Chem. 1998;273:13230–13235. doi: 10.1074/jbc.273.21.13230. [DOI] [PubMed] [Google Scholar]
- 10.Maruta F, Parker AL, Fisher KD, Hallissey MT, Ismail T, Rowlands DC, Chandler LA, Kerr DJ, Seymour LW. Identification of FGF receptor-binding peptides for cancer gene therapy. Cancer Gene Ther. 2002;9:543–552. doi: 10.1038/sj.cgt.7700470. [DOI] [PubMed] [Google Scholar]
- 11.Johnston CL, Cox HC, Gomm JJ, Coombes RC. bFGF and aFGF induce membrane ruffling in breast cancer cells but not in normal breast epithelial cells: FGFR-4 involvement. Biochem. J. 1995;306(Pt 2):609–616. doi: 10.1042/bj3060609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ying W, Martineau D, Beitz J, Lappi DA, Baird A. Anti-B16-F10 melanoma activity of a basic fibroblast growth factor-saporin mitotoxin. Cancer. 1994;74:848–853. doi: 10.1002/1097-0142(19940801)74:3<848::aid-cncr2820740310>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 13.Beitz JG, Davol P, Clark JW, Kato J, Medina M, Frackelton AR, Jr, Lappi DA, Baird A, Calabresi P. Antitumor activity of basic fibroblast growth factor-saporin mitotoxin in vitro and in vivo. Cancer Res. 1992;52:227–230. [PubMed] [Google Scholar]
- 14.Kandel E. Tumor-associated oncogenes go on (phage) display. Oncotarget. 2010;1:84–85. doi: 10.18632/oncotarget.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ionov Y. A high throughput method for identifying personalized tumor-associated antigens. Oncotarget. 2010;1:148–155. doi: 10.18632/oncotarget.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrieu-Weisbuch C, Michel S, Collomb-Clerc E, Pothion C, Deléage G, Jolivet-Reynaud C. Characterization of prostate-specific antigen binding peptides selected by phage display technology. J. Mol. Recognit. 2006;19:10–20. doi: 10.1002/jmr.762. [DOI] [PubMed] [Google Scholar]
- 17.Knights V, Cook SJ. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol. Ther. 2010;125:105–117. doi: 10.1016/j.pharmthera.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int. J. Cancer. 2003;104:527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 19.Nesbit M, Nesbit HK, Bennett J, Andl T, Hsu MY, Dejesus E, McBrian M, Gupta AR, Eck SL, Herlyn M. Basic fibroblast growth factor induces a transformed phenotype in normal human melanocytes. Oncogene. 1999;18:6469–6476. doi: 10.1038/sj.onc.1203066. [DOI] [PubMed] [Google Scholar]
- 20.Lazar-Molnar E, Hegyesi H, Toth S, Falus A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine. 2000;12:547–554. doi: 10.1006/cyto.1999.0614. [DOI] [PubMed] [Google Scholar]
- 21.Lefevre G, Babchia N, Calipel A, Mouriaux F, Faussat AM, Mrzyk S, Mascarelli F. Activation of the FGF2/FGFR1 autocrine loop for cell proliferation and survival in uveal melanoma cells. Invest. Ophthalmol. Vis. Sci. 2009;50:1047–1057. doi: 10.1167/iovs.08-2378. [DOI] [PubMed] [Google Scholar]
- 22.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 23.Welsh CF, Roovers K, Villanueva J, Liu Y, Schwartz MA, Assoian RK. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat. Cell Biol. 2001;3:950–957. doi: 10.1038/ncb1101-950. [DOI] [PubMed] [Google Scholar]
- 24.Pagès G, Lenormand P, L'Allemain G, Chambard JC, Meloche S, Pouysségur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl. Acad. Sci. U. S. A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae DG, Kim TD, Li G, Yoon WH, Chae CB. Anti-flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin. Cancer Res. 2005;11:2651–2661. doi: 10.1158/1078-0432.CCR-04-1564. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, Zhao H, Zheng Y, Gu Q, Ma J, Xu X. A novel antiangiogenic peptide derived from hepatocyte growth factor inhibits neovascularization in vitro and in vivo. Mol. Vis. 2010;16:1982–1995. [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Edwards DS. 99mTc-Labeled Small Peptides as Diagnostic Radiopharmaceuticals. Chem. Rev. 1999;99:2235–2268. doi: 10.1021/cr980436l. [DOI] [PubMed] [Google Scholar]
- 28.Posch C, Ortiz-Urda S. NRAS mutant melanoma--undrugable? Oncotarget. 2013;4:494–495. doi: 10.18632/oncotarget.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun J, Blaskovich MA, Knowles D, Qian Y, Ohkanda J, Bailey RD, Hamilton AD, Sebti SM. Antitumor efficacy of a novel class of non-thiol-containing peptidomimetic inhibitors of farnesyltransferase and geranylgeranyltransferase I: combination therapy with the cytotoxic agents cisplatin, Taxol, and gemcitabine. Cancer Res. 1999;59:4919–4926. [PubMed] [Google Scholar]