

Abstract
The mucosal epithelia together with adaptive immune responses, such as local production and secretion of dimeric and polymeric immunoglobulin A (IgA), are a crucial part of the first line of defense against invading pathogens. IgA is primarily secreted as SIgA and plays multiply roles in mucosal defense. The study of SIgA-mediated protection is an important area of research in mucosal immunity but an easy, fast and reproducible method to generate pathogen-specific SIgA in vitro has not been available. We report here a new method to produce SIgA by co-purification of dimeric IgA, containing J chain, and recombinant human SC expressed in CHO cells. We previously reported the generation, production and characterization of the human recombinant monoclonal antibody IgA2 b12. This antibody, derived from the variable regions of the neutralizing anti-HIV-1 mAb IgG1 b12, blocked viral attachment and uptake by epithelial cells in vitro. We used a cloned CHO cell line that expresses monomeric, dimeric and polymeric species of IgA2 b12 for large-scale production of dIgA2 b12. Subsequently, we generated a CHO cell line to express recombinant human secretory component (rhSC). Here, we combined dIgA2 b12 and CHO-expressed rhSC via column chromatography to produce SIgA2 b12 that remains fully intact upon elution with 0.1M Citric acid, pH 3.0. We have performed biochemical analysis of the synthesized SIgA to confirm the species is of the expected size and retains the functional properties previously described for IgA2 b12. We show that SIgA2 b12 binds to the HIV-1 gp120 glycoprotein with similar apparent affinity to that of monomeric and dimeric forms of IgA2 b12 and neutralizes HIV-1 isolates with similar potency. An average yield of 6 mg of SIgA2 b12 was achieved from the combination of 20 mg of purified dIgA2 b12 and 2 L of rhSC-containing CHO cell supernatant. We conclude that synthesized production of stable SIgA can be generated by co-purification. This process introduces a simplified means of generating a variety of pathogen-specific SIgA antibodies for research and clinical applications.
Keywords: IgA, Secretory IgA, mucosal IgA, affinity chromatography, antibody
1. Introduction
1.1 Structural and immunological background of SIgA
Secretory IgA (SIgA) plays an important role in mucosal immunity and homeostasis by clearing antigens and pathogenic microorganisms. Because of its capabilities independent of the antibody variable region, it can be considered a part of the innate immune system [1]. Human infants receive maternal IgG via placental transfer during gestation, but neonates depend on SIgA antibodies in breast milk to supply an adequate mucosal barrier from infection [2] that may also include a crucial antimicrobial component [3]. Most of the body’s activated B cells are found within the mucosae and exocrine glands where antibody-secreting plasma cells (PCs) produce polymeric IgA (pIgA) consisting of dimers and some trimers of IgA [4, 5]. Human mucosal IgA-producing PCs synthesize J chain [4–6] that is crucial for interaction with the polymeric Ig receptor (pIgR) basolaterally expressed on epithelial cells [7, 8]. The pIgR/pIgA/J chain complex is stabilized by disulphide bonds between the Cα2 domain of the antibody and domain 5 of the receptor [7, 8] enabling transport to the mucosal lumen that is facilitated by the pIgR [9]. For transport, the complex is internalized and actively transcytosed within vesicles through the epithelia cell and directed to the apical surface where the extracellular portion of pIgR is proteolytically cleaved [10]. The cleaved fragment remains disulphide bonded to pIgA forming the newly generated SIgA that is released at the mucosal surface. Any unbound pIgR that has been transcytosed to the lumen is released as free secretory component (SC) [11]. Thus, in contrast to the serum IgA that is mostly monomeric, SIgA, originates from two different cell types to form a multi-polypeptide complex [7].
Moreover, serum and SIgA carry out different physiological functions and these differences may be primarily attributable to the high degree of glycosylation of SC [12]. The extensive glycosylation on both the IgA heavy chain (Fcα) and SC allow for effective interactions with pathogens that competitively inhibit pathogen binding to host cells [12–14]. IgA dimer formation is dependent upon the N-glycan site to provide the correct J Chain conformation [15, 16]. The glycans on SC are also known to be involved in anchoring the SIgA to mucus lining the epithelial surface through carbohydrate residues [17].
1.2 Objectives of the work
The HIV-1 specific neutralizing mAb, IgG1 b12, protects rhesus macaques against vaginal challenge with Simiam/Human Immunodeficiency Virus (SHIV) following either topical application or systemic intravenous delivery [18–20]. In a separate study, we generated and characterized an isotyped switched IgA2 b12 molecule [21]. Our objective in the present study was to develop a method by which we could generate SIgA2 b12 that could potentially be used in passive protection studies in the macaque SHIV challenge model. Humans express two subclasses of IgA, IgA1 and IgA2, that carry distinct heavy chain constant regions [22]. The hinge of IgA2 is 13 residues shorter than the IgA1 hinge and is uniquely resistant to a specific group of microbial proteases that cleave IgA1 but not IgA2, making it particularly valuable in the distal gut [23]. The extended 23 amino acid hinge region of IgA1 contains nine potential O-glycosylation sites [24]. As noted above, this variety of sugar epitopes on IgA subclasses and SC provide the capability to bind and compete with adhesins found on the surface of many pathogens thus increasing the role in innate immunity [12]. Differing from the T-shape formed by the Fab arms of SIgA1, the abbreviated hinge sequence of SIgA2 results in a unique nonplanar structure [25]. A graphical representation of the various forms of human IgA that includes a depiction of the variations in carbohydrate moieties is shown in Figure 1. About 62% of the IgA in the colon is SIgA2, but SIgA2 is also prominent in mammary and salivary glands and in the female urogenital tract [26]. Thus, we anticipated that SIgA2 b12 could potentially be a valuable tool to assess immune exclusion against HIV-1 mucosal exposure.
Figure 1. Schematic representation of the molecular forms of human IgA.

The constant regions of the heavy chain are shown in blue and the variable heavy chain domains are shown in yellow. Variable light chain domains are shown in gray with constant regions shown in orange. J chain is shown in green on the dimeric and secretory forms of the antibody. The 5-domain secretory component is shown in pink. N-glycans are shown in black as Y shapes; O-glycans are shown as black circles. Symbols and placement of glycans are adapted from Royle, et al. 2003 [12].
1.3 Rationale for innovating in vitro methods to produce SIgA
Early immunological studies of SIgA required tedious purification methods to isolate SIgA and free SC from human colostrum yielding small amounts and non-pathogen specific antibodies [27]. Subsequently, co-culturing pIgR-expressing with IgA-producing cells yielded SC-dIgA complexes that were successfully transported from the basolateral into the lumenal compartment [28]. Soon thereafter, in vitro combination of purified dIgA and free SC was achieved demonstrating that the rhSC could re-associate with dIgA purified from hybridoma cells [29]. Berdoz and colleagues later reported the in vitro synthesis of SIgA by CHO cells sequentially transfected with four different genetic elements [30]. Their approach suggested it was possible to produce larger quantities of SIgA for immunological research or clinical applications. Most recently, production of SIgA and free SC from colostrum was improved by modifying the technique to remove lactoferrin, a problematic contaminant in earlier methods [1, 31].
For a separate purpose, we had successfully produced large quantities of dIgA2 b12 with associated J chain. In anticipation of producing SIgA2 b12, we subsequently cloned a CHO cell line secreting rhSC. We reasoned that since others have shown that SIgA can be produced by combining dIgA and SC in vitro, other methods of in vitro combination were likely possible. Thus, we innovated the combination process by allowing direct contact between secreted rhSC in CHO cell supernatant with purified dIgA immobilized on a Protein L affinity matrix. Our goal was to avoid multiple purifications of rhSC and in vitro combinations needed to obtain the large quantities required for in vivo experiments. Naturally occurring hybrid SIgA antibodies are remarkably stable with the SC disulfide linked to one of the IgA subunits. We discovered that once bound to the dIgA2 b12 on the Protein L column, the rhSC in the CHO cell supernatant was not disrupted during a pH-lowering elution step, thus providing a one-step purification process yielding recombinant SIgA2 b12 macromolecules.
2. Materials and methods
2.1 Antibodies
IgG b12 is a human antibody (IgG1, kappa) that recognizes an epitope overlapping the CD4 binding site of gp120 [18]. The generation of an IgA2 m(1) version of IgG1 b12 has been fully described elsewhere [21]. Recombinant IgG1 b12 and dIgA2 b12 were expressed in CHO-K1 cells and purified as previously described [20, 21].
2.2 Purification of dIgA2 b12
Briefly, supernatant from IgA2 b12/J-chain producing CHO cells [21] was pooled and sterile filtered before polymeric IgA2 antibodies (pIgA2 b12) were purified by Protein L affinity chromatography (Thermo Fisher Scientific, Rockford, IL). Purified pIgA2 b12 was concentrated and dimeric IgA2 b12 was further isolated by size exclusion chromatography as previously described [21] (AKTA FPLC; GE Healthcare, Piscataway, NJ). Final yields indicated that pIgA2 b12 contained approximately 30% dIgA2 b12.
2.3 Generation of recombinant human secretory component
A plasmid expressing the human secretory component was kindly provided by Dr. Jenny Woof and fully described elsewhere [32]. CHO cells were transfected with the pcL-2 rhSC plasmid and grown in RPMI supplemented with 10% FCS, 100units/ml of Penicillin, 100μg/ml of Streptomycin, 2mM L-glutamine and 200 μg/ml of Hygromycin B.
2.4 ELISAs
2.3.1 Binding of b12 antibodies to HIV-1 gp120 antigen
Binding of IgA b12 antibodies to HIV-1JRFL gp120 was measured by coating ELISA plates (Corning Life Sciences, Lowell, MA) with HIV-1JRFL gp120 at 2 μg/ml overnight at 4°C. Plates were blocked with 3% BSA for 30 min before serial dilutions of the IgA variants were incubated for 1h at room temperature. Binding was detected with an AP conjugated goat anti-IgA α-chain antibody (Sigma Aldrich, St Louis, MO) and a phosphatase substrate (Sigma Aldrich, St Louis, MO).
2.3.2 Anti-SC capture/anti-IgA detection ELISA
The generation of SIgA2 b12 was evaluated by coating ELISA plates (Corning Life Sciences, Lowell, MA) with an rabbit anti-SC antibody (Dako, Carpinteria, CA) overnight at 4°C. Plates were blocked with 3% BSA for 1h before serial dilutions of IgA antibodies were incubated for 1.5h at room temperature. Binding of SIgA was detected with an AP conjugated goat anti-human IgA α-chain antibody (Sigma, St Louis, MO) and a phosphatase substrate (Sigma Aldrich, St Louis, MO).
2.5 Gel and Western blot
Antibodies were analyzed by SDS-PAGE on 4–15% Mini-Protean TGX polyacrylamide gels (Biorad, Hercules, CA). 2–4 μg of each antibody were used and when indicated reduced using 100 mM dithiothreitol (DTT). Gels were stained using SimplyBlue (Invitrogen, Carlsbad, CA). The Precision Plus Protein standard (Biorad, Hercules, CA) was included to estimate the size of the bands.
For western blots, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Biorad, Hercules, CA) and the membrane was blocked in 1:1 v/v of Odyssey blocking buffer (Li-cor, Lincolm, NE) and PBS. The membrane was incubated for 1h with a mixture of mouse anti-IgA2 α-chain (Southern Biotech, Birmingham, AL) and goat anti-SC (Sigma Aldrich, St Louis, MO) followed by a mixture of goat anti-mouse IgG Alexa Fluor 680 (Invitrogen, Carlsbad, CA) and rabbit anti-goat-IgG Dylight 800 (Rockland Immunochemicals, Inc. Gilbertsville, PA). Membrane was scanned using the Odyssey Infrared Imager (Li-cor, Lincolm, NE). The Precision Plus Protein standard (Biorad, Hercules, CA) was included to estimate the size of the bands.
2.6 Neutralization assays
The pseudovirus based neutralization assay was done as previously described [33]. In brief, HIV-1 envelope pseudovirus was generated by cotransfection of 293T cells with HIV-1 Env-expressing plasmid (pSVIII) and pSG3ΔEnv. Serial dilutions of b12 antibodies and control antibodies were preincubated with a dose of 250 TCID50 pseudovirus for 1 h at 37°C before being added to TZM-bl cells. Luciferase reporter gene expression was evaluated 48 hours later. The antibody and plasma dilution resulting in 50% reduction (IC50) was calculated by regression analysis using GraphPad Prism.
3. Approach and results
3.1 Loading of Protein L column with dIgA2 b12
After purification of dIgA2 b12 by size exclusion chromatography and experiments to establish the optimal ratio of dIgA2 b12 per ml of Protein L to generate SIgA2 b12 (data not shown), we loaded 20 mg of purified dIgA2 b12 onto a column containing 5 ml of Protein L resin (Thermo Fisher Scientific, Rockford, IL) for each batch of purified SIgA2 b12. In order to allow full exposure to the Protein L and to promote efficient binding, we incubated the beads and protein for a total of 4 hours with gentle rocking at 4°C. We then allowed the resin to settle in an upright position before allowing the unbound dIgA to flow through the column, which was then collected and retained. We note that Protein L is specific for kappa chain containing antibodies and the procedure must be adjusted for lambda chain containing antibodies.
3.2 Purification of SIgA2 b12
For large-scale production, we expressed rhSC in Corning hyperflask cultures from pcl-2 transfected CHO cells. Two liters of media containing secreted rhSC were pooled from 4 hyperflasks and centrifuged to pellet any cells contained in the supernatant that was then 0.22 μm filtered prior to loading onto the dIgA-bound Protein L column. We flowed the supernatant over the column using peristaltic pumps (BioRad, Hercules, CA) set at a rate of 0.5 ml/min. The generated SIgA was eluted with 0.1 M citric acid, pH 3.0 and immediately neutralized to pH 7.0 with Tris base, pH 9.0. An elution profile of collected 1 ml fractions was evaluated and the peak fractions containing SIgA were pooled (data not shown) for dialysis and concentration before being 0.22 μm filtered for further analyses. All steps were performed under sterile conditions. Average yield was 6 mg, ranging from 4–9 mg per batch (20 mg dIgA + 2L pcl-2 supernatant). According to standard procedure for antibody preparations for animal studies, batches were assayed for the presence of endotoxin using the Limulus Amebocyte Lysate assay (Lonza, Walkersville, MD). Columns were washed and re-equilibrated with 250–300 ml 1x PBS prior to and after elution and stored in 1xPBS/0.05% sodium azide.
3.3 Analysis of purified antibody
In order to determine if the eluted antibody was of the expected size, we examined samples of mIgA2 b12, dIgA2 b12 and SIgA2 b12 by SDS-PAGE under reducing and non-reducing conditions (Figure 2A). The reduced samples each show identical bands at 60 kDa for heavy chain and 25 kDa for light chain representing the polypeptide chains that comprise each antibody. J chain is not visible at 15 kDa for dIgA2 and SIgA2 probably due to the extremely small quantity in the loaded amount of protein and the sensitivity of the SimplyBlue stain. However, in earlier studies we have confirmed the expression of J chain in the CHO supernatant and the purified pIgA2 and dIgA2 preparations by Western blot and gel filtration [21]. IgG1 b12 was included for comparison and resolves in two bands (50 kDA for heavy chain and 25 kDa for light chain). The molecular mass of SC is 79.6 kDa containing five immunoglobulin variable domains of 100–110 residues each. Notably, a band at approximately 80 KDa was observed only in SIgA2 b12 that corresponds to rhSC. Under non-reduced conditions mIgA2 b12, dIgA2 b12, and SIgA2 b12 were expected to run at approximately 170 kDa (H2L2), 360 kDa (H4L4J) and 440 kDa (H4L4JSC), respectively. Under non-reducing conditions, IgA is often seen in multiple bands as a ‘smear’ [21, 31, 32]. A separate band corresponding to rhSC was not detected. A band at 50 kDA was present in all IgA preparations, which corresponds to light chain dimers unique to the IgA2m(1) allotype. IgG1 b12 was included for comparison and is seen as a single band at 160 kDa (MW 150kDa).
Figure 2. Analysis of SIgA2 b12 by SDS-PAGE and western blot.
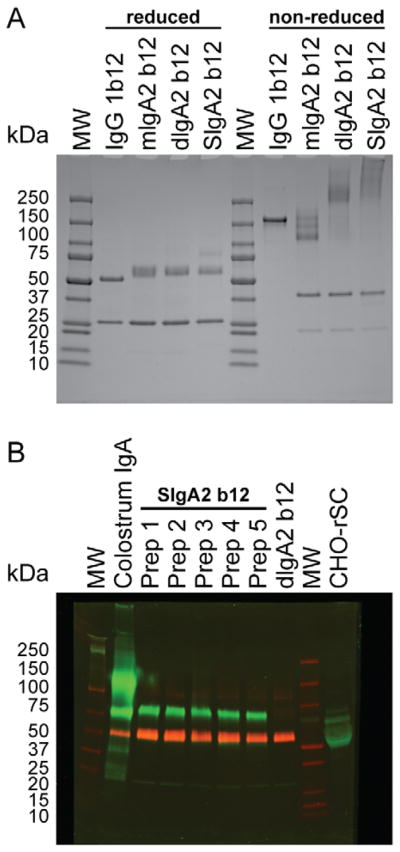
(A) Reduced and non-reduced SDS-PAGE on 4–15% gradient Tris-HCl gels of IgG1 b12, mIgA2 b12, dIgA2 b12 and SIgA2 b12. Lanes are labeled with sample identification. Protein molecular weight markers are indicated with MW. (B) Anti-secretory component and anti-IgA2 α-chain western blot. Under reduced conditions, five separate preparations of SIgA2 b12 (Prep 1 to 5) were compared to human colostrum IgA, dIgA2 b12 and supernatant from the secretory component producing CHO cell line (CHO-rhSC). Samples were probed with anti-SC (green) and anti-human IgA α-chain (red) antibodies. Protein molecular weight markers are indicated with MW.
We examined purified batches of SIgA2 b12 in Western blot to visualize SC as part of the eluted antibody and to compare batch-to-batch reproducibility (Figure 2B). Under reduced conditions, five separate preparations of SIgA2 b12 (Prep 1 to 5) were compared to human colostrum IgA, dIgA2 b12 and supernatant from the secretory component producing CHO cell line (CHO-rhSC). Incorporation of rhSC was observed for the SIgA2 b12 preparations (green band at ~80kDa). As expected, the band was also present in colostrum IgA and CHO-rhSC but not in dIgA2 b12. The red band corresponds to IgA2 heavy chain (not present in CHO-rhSC).
In order to verify that intact SIgA2 b12 had been eluted from the column, we performed a secretory component specific IgA ELISA (Figure 3A). SIgA2 b12 was captured as efficiently as human colostrum IgA. However, no capture was observed with dIgA2 b12. CHO cell supernatant containing recombinant secretory component (CHO-rhSC) was included as an assay negative control.
Figure 3. Detection of secretory component and binding to HIVJRFL gp120.
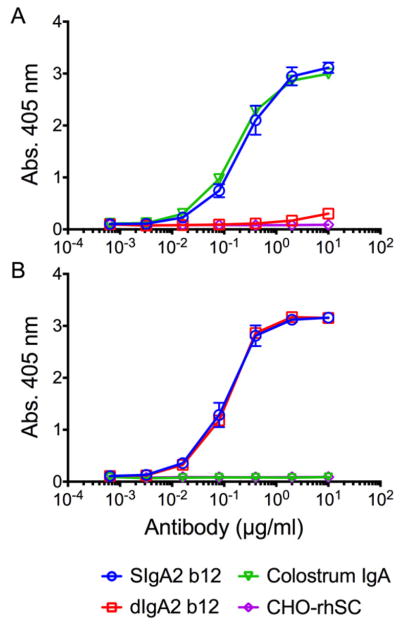
(A) Secretory component specific IgA ELISA. Serially diluted samples were captured onto ELISA plates pre-coated with anti-SC antibody. SIgA binding was detected with AP-conjugated anti-human IgA α-chain. (B) HIVJRFL gp120 ELISA. SIgA2 b12 retained specificity for HIVJRFL gp120 with similar apparent affinity as dIgA2 b12. HIVJRFL gp120 was pre-coated on ELISA plates before incubation with serially diluted samples. Binding was detected with AP-conjugated anti-human IgA α-chain. Human colostrum IgA and CHO-rhSC did not bind HIVJRFL gp120. Note that for both (A) and (B) CHO-rSC is unpurified supernatant from a secretory component producing CHO cell line and values for CHO-rSC are not the actual concentration. Individual colors and shapes are used to distinguish samples. Values are means and SDs of duplicate wells.
We also tested the antigen specificity of the SIgA2 b12 in an HIV-1 gp120 ELISA (Figure 3B). SIgA2 b12 retained specificity for HIVJRFL gp120 with similar apparent affinity compared to dIgA2 b12 whereas human colostrum IgA did not bind HIVJRFL gp120. CHO cell supernatant containing recombinant secretory component CHO-rhSC was included as an assay negative control.
Finally, to ensure that the synthesized SIgA2 b12 retained the ability to neutralize HIV-1, we tested the purified SIgA2 b12 in a pseudovirus neturalization assay (Figure 4). The results demonstrated that SIgA2 b12 retained a comparable neutralization potency as dIgA2 b12 and mIgA2 b12 against HIV-1JRFL and HIV-1JRCSF, subtype B viruses that are efficiently neutralized by IgG1 b12. Although not directly shown here, the IC50 of IgG1 b12 against many HIV-1 isolates is well-established. In addition to the data in Figure 4, a comparison of IC50 data based on several repetitions of the neutralization assay are as follows:
Figure 4.

Neutralization of HIVJRFL and HIVJRCSF by SIgA2 b12. Neutralization potency of the various forms of IgA2 b12 was evaluated in the TZMbl-based pseudovirus assay against (A) HIVJRFL and (B) HIVJRCSF. No neutralization was observed using the anti-dengue antibody DEN3. Individual colors and shapes are used to distinguish samples. Values are means and SDs of triplicate wells.
JRFL: IgG 0.02ug/ml, SIgA 0.1ug/ml, dIgA 0.08ug/ml, mIgA 0.09ug/ml
JRCSF: IgG 0.2ug/ml, SIgA 0.6ug/ml, dIgA 0.7ug/ml, mIgA 1.0ug/ml
3.4 Summary
In our earlier work [21], we characterized the newly engineered antibody, dIgA2 b12, by size exclusion chromatography and SDS-PAGE [34]. Here, we endeavored to synthesize functional SIgA2 b12 of high purity by developing an uncomplicated in vitro scheme for combination that would provide large yields for possible in vivo studies. Although several studies have reported successful in vitro combination of rhSC and dIgA [28, 29, 32, 35], there was evidence that not all SC binds covalently in SIgA2 [31]. We therefore proceeded cautiously to ensure that we could produce the correctly combined hybrid molecule, and we show here that we were able to produce SIgA2 b12 with bound rhSC. Our combination process yielded an average of 6 mg of purified SIgA2 b12 per purification batch. Afinity purifying intact SIgA, including a step of acid elution, was possible due to the bonding, whether covalent or non-covalent, between rhSC and the α-chain of dIgA that make it naturally well-suited to resist acid denaturing. We have demonstrated that the co-purified SIgA2 b12 retains all of the antigen binding and neutralization properties of the parent IgG1 b12 and the precursor dIgA2 b12 antibodies.
4. Discussion
Relatively few mucosal vaccines have been approved for humans, but the large numbers of mucosal infections represent a global challenge for vaccine development to prevent diseases that result from the colonization of surface epithelium. Inducing specific SIgA antibodies that could elicit immunological and systemic memory against pathogens leading to gastrointestinal, respiratory and genital tract infections are highly desirable goals. Our initial objective in developing this new method of producing SIgA was to generate the antibody in both the quantity and quality required for use in topical pre-clinical studies to further our HIV-1 antibody protection and vaccine-related knowledge. We have shown that SIgA2 b12 can potentially block early events of HIV-1 transit [21], and we have shown that IgG1 b12, when applied topically in the vagina of female rhesus macaques, can block infection [19]. Therefore, topically applying SIgA2 b12 in a similar experiment may be a practical next step. Nevertheless, we view the topical administration of SIgA2 b12 in nonhuman primates only in terms of a proof-of-concept experiment. If SIgA2 b12, in this experimental context, successfully protects against vaginal mucosal challenge, then it is reasonable to predict that a vaccine eliciting a protective SIgA could be possible. Therefore, for mucosally transmitted pathogens such as HIV where it is believed that SIgA can block the earliest events in HIV-1 transit across mucosal surfaces, critical research could be accelerated by simplifying the production of SIgA antibodies. We discovered that because of the remarkable stability of SIgA and its ability to maintain functional activity for extended periods in hostile environments including the gut and mouth, it can also withstand an acid elution step and remain an intact macromolecule. This characteristic allows for a unique opportunity for protein purification that would be prohibitive for most hybrid molecules. Using the methods described here, existing cell lines expressing antigen specific pIgA antibodies secreted in cell culture supernatants could easily be combined or possibly co-purified to produce functional SIgA molecules that could then be evaluated in vitro or in vivo for efficacy against a variety of pathogens.
Highlights.
Efficient method to synthesize SIgA using column chromatography
Synthesized SIgA remains stable during mild acid elution
Synthesized SIgA retains functional properties of parent antibody
Acknowledgments
We thank Diane Kubitz and staff at the Antibody Production Core at The Scripps Research Institute for purification of dIgA2 b12. We thank Christina Corbaci for graphic design and artwork. This work was supported by the National Institutes of Allergy and Infectious Diseases grant R01 AI33292 (DRB), Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery grant UM1AI100663 (DRB), and the Lundbeck Foundation (BM).
Abbreviations
- SC
secretory component
- rhSC
recombinant human secretory component
- mAb
monoclonal antibody
- IgA
immunolglobulin A
- IgG
immunolglobulin G
- dIgA
dimeric IgA
- mIgA
monomeric IgA
- SIgA
secretory IgA
- J chain
joining chain
- pIgR
polymeric Ig receptor
- SHIV
Simian/Human Immunodeficiency Virus
- PCs
plasma cells
- CHO
chinese hamster ovary
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mantis NJ, Rol N, Corthesy B. Mucosal immunology. 2011;4:603–611. doi: 10.1038/mi.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lancet. 1994;344:1239–1241. [PubMed] [Google Scholar]
- 3.Dickinson EC, Gorga JC, Garrett M, Tuncer R, Boyle P, Watkins SC, Alber SM, Parizhskaya M, Trucco M, Rowe MI, Ford HR. Surgery. 1998;124:284–290. [PubMed] [Google Scholar]
- 4.Brandtzaeg P, Johansen FE. Immunological reviews. 2005;206:32–63. doi: 10.1111/j.0105-2896.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 5.Brandtzaeg P, Farstad IN, Johansen FE, Morton HC, Norderhaug IN, Yamanaka T. Immunological reviews. 1999;171:45–87. doi: 10.1111/j.1600-065X.1999.tb01342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandtzaeg P. Nature. 1974;252:418–420. doi: 10.1038/252418a0. [DOI] [PubMed] [Google Scholar]
- 7.Norderhaug IN, Johansen FE, Schjerven H, Brandtzaeg P. Critical reviews in immunology. 1999;19:481–508. [PubMed] [Google Scholar]
- 8.Johansen FE, Braathen R, Brandtzaeg P. Scandinavian journal of immunology. 2000;52:240–248. doi: 10.1046/j.1365-3083.2000.00790.x. [DOI] [PubMed] [Google Scholar]
- 9.Strugnell RA, Wijburg OL. Nature reviews Microbiology. 2010;8:656–667. doi: 10.1038/nrmicro2384. [DOI] [PubMed] [Google Scholar]
- 10.Gibson A, Futter CE, Maxwell S, Allchin EH, Shipman M, Kraehenbuhl JP, Domingo D, Odorizzi G, Trowbridge IS, Hopkins CR. The Journal of cell biology. 1998;143:81–94. doi: 10.1083/jcb.143.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaetzel CS. Immunological reviews. 2005;206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 12.Royle L, Roos A, Harvey DJ, Wormald MR, van Gijlswijk-Janssen D, Redwanel RM, Wilson IA, Daha MR, Dwek RA, Rudd PM. The Journal of biological chemistry. 2003;278:20140–20153. doi: 10.1074/jbc.M301436200. [DOI] [PubMed] [Google Scholar]
- 13.Mestecky J, Moldoveanu Z, Smith PD, Hel Z, Alexander RC. Journal of reproductive immunology. 2009;83:196–200. doi: 10.1016/j.jri.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrier C, Sprenger N, Corthesy B. The Journal of biological chemistry. 2006;281:14280–14287. doi: 10.1074/jbc.M512958200. [DOI] [PubMed] [Google Scholar]
- 15.Johansen FE, Braathen R, Brandtzaeg P. J Immunol. 2001;167:5185–5192. doi: 10.4049/jimmunol.167.9.5185. [DOI] [PubMed] [Google Scholar]
- 16.Vaerman JP, Langendries A, Giffroy D, Brandtzaeg P, Kobayashi K. Immunology. 1998;95:90–96. doi: 10.1046/j.1365-2567.1998.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phalipon A, Cardona A, Kraehenbuhl JP, Edelman L, Sansonetti PJ, Corthesy B. Immunity. 2002;17:107–115. doi: 10.1016/s1074-7613(02)00341-2. [DOI] [PubMed] [Google Scholar]
- 18.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PW, Sawyer LS, Hendry RM, Dunlop N, Nara PL, et al. Science. 1994;266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 19.Veazey RS, Shattock RJ, Pope M, Kirijan JC, Jones J, Hu Q, Ketas T, Marx PA, Klasse PJ, Burton DR, Moore JP. Nature medicine. 2003;9:343–346. doi: 10.1038/nm833. [DOI] [PubMed] [Google Scholar]
- 20.Parren PW, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, Moore JP, Burton DR. Journal of virology. 2001;75:8340–8347. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantis NJ, Palaia J, Hessell AJ, Mehta S, Zhu Z, Corthesy B, Neutra MR, Burton DR, Janoff EN. J Immunol. 2007;179:3144–3152. doi: 10.4049/jimmunol.179.5.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kerr MA. The Biochemical journal. 1990;271:285–296. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kett K, Brandtzaeg P, Radl J, Haaijman JJ. J Immunol. 1986;136:3631–3635. [PubMed] [Google Scholar]
- 24.Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, Rudd PM, Woof JM, Dwek RA. The Journal of biological chemistry. 1998;273:2260–2272. doi: 10.1074/jbc.273.4.2260. [DOI] [PubMed] [Google Scholar]
- 25.Bonner A, Furtado PB, Almogren A, Kerr MA, Perkins SJ. J Immunol. 2008;180:1008–1018. doi: 10.4049/jimmunol.180.2.1008. [DOI] [PubMed] [Google Scholar]
- 26.Prigent-Delecourt L, Coffin B, Colombel JF, Dehennin JP, Vaerman JP, Rambaud JC. Clinical and experimental immunology. 1995;99:221–225. doi: 10.1111/j.1365-2249.1995.tb05536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loomes LM, Stewart WW, Mazengera RL, Senior BW, Kerr MA. Journal of immunological methods. 1991;141:209–218. doi: 10.1016/0022-1759(91)90147-8. [DOI] [PubMed] [Google Scholar]
- 28.Hirt RP, Hughes GJ, Frutiger S, Michetti P, Perregaux C, Poulain-Godefroy O, Jeanguenat N, Neutra MR, Kraehenbuhl JP. Cell. 1993;74:245–255. doi: 10.1016/0092-8674(93)90416-n. [DOI] [PubMed] [Google Scholar]
- 29.Rindisbacher L, Cottet S, Wittek R, Kraehenbuhl JP, Corthesy B. The Journal of biological chemistry. 1995;270:14220–14228. doi: 10.1074/jbc.270.23.14220. [DOI] [PubMed] [Google Scholar]
- 30.Berdoz J, Blanc CT, Reinhardt M, Kraehenbuhl JP, Corthesy B. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3029–3034. doi: 10.1073/pnas.96.6.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Almogren A, Senior BW, Kerr MA. Immunology. 2007;120:273–280. doi: 10.1111/j.1365-2567.2006.02498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis MJ, Pleass RJ, Batten MR, Atkin JD, Woof JM. J Immunol. 2005;175:6694–6701. doi: 10.4049/jimmunol.175.10.6694. [DOI] [PubMed] [Google Scholar]
- 33.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Journal of virology. 2005;79:10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morton HC, Atkin JD, Owens RJ, Woof JM. J Immunol. 1993;151:4743–4752. [PubMed] [Google Scholar]
- 35.Lullau E, Heyse S, Vogel H, Marison I, von Stockar U, Kraehenbuhl JP, Corthesy B. The Journal of biological chemistry. 1996;271:16300–16309. doi: 10.1074/jbc.271.27.16300. [DOI] [PubMed] [Google Scholar]