

Abstract
Rett syndrome (RTT) is a rare genetic disorder within the autistic spectrum. This study compared socio-demographic, clinical and genetic characteristics of the international database, InterRett, and the population based Australian Rett syndrome database (ARSD). It also explored the strengths and limitations of InterRett in comparison with other studies. A literature review compared InterRett with RTT population-based and case-based studies of thirty or more cases that investigated genotype and/or phenotype relationships. Questionnaire data were used to determine case status and to investigate the comparability of InterRett and ARSD. Twenty four case series, five population based studies and a MECP2 mutation database were identified of which twenty one (70%) collected phenotype and genotype data. Only three studies were representative of their underlying case population and many had low numbers. Of one thousand one hundred and fourteen InterRett subjects, nine hundred and thirty five born after 1976 could be verified as Rett cases and compared with the two hundred and ninety five ARSD subjects. Although more InterRett families had higher education and occupation levels and their children were marginally less severe, the distribution of MECP2 mutation types was similar. The InterRett can be used with confidence to investigate genotype phenotype associations and clinical variation in RTT and provides an exemplary international model for other rare disorders.
Keywords: Rett syndrome, international database, rare disorder, MECP2, phenotype
Rett syndrome (RTT) is characterised by severe intellectual and physical disability and affects 1 in 8500 females by the age of 15 years (B. Hagberg, 1985; Laurvick et al., 2006). In general, the most notable characteristics of RTT are the development of stereotypic hand movements and a regression in motor and cognitive function usually after a period of normal development (Trevathan, 1988). Other features include development of motor impairments including apraxia, epilepsy, scoliosis and breathing abnormalities.
Criteria for classical Rett syndrome were originally developed in 1988 to help clinicians diagnose the disorder (Trevathan, 1988) and included the following: normal fetal and postnatal development; a normal head circumference at birth often followed by deceleration of head growth; loss of hand skills; impaired language skills; development of stereotypic hand movements; the development of an impaired gait. However in the following decade it became clear that there was considerable clinical variability in this disorder and consequently the term variant was used to describe these phenotypes which did not conform exactly to the original criteria (Hagberg & Skjeldal, 1994). Examples of these were the “preserved speech” variant (Renieri et al., 2008) where individuals retained some words or speech, the “forme fruste” variant where the clinical features were milder overall (Hagberg & Skjeldal, 1994) and the congenital variant where symptoms were present from birth (Ariani et al., 2008).
In 1999 the link between Rett syndrome and the methyl CpG binding protein 2 (MECP2) was identified (Amir et al., 1999). It subsequently became apparent that pathogenic MECP2 mutations were indeed present in many individuals who did not meet the criteria for the classical form of the disorder, e.g. in those without normal early development or without demonstrated head growth deceleration. Consequently the diagnostic criteria were amended (B. Hagberg, 2002) to account for this broader phenotype.
The presence of a MECP2 mutation often provides confirmatory evidence of RTT (B. Hagberg, 2002) and testing for MECP2 mutations has been available in North America (Percy et al., 2007) and countries such as UK (Kerr & Prescott, 2005), Israel (Yaron et al., 2002), Australia (Colvin et al., 2004), and in European countries such as Spain (Monros et al., 2001), France (Bienvenu et al., 2000), Germany (Huppke, Held, Hanefeld, Engel, & Laccone, 2002) and Italy (Vacca et al., 2001), at least in a research capacity, since 2000. Over 200 different pathogenic MECP2 mutations have been identified and one focus of recent research has been to investigate whether there is a relationship between specific clinical features (phenotype) and specific genetic mutations (genotype) (Bebbington et al., 2008; Chae, Hwang, Hwang, Cheong, & Kim, 2004; Charman et al., 2005; Colvin et al., 2004; Hoffbuhr et al., 2001; Huppke, Held, Laccone, & Hanefeld, 2003; Monros et al., 2001; Neul et al., 2008; Nicolao et al., 2001).
Early genotype and phenotype studies were often based on small sample sizes (Amir et al., 2000; Cheadle et al., 2000; Monros et al., 2001; Nicolao et al., 2001) and lacked the power necessary to investigate specific MECP2 mutations separately and differentiate between similar phenotypes. Even where data have been pooled from different sources the number of cases for specific mutations has still been relatively small. An example includes a study in which data from three countries, Japan, Australia and the United Kingdom, were combined to enable the phenotype of one specific MECP2 mutation, p.R133C, to be compared with that of other mutations (Leonard et al., 2003).
The Australian Rett syndrome Database (ARSD), initially established in 1993, is the only ongoing RTT population based study worldwide (Laurvick et al., 2006; Leonard, Bower, & English, 1997; Moore, Leonard, Fyfe, De Klerk, & Leonard, 2005). Potential RTT cases are ascertained by the ARSD through several sources including the Australian Paediatric Surveillance unit (a national network which facilitates the reporting of rare disorders by paediatricians) and the parent support group, the Rett Syndrome Association of Australia (Leonard, Bower, & English, 1997). Ascertainment has been estimated as 93% complete (Leonard, Bower, & English, 1997) and by the end of 2007 the ARSD contained 311 diagnosed cases of RTT (Downs et al., 2008).
InterRett, an international database of RTT cases, was set up in 2002 to contribute larger case numbers for genotype and phenotype investigations than could be provided by the ARSD which is based on a national population of just over 21 million people (Bebbington et al., 2008; Fyfe, Cream, de Klerk, Christodoulou, & Leonard, 2003; Leonard et al., 2005; Moore, Leonard, Fyfe, De Klerk, & Leonard, 2005). Unlike the ARSD, InterRett participants are determined by a variety of mechanisms including bulk submission of data from a number of countries, parent listservs and advertisement in newsletters and parent support associations. InterRett data are collected worldwide from family members with a child with RTT and by clinicians who have a patient with RTT via web and paper based questionnaires. Family (FQ) questionnaires were based on those already used by the ARSD (Laurvick et al., 2006) and clinician (CQ) questionnaires were developed by an international reference panel consisting of families with a child with RTT, clinicians, therapists and researchers. These questionnaires have been translated into French, Spanish, German, Italian and Mandarin. Data have also been collected from Spain, France, China, Israel and Canada (using the mechanisms described in table 1) and cases from these sources represent over half of the 1293 non-ARSD cases currently in the database (Anderson, personal communication, July 2008).
Table 1. Mechanism used to collect and submit bulk data to InterRett.
| Source | Mechanism of data collection &/or data submission | n |
|---|---|---|
| Spain | A FileMaker database was provided to clinician A Clinician A entered cases based on own protocol. | 318 |
| Israel | Clinician B extracted data from clinical records and entered in online questionnaire | 74 |
| OR Physiotherapist X translated questionnaires into Hebrew and administered to families. Questionnaires then entered online in English. | 13 | |
| Canada | Data from clinical records of clinician C patients extracted and entered data online. | 71 |
| China | Clinician D translated family questionnaires into English. Questionnaires administered families attending D's clinic. Responses translated back into English and then returned to InterRett in paper form. | 31 |
| France | Clinician E adapted family questionnaire and added new questions to E's area of interest. Questionnaires administered through a family association. Responses returned to InterRett in form of de identified excel spreadsheet. | 232 |
| Total | 739 |
In 2006, an investigation of genotype and phenotype relationships was carried out using the InterRett database(Bebbington et al., 2008). The study demonstrated that p.R270X and p.R255X MECP2 mutations were associated with a more severe phenotype and p.R133C and p.R294X with a milder phenotype. At the time of publication there was no other known RTT study which had used an international dataset as large to examine specific genotype and phenotype relationships. While InterRett is a large dataset, it does contain data from diverse sources and is not population-based.
This study aimed to:
examine the strengths and limitations of InterRett in relation to other registers, databases, or studies involving approximately 30 or more cases.
to determine whether the socio-demographic, clinical and genetic characteristics of InterRett subjects are congruent with those of a population-based study (the Australian Rett syndrome database – ARSD).
Methods
a) Literature Review
A review of the literature identified studies published in English speaking journals investigating Rett syndrome genotypes and/or phenotypes. Articles published between 2000 and 2008 (inclusive) were selected from a Medline search using the search terms “Rett syndrome” and “phenotype” or “genotype”. A broad search of RTT population based studies and case series was conducted for studies before 2000.
Parameters such as sample size, data source and case validation method were then compared. Estimated RTT cases per year were determined for studies that appeared to collect cases from a specific catchment area by dividing the catchment area population by 1000, multiplying by the birth rate and then multiplying the reciprocal of the sex birth-ratio plus one. Population data were extracted from the CIA world fact book (The Central Intelligence Agency, 2008). The catchment specific RTT birth estimate was then divided by the RTT incidence in Australia as the ARSD is the only current RTT population based study.
b) Comparison of socio-demographic, clinical and genetic characteristics of InterRett and ARSD subjects
Data source
Participants
Data on a total of 1114 cases (12 of whom were deceased) on whom information had been submitted to InterRett from 2003 to June 2007, were available for analysis. Data from 312 validated ARSD [3] cases, born after 1976, including 39 deceased, were available for comparison. InterRett and ARSD cases were mutually exclusive.
Data management
Case status was defined using a uniform verification system applicable to both databases (Fig 1). This was devised to account for the differing data content provided by the various sources. Data could be available for both InterRett and ARSD participants from a questionnaire completed by a family member (FQ only), clinician (CQ only) or both (FQ & CQ). Included amongst the InterRett participants were the Spanish (n=314) and French (n=232) cases (Table 2). Participants with a clinician's diagnosis were coded as clinically definite, those without a clinician's diagnosis but who met Hagberg's revised criteria (B. Hagberg, Hanefeld, Percy, & Skjeldal, 2002) were classified as phenotypically definite and those with insufficient information to meet either of these criteria but with a known MECP2 mutation were classified as genotypically definite. Subjects who met any of these three classifications were regarded as verified RTT cases for this study (Fig 1). Socio-demographic, clinical (level of severity) and genetic (genetic testing and results) variables were selected for analysis (Table 2).
Figure 1. Flow diagram of methodology used to verify a subject's cases status.

Table 2. Percentage of variables for which data where provided from different data sources.
| ARSD (n=295) |
InterRett (n=1004) |
||||||
|---|---|---|---|---|---|---|---|
| FQ (n=160) |
CQ (n=132) |
FQ & CQ (n=188) |
French (n=213) |
Spanish (n=311) |
|||
| SOCIO-DEMOGRAPHIC | Age at Diagnosis | 89.8 | 95.0 | NA | 98.4 | 89.2 | 99.0 |
| Subject's age at questionnair e completion | 95.3 | 100.0 | 91.7 | 100.0 | 98.6 | 98.1 | |
| Mother's Age at Subject's Birth | 93.6 | 96.3 | NA | 89.9 | 92.0 | NA | |
| Mother's Occupation | 99.7 | 97.5 | NA | 100.0 | 97.2 | NA | |
| Father's Occupation | 99.7 | 97.5 | NA | 100.0 | 98.1 | NA | |
| Mother's Education | 90.8 | 97.5 | NA | 93.6 | NA | NA | |
| Father's Education | 83.1 | 96.3 | NA | 91.5 | NA | NA | |
| Birth Order | 72.9 | 53.8 | NA | 60.1 | 98.1 | NA | |
| GENETIC | MECP2 test performed | 98.3 | 94.4 | 97.0 | 100.0 | 99.1 | 96.8 |
| Results Of MECP2 test | 99.2+ | 73.4+ | 98.4+ | 77.3+ | 59.9+ | 99.0+ | |
| MECP2 mutation identified | 100.0* | 100.0* | 100.0* | 100.0* | 100.0* | 100.0* | |
| CLINICAL | Severity | 80.0ˆ | 62.4ˆ | 31.3ˆ | 96.0ˆ | NA | 94.2ˆ |
Results known given a family and clinician indicated MeCP2 test was performed
Percentage calculated given that family/ clinician indicated yes results form MeCP2 test was performed
Percentage based on MeCP2 positive subjects only
NA -Data not collected
Variables relating to the socio-demographic characteristics of a subject and/or their family could only be assessed from family questionnaires. Follow-up questionnaires provided additional information on parents' occupation, employment and/or birth order (2000, 2002 and 2004) that were not provided in original ARSD family questionnaires (Table 2). Variables relating to clinical characteristics could be assessed from both family and/or clinician questionnaires. Genetic testing and results were assessed from both family and clinician questionnaires (table 2).
Data coding
Socio-demographic variables
Education was categorised using mothers' and fathers' highest educational qualifications [20]. An additional category, “Some Schooling” was created to account for parents who had completed some high school. The Australian and New Zealand Standard for coding of Occupation (ANZSCO) (Australian Bureau of Statistics, 2006) was used to code most recent occupation type.
Clinical severity
For clinical severity a modified version of the Pineda scale, first defined by the Spanish group (Monros et al., 2001) and later used to assess Australian (Colvin et al., 2004) and international (Bebbington et al., 2008) data, was used. Items within the Pineda scale assess phenotype severity using mainly developmental characteristics including onset of hand stereotypies and age milestones for crawling, sitting and walking. A maximum score of 31 is possible from the assessment of 10 items within the Pineda scale with higher score indicating greater severity.
Mutation type
MECP2 mutation type was coded as follows; the eight most common missense and nonsense MECP2 mutations (p.R106W, p.R133C, p.T158M, p.R168X, p.R255C, p.R270X, p.R294X and p.R306C), large deletions and other (e.g. frameshift).
Statistical Analysis
STATA version 9.2 (StataCorp, 2005) was used for analysis. Continuous data were compared between the ARSD and InterRett using an independent sample t-test and categorical information was assessed by Chi squared tests. Multinomial logistic regression reporting relative risk ratios (RRR) was used to adjust for confounders, specifically mother's age at questionnaire completion. Analysis was restricted to subjects born within and after 1976.
Results
Twenty four studies using case series, 5 studies using population based data and 1 MECP2 mutation database met the inclusion criteria for the literature search (Table 3). Of the studies 1 (3.3 %) provided only phenotypic information, 7 only genotypic information (23.3%) and 21 (70.0%) both phenotypic and genotypic information. Only for the population-based studies was there sufficient information to complete all the parameters with information generally poorly provided on case source and country of origin of participants. Only 2 parameters (18.2%) could be completed for the MECP2 mutation database (“RettBASE: IRSA MECP2 Variation Database”). This was largely attributed to the difficulty of determining the exact number of RTT cases as there appeared to be no mechanism in place to identify duplicated cases and the variable nomenclature used to describe the same mutation.
Table 3. Summary of articles, concerning RTT phenotypes and/or genotypes identified from literature search.
| Publication year | Author | N (age distribution in years if available) | Origin of Subjects | Estimate d cases per birth year | Source of cases | Case validation method | Phenotype data (n, % complete data) | Genotype data (n (%), % MECP2 positive) | Data source | Disease severity method | Results of genotype and phenotype associations |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Population-based studies | |||||||||||
| 2008 | Downs et al | 240 (2-29 years) in 2004 | Australia (83% of cases in ARSD) | 14 | ARSD (Australian Paediatric Surveillance unit and Rett Syndrome Association of Australia) | Hagberg 2002 | Information on fractures (234, 98.3%) | 215 (91.9%), 76.3% | Family and/or clinicians | Fractures | Fracture risk was increased specifically in cases with p.R270X mutations and in cases with p.R168X mutations. |
| 2007 | Jian et al. | 162 (2-25 years) in 2000 | Australia (81% of live cases in ARSD) | 14 | ARSD (Australian Paediatric Surveillance unit and Rett Syndrome Association of Australia) | Hagberg 2002 | Information on seizure frequency in 2000 (162,100%) | 154 (95.1%), 72.6% | Family and/or clinicians | Seizure rate | Seizure rates lower in those with p.R294X, p.R255X mutations and C terminal mutations. |
| 2006 | Jian et al | 288 (2-29 years) in 2004 | Australia | 14 | ARSD (Australian Paediatric Surveillance unit and Rett Syndrome Association of Australia) | Hagberg 2002 | Information on onset of seizures (275, 95.5%) | 254 (88.2%), 73.2% | Family and/or clinicians | Onset of seizures | p.R168X mutation conferred a protective effect for seizure onset within the first 4 years, as did p.R294X and C-terminal mutations |
| 2004 | Colvin et al | 247 (2-25 years) | Australia | 14 | ARSD (Australian Paediatric Surveillance unit and Rett Syndrome Association of Australia) | Trevathan 1988 | 295, 100% | 196 (79.3 %), 66% | Family and/or clinicians | Pineda, Percy and Kerr severity scales | Mutations in NLS of TRS associated with more severe phenotype p.R133C associated with milder phenotypes while p.R270X associated with most severe phenotypes |
| 1993 | Kozinetz et al | 103 (2-18 years) | Texas, USA | 17 | Health and education agencies, services and facilities | Trevathan 1988 | 103,100% | Not available, study prior to MECP2 testing | Physician or non-physician based | NA | NA |
| Case series | |||||||||||
| 2008 | Neul et al | 245 | USA | Blue Bird Circle Rett Centre at Texas Children's Hospital or UAB Rett Centre between 1990-2004 | Hagberg 2002 | 245, 100% | 245 (100%), 96.3% | Clinical evaluation and medical history | Clinical Severity Score | p.R168X associated with a more severe phenotype than p.R294X and p.R133C mutations p.R168X less likely to walk, use hands or use words | |
| 2008 | Nectoux et al | 81 (5-50 years) | France | 47 | Not specified | Trevathan 1988 | 81,100% | 81 (100%), 93.8% | Clinical examination (n=70) and questionnair es (n=11) | Kerr | Missense mutations associated with more severe epileptic phenotypes and Nonsense and late truncating mutation associated with less severe phenotype regarding walking |
| 2008 | Bebbington et al | 346 (2-45years) | Spain, France, Israel, USA, UK and other | Parent listserv Rettnet, advertisemt in newsletters of parent support associations and presentation s at meetings as well as bulk data (see Table 1) | Pathogenic MECP2 mutation | 346, 100% | 346 (100%), 100% | Family and/or clinician | Modified Pineda Percy and Kerr severity scale. | Overall p.R270X and p.R255X associated with most severe phenotype. p.R133C and p.R294X mutations were associated with the least severe phenotypes. | |
| 2007 | Zahorkova et al | 87 | Czech & Slovak Republics & Ukraine | Czech:5 Slovakia3 Ukraine 25 | Neurologica l, genetic and paediatric departments | Trevathan 1988 | Not specified | 87 (100%), 78.2% | Not specified | Not assessed | NA |
| 2007 | Archer et al+ | 83 | Australia UK | ARSD, Cardiff Rett & University of Glasgow Rett studies | Hagberg 2002 | 83,100% | 83 (100%), 100% | Family and/or clinicians | Kerr and Pineda scales | Statistically significant reduction in clinical severity for the two mutations (p.T158M and p.R168X) associated with skewed X inactivation | |
| 2007 | Percy et al | 1928 | USA and Canada | USA:251 Canada:2 0 | IRSA members USA and Canada | Clinical diagnosis | Not collected | 1165 (60.4%), 78.5% | Data provided by family indicating, dob, diagnosis, mutation testing and testing results | Not assessed | N/A |
| 2007 | Scala et al [44] | 77 (∼2-33years) | Italy | 28 | Italian RTT database and biobank | Hagberg 2002 | 64, 83.1% | 77 (100%), 23.4% | Not specified | Modified version of Kerr score | Large deletions associate with classic RTT phenotype |
| 2007 | Li et al | 121 (oldest: 24 years) | Chinese | 1017 | Not specified | Hagberg 2002 | Not Specified | 121 (100%), 84.3%. | Not specified | Not assessed | N/A |
| 2007 | Sampieri et al | 219 | Italy | 28 | Medical Genetics Unit of the University Hospital of Siena | Hagberg 2002 | Not collected | 187 (100%), 74.9% | Genetic test results | Not assessed | N/A |
| 2006 | Bienvenu et al | 424 (4-15 years) | France | 47 | Association Francaise du Syndrome Rett | MECP2 mutation testing | Not collected | 424 (100%), 100% | Eight French genetic laboratories | Clinical severity not assessed | NA |
| 2005 | Jian et al + | 524 n=174 (2-29) n=350 (1-54) | Australia UK | ARSD, BIRS and Cardiff study | Trevathan 1988 MECP2 mutation testing | Not applicable to this study-mortality study | 524 (100%), 100% | Family and/or clinicians | Clinical severity not assessed | Mortality was significantly increased in those with p.R270X compared with other mutations | |
| 2005 | Leonard et al.+ | 235 (2-27 years) 85 (3-41 years) | Australia China, UK & US | ARSD InterRett | Trevathan 1988 Reported clinical diagnosis or MECP2 mutation testing | 320,100% | 263 (82%), 68.8% | Family and/or clinicians | Scoring system developed to assess characteri stics in perinatal period and early developm ent | Infants with p.R294X and p.R133C (PZ0.03) were less likely than those with p.R255X to have problems in the perinatal period. The most severe profile overall for early development was associated with mutations p.R255X and p.R270X. | |
| 2005 | Kerr & Prescott | 886 | UK | UK: 37 | British Paediatric Surveillance and pre existing cohort data from 1982 | Clinical examination | 834, 94.1% | 440 (49.7%), 100% | Clinical examination s, reports and postal questionnair es | British Isles scoring system | p.T158M, p.R255X and p.R168X associated with more severe phenotype and p.R306C and p.R133C associated with less severe phenotype. |
| 2004 | Schanen et al | 85 | Not Specified | By referral to study or through contact in clinical setting | 52/85 clinical examination | 85, 100% | 85 (100%), 100% | Medical records, parent report and physical examination | Modified scale by Monros and Amir | Missense mutations associated with less severe phenotypes. No difference in the severity of subjects with mutation located in MBD or TRD. | |
| 2003 | Smeets et al | 30 (women born between 1941 to 1987) | Not specified | Patients seen by first author | Clinically diagnosed | 30, 100% | 30 (100%) , 80% | Medical examination | 2 scoring systems Kerr 2001 & a simplified scoring system devised for study | Generally missense mutations associated with milder phenotypes when compared to early truncating mutations Most severe mutations p.R255X and p.R298X. | |
| 2003 | Chae et al | 42 | Korea | 27 | Not specified | Trevathan 1988 | 42,100% | 42 (100%), 71.4% | Clinical examination | Based on hand and motor performan ce an language and skill at 5 years | Nonsense mutations associated with more severe language retardation and earlier regression than missense mutations |
| 2002 | Huppke et al | 123 | Germany | 38 | Not specified | Clinical diagnosis | 123,100%, | 123 (100%), 95.1% | Clinical data | Own protocol | C deletion group associated with lower severity. Mutations within the truncating NLS associated with higher severity when compared to other truncating mutations |
| 2001 | Hoffbuhr et al | 116 (2-34 years) | USA | Rett syndrome clinic at Kennedy Krieger Institute | Clinical examination | 104, 89.7% | 116 (100%), 62.9% | Clinical examination and telephone interviews OR review of medical history | Severity based on 5 clinical features | Overall c-terminal and nonsense mutations in TRD associated with less sever phenotype. | |
| 2001 | Giunti et al | 75 | Italy | 28 | Rett syndrome research project | Clinical Evaluation | Not specified | 75 (100%), 85.3% | DNA bank of Genetics and Molecular Medicine Unit, Dept of Paediatrics, Uni of Florence | NA | N/A |
| 2001 | Vacca et al | 62 | UK and Italy | Not specified | Trevathan 1988 | Not specified | 62 (100%), 69.4% | Not specified | Severity not measured | N/A | |
| 2001 | Monros et al | 46 +1 male | Spain | 23 | Not specified | Trevathan 1988 | 47,100% | 46 (100%), 100% | Not specified | Pineda severity scale | Truncating mutations associated with more severe phenotype and missense mutations with less severe phenotypes. Large deletions identified in subjects with both. |
| 2000 | Cheadle et al | 60 | Not specified | N/A | UK Rett syndrome Association Genetic bank and samples taken from combined metabolic and genetic studies | Adaptations of Trevathan 1988 and Hagberg 1993 | Not specified | 60 (100%), 75% | Not specified | Derived from assessing hand use, speech and walking | Missense mutations and late truncating mutations associated with more severe phenotypes compared to truncating and early mutations respectively. |
| 2000 | Amir et al | 78 | Texas | Blue Bird Circle Rett Centre | Hagberg 1995 | 48,61.5% | 78 (100%), 71.8% | Most recent Clinical profile | Clinical Severity Score | Scoliosis associated with missense mutations | |
| MECP2 mutation database | |||||||||||
| 2008 (last updated) | http://mecp2.chw.edu.au/ | 33931 | Not Specified | Merging published and unpublished mutation and Polymorphi sm data | Not specified | Not specified | Not specified | Published and unpublished data from international collaborators | Not assessed | NA | |
Exact number of cases could not be determined as RTT cases from published papers were not identified.
Studies include population data and are also supplemented by cases series of another country of origin
Using the number of potential RTT cases within a defined catchment area as an estimate of RTT cases in the study, only the Texan (Kozinetz et al., 1993), Australian (Colvin et al., 2004) and French (Bienvenu et al., 2006) studies could be considered representative of their underlying population of cases. Of the remaining studies where sufficient information was provided, case numbers were all substantially less than would be expected based on the estimated RTT cases born per year. For example Huppke et al (Huppke, Held, Hanefeld, Engel, & Laccone, 2002) reported on 123 participants, approximately only the number that would be expected to be born in Germany over three years, although it was clear that the age range would have been much greater. Moreover age distribution of cases wasn't available for many of the studies.
Overall, the North American study by Percy et al. (2007) contained the most participants (n=1928), InterRett the most culturally diverse subjects (with greater than six countries represented within the sample), and the most common data source was from family and clinicians combined (33.3%). Of the severity scales used the four main types of scales were Percy (also known as the Clinical Severity Score(Neul et al., 2008); used by 20.3 %), Kerr (25 %), Pineda (20.5%), British Isles Rett Syndrome questionnaire (8.3 %) while Leonard et al (Leonard et al., 2005), Smeets et al (Smeets et al., 2003), Hoffbuhr et al (Hoffbuhr et al., 2001), Huppke et al (Huppke, Held, Hanefeld, Engel, & Laccone, 2002), Cheadle et al (Cheadle et al., 2000) and Chae et al (Chae, Hwang, Hwang, Cheong, & Kim, 2004) also used their own specific scales.
Only 21 (70.0 %) of the 30 identified studies investigated phenotype and genotype associations. Overall there was general agreement that pRI33C and C-terminal deletions were associated with milder RTT phenotypes (Bebbington et al., 2008; Charman et al., 2005; Colvin et al., 2004; Hoffbuhr et al., 2001; Huppke, Held, Hanefeld, Engel, & Laccone, 2002; Neul et al., 2008)and that p.R270X, p.R168X and p.R255X mutations were associated with more severe RTT phenotypes (Bebbington et al., 2008; Colvin et al., 2004; Kerr & Prescott, 2005; Neul et al., 2008; Smeets et al., 2003). Inconsistent findings were reported on the influence of late truncating mutations, missense and nonsense mutations. An early study (Cheadle et al., 2000) reported that late truncating mutations were associated with a more severe RTT phenotype, but this was refuted in two later studies (Charman et al., 2005; Nectoux et al., 2008) which found that late truncating mutations were associated with milder RTT phenotypes. The same early study (Cheadle et al., 2000) reported that missense mutations were associated with a more severe phenotype while four subsequent studies (Chae, Hwang, Hwang, Cheong, & Kim, 2004; Monros et al., 2001; Schanen et al., 2004; Smeets et al., 2003) reported the association of missense mutations with a less severe phenotype. In addition, the sample sizes reported generally increased over time.
Participants
Of the 1114 individuals contacted by InterRett, 1004 (90.1%) could be verified as RTT cases by the study protocol (Figure 1). Of the 1004 InterRett participants, 935 were born within and after 1976. Five hundred and forty nine cases (58.7%) were validated by a clinician's diagnosis (clinically definite), a further 369 cases (39.5%) were categorised by the revised criteria (phenotypically definite) and 17 (1.8%) were verified only by the presence of a pathogenic MECP2 mutation (genotypically definite). Geographically the majority of InterRett cases originated from Southern Europe (29.3%), Western Europe (22.4%) and Northern America (20.9%).
Of the 312 cases in the ARSD, 295 (94.6%) could be verified by the study protocol. One hundred and ninety cases (64.4%) were categorised as clinically definite by a clinician's diagnosis, a further 92 cases (31.2%) by the revised criteria only (phenotypically definite) and 13 (4.4%) only by the presence of a pathogenic MECP2 mutation (genotypically definite).
Genetic and Clinical Characteristics
As seen in Table 4 InterRett participants (born 1976 and subsequently) were older at time of questionnaire completion (p<0.001) but similar at age of diagnosis (p=0.697), compared with ARSD participants.
Table 4. Continuous Socio-demographic Characteristics of InterRett Subjects and Families compared with the ARSD.
| ARSD | InterRett (complete) |
InterRett (≥1976) |
||
|---|---|---|---|---|
| Mother's age at subject's birth | n | 276 | 519 | 479 |
| Mean ± Std dev | 28.04±5.49 | 29.76±5.26 | 30.03±5.23 | |
| 95%CI | (27.39,28.69) | (29.30,30.21) | (29.56,30.50) | |
| p value | <0.001a | <0.001a | ||
| Age of subject at questionnaire completion | n | 281 | 987 | 922 |
| Mean ± Std dev | 7.92±5.01 | 12.17±8.77 | 10.62±6.64 | |
| 95%CI | (7.33,8.51) | (11.62,12.71) | (10.19,11.05) | |
| p value | <0.001a | <0.001a | ||
| Age of subject at diagnosis | n | 265 | 838 | 790 |
| Mean ± Std dev | 4.88±3.47 | 5.90±5.70 | 4.97±3.88 | |
| 95%CI | (4.46,5.30) | (5.51,6.29) | (4.70,5.24) | |
| Median | 3.83 | 4 | 3.5 | |
| p value | <0.001a | 0.697a | ||
when compared to the ARSD
Despite being older InterRett participants had marginally lower severity than ARSD participants on the modified Pineda scale (p=0.043) (Table 5 and Fig 2). The distribution of mutations in the 2 groups was similar (p=0.385) with the most common 8 mutations accounting for 64.1% of the InterRett and 64.4% of the ARSD mutations. Of the 8 most common mutations p.T158M (12.4%) was the most frequently occurring mutation in InterRett and p.R168X (11.2%) in the ARSD. In both InterRett (3.5%) and the ARSD (3.9%) p.R106W was the least common of this group (Table 5).
Table 5. Clinical and Genetic characteristics of InterRett Subjects compared with the ARSD.
| ARSD | InterRett (complete) |
InterRett (≥1976) |
||
|---|---|---|---|---|
| Clinical severity | n | 164 | 372 | 354 |
| Median | 16 | 15 | 15 | |
| p value | 0.039a | 0.043a | ||
| MECP2 Genetic test performed | n | 290 | 982 | 915 |
| Yes (%) | 90.3 | 91.2 | 91.4 | |
| No (%) | 9.7 | 8.8 | 8.6 | |
| Overall χ2 p value | 0.638a | 0.594a | ||
| Results of MECP2 test | n | 260 | 922 | 723 |
| Negative (%) | 21.2 | 22.9 | 23.6 | |
| Positive (%) | 78.8 | 77.1 | 76.4 | |
| Overall χ2 p value | 0.560a | 0.412a | ||
| Types of MECP2 mutations present | n | 205 | 596 | 552 |
| R106W (%) | 3.9 | 3.5 | 3.4 | |
| R133C (%) | 7.8 | 6.4 | 6.3 | |
| R168X (%) | 11.2 | 10.1 | 10.5 | |
| R255X (%) | 7.3 | 11.1 | 11.8 | |
| T158M (%) | 10.7 | 12.4 | 12.3 | |
| R306C (%) | 6.8 | 7.1 | 6.2 | |
| R294X (%) | 8.8 | 5.7 | 5.6 | |
| R270X (%) | 8.8 | 7.7 | 8.0 | |
| Large Del (%) | 6.3 | 3.4 | 3.6 | |
| Other (%) | 29.3 | 32.7 | 32.3 | |
| Overall χ2 p value | 0.358a | 0.385a | ||
when compared to the ARSD
Figure 2. The distribution of ARSD and InterRett severity scores using a modified version of the Pineda severity score among MECP2 mutation positive subjects.
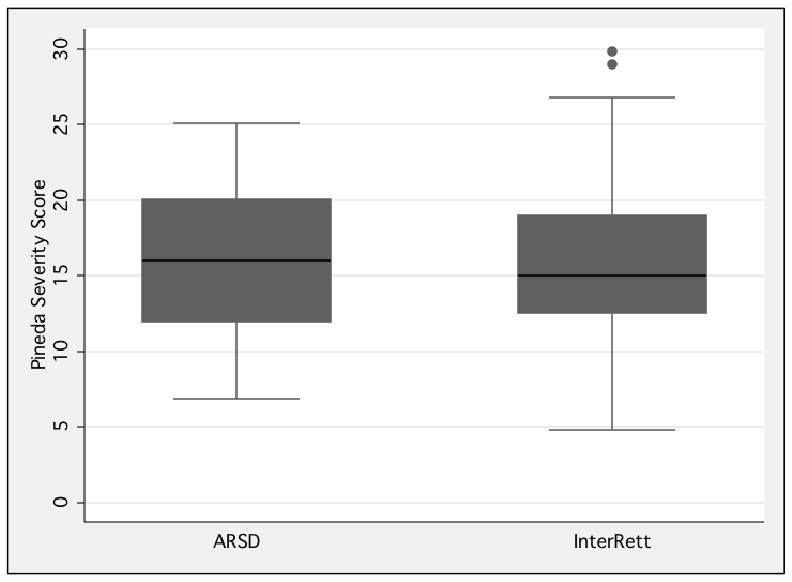
Socio-demographic Characteristics of Subjects and Families
There were differences in the socio-demographic characteristics of InterRett and ARSD subjects and families available for analysis (Tables 4 and 6). InterRett mothers were on average, about 2 years older at subject's birth than ARSD mothers (p<0.001) (Table 4). InterRett mothers were more likely to have fewer children after having a child with RTT than those in the ARSD (p=0.027). InterRett participants were more likely to be an only (16.5%) or youngest child (44.0%) compared with ARSD participants (10.7% and 34.9% respectively).
Table 6. Categorical Socio-demographic Characteristics of InterRett Subjects and Families compared with the ARSD.
| ARSD | InterRett (complete) |
InterRett (≥1976) |
RRR | 95%CI | ||
|---|---|---|---|---|---|---|
| Birth Order | n | 215 | 408 | 370 | ||
| Only (%) | 10.7 | 16.5 | 16.4 | 1.89* | (1.02, 3.48) | |
| 1st (%) | 33.0 | 25.8 | 24.6 | 0.93 | (0.59, 1.47) | |
| 2nd (%) | 15.4 | 10.1 | 10.2 | 0.74 | (0.41, 1.35) | |
| 3rd (%) | 4.6 | 2.8 | 2.8 | 0.52 | (0.18, 1.45) | |
| ≥4th (%) | 1.4 | 1.0 | 1.0 | 0.42 | (0.07, 2.68) | |
| Youngest (%) | 34.9 | 43.8 | 45.0 | |||
| Overall χ2 p value | 0.041a | 0.027 a | ||||
| Mother's Education | n | 268 | 332 | 299 | ||
| Some High School (%) | 36.5 | 6.8 | 6.5 | |||
| School Accreditation (%) | 17.2 | 26.3 | 24.4 | 7.49* | (3.83, 14.65) | |
| Vocational Education and | 28.0 | 18.3 | 18.7 | 3.29* | (1.69, 6.43) | |
| Training (%) | ||||||
| Higher Education (%) | 18.3 | 48.6 | 50.4 | 12.34* | (6.49, 23.44) | |
| Overall χ2 p value | <0.001 a | <0.001a | ||||
| Father's Education+ | n | 245 | 326 | 295 | ||
| Some High School (%) | 21.6 | 9.6 | 9.6 | |||
| School Accreditation (%) | 18.4 | 21.8 | 21.4 | |||
| Vocational Education and | 43.3 | 19.4 | 18.8 | |||
| Training (%) | ||||||
| Higher Education (%) | 16.7 | 49.2 | 50.2 | |||
| Overall χ2 p value | <0.001 a | <0.001a | ||||
| Mother's Occupation | n | 294 | 551 | 505 | ||
| Managers (%) | 5.8 | 18.2 | 18.2 | 15.65* | (3.87, 63.36) | |
| Professional (%) | 14.6 | 25.4 | 25.4 | 9.69* | (2.55, 36.85) | |
| Trade & Technician (%) | 5.4 | 8.9 | 8.7 | 8.54* | (2.07, 35.30) | |
| Community & Personal | 12.3 | 10.5 | 10.9 | 4.89* | (1.25, 19.07) | |
| Service (%) | ||||||
| Clerical & Admin (%) | 19.4 | 7.8 | 8.1 | 2.79 | (0.72, 10.80) | |
| Sales (%) | 7.8 | 3.4 | 3.2 | 3.97 | (0.94, 16.71) | |
| Machine Operator & Driver (%) | 0.0 | 0.4 | 0.2 | 0 | - | |
| Labourers (%) | 5.8 | 0.5 | 0.6 | |||
| Miscellaneous (%) | 28.9 | 24.9 | 24.7 | |||
| Overall χ2 p value | <0.001 a | <0.001a | N/A | |||
| Father's Occupation+ | n | 295 | 553 | 506 | ||
| Managers (%) | 18.0 | 16.8 | 16.8 | |||
| Professional (%) | 13.6 | 34.2 | 34.2 | |||
| Trade & Technician (%) | 23.1 | 14.5 | 14.0 | |||
| Community & Personal | 4.4 | 6.2 | 6.7 | |||
| Service (%) | ||||||
| Clerical & Admin (%) | 1.4 | 6.5 | 6.9 | |||
| Sales (%) | 3.4 | 3.3 | 3.2 | |||
| Machine Operator & Driver (%) | 8.2 | 4.3 | 4.4 | |||
| Labourers (%) | 10.2 | 1.4 | 1.4 | |||
| Miscellaneous (%) | 17.7 | 12.8 | 12.4 | |||
| Overall χ2 p value | <0.001 a | <0.001a | ||||
Multinomial logistic regression was not performed
when compared to the ARSD
RRR: p<0.05
Both mothers (p<0.001) and fathers (p<0.001) of participants in InterRett were more highly educated than those of the ARSD. After adjustment for maternal age, mothers of children in InterRett were over 7 times as likely (RRR=7.49 [CI: 3.83, 14.65]) to have school accreditation; more than 3 times as likely (RRR=3.29 [CI: 1.69, 6.43]) to have a vocational education and 12 times as likely (RRR=12.34 [CI: 6.49, 23.44) to have a higher education, compared to mothers of cases in the ARSD (Table 6). Of the 229 InterRett fathers for whom this information was available, 115 (50.2%) had a higher educational qualification compared with 16.7 % (41/245) of ARSD fathers (Table 6). InterRett mothers (p<0.001) and fathers (p<0.001) were also more likely to have occupations requiring a higher skill level (Table 6).
Discussion
Much research has attempted to investigate genotype and phenotype relationships with small studies, which consequently have been insufficiently powered to detect the real differences. Therefore findings have at times been conflicting (Charman et al., 2005; Cheadle et al., 2000; Nectoux et al., 2008). Often investigators, particularly in early studies, have attempted to group mutations to increase power. However, in the grouping, mutations with directly opposing characteristics (such as p.R294X and p.R270X, both nonsense mutations) may have been combined, thus nullifying effects. Our results clearly demonstrate that with time and larger case numbers, which include international collaborations, the true relationship between genotype and phenotype is gradually defined better. Furthermore international collaboration is also needed to investigate and draw conclusions about less frequently occurring clinical characteristics of RTT such as gall bladder disease where national studies may also be inadequately powered.
X inactivation has been demonstrated to influence the variability of RTT phenotypes (Archer et al., 2007) but it is not known whether this effect might vary by mutation. Such an analysis would require very large sample sizes. Only a minority of projects included X inactivation data on all (Archer et al., 2007) or some (Hoffbuhr et al., 2001; Leonard et al., 2003; Nectoux et al., 2008; Neul et al., 2008) of the data. In most countries these studies, which also require parental samples, are not included as part of routine testing. Therefore their inclusion would require special sample collection and specific funding and is not appropriate for InterRett which generally does not instigate genetic investigations but instead takes advantage of the power of combining together existing data from different sources.
Early studies investigating genotype phenotype relationships in RTT were generally based on small numbers (Hoffbuhr et al., 2001; Monros et al., 2001; Nicolao et al., 2001) and the appropriateness of generalising results were not considered. However for a study to be valid it must be possible to use its findings to make inferences beyond the study sample. The representativeness of the study sample and the nature of the population, from which it is drawn are two important characteristics that need to be taken into account when considering study validity. To provide such a population the ARSD has been maintained and nurtured over the past 15 years and in any study using these data we have always strived to relate the study participants back to the whole population in terms of their representativeness. However this is the first time the characteristics of study subjects in another Rett syndrome research sample, in this case InterRett, have been compared with those of the ARSD population-based cohort.
We found that the greatest differences between the InterRett sample and ARSD related to socio-demographic characteristics, with InterRett participants more likely to be an only or youngest child with older, more highly educated and more highly skilled parents. The question is whether the preferential inclusion of a more educated group of parents who directly report data to InterRett will result in data which are biased in some way. It is possible that higher parental socio-economic status may influence children's access to treatment and thus the InterRett group could be more advantaged with respect to some aspects of their health status (Halldorsson, Kunst, Kohler, & Mackenbach, 2002; Leonard, Fyfe, Leonard, & Msall, 2001). This may account for their severity being slightly less than expected and may be a factor to take into consideration when planning future clinical trials in Rett syndrome.
In terms of mutation distribution however, the InterRett dataset was comparable to the ARSD. Although InterRett participants were marginally less severe than ARSD individuals the distribution of severity scores was similar in both groups. This is an important finding as mutation type is an increasing focus for differentiating between subjects and the link to phenotype provides important information to parents and clinicians about the likely outcomes of this disorder for the child. Knowing that mutation distribution and phenotypic severity can be generalised gives confidence in the genotype phenotype association found in the InterRett dataset and validates the findings from a phenotype-genotype study based on InterRett data (Bebbington et al., 2008).
Strong international collaborations particularly with Spain, Israel (Bebbington et al., 2008) and France have helped build the links which have led to the submission of bulk data and made a major contribution to the total number of subjects within the InterRett sample. This is the most resource efficient mechanism for data inclusion but is dependent on the strength, trust and good will of the collaborative relationship and the capacity to ensure that data submitters are offered the opportunity to be involved in subsequent research output. Data that are submitted in bulk are also much more likely to be representative of the underlying population than individually-provided data as they are more likely to be systematically collected from this population. Therefore, they impact positively on the total representativeness of the InterRett dataset especially when, as they do, they comprise a large proportion (66.3%) of the total sample.
The ability to use different sources of data generally expands, the quality of data available for research and the number of RTT cases (Kozinetz et al., 1995). The submission of data from two independent sources (family and clinician) ensures that data within InterRett are reflective of a child's optimal functioning and not limited by their performance in a clinical assessment at a specific point in time. In addition by collecting data from two sources InterRett is able to ensure that families of RTT cases, where clinician data are not available, can still participate and provide valuable information to the study.
InterRett collects data from different sources around the world and in some cases may be dependent on receiving the data in the format which it has been originally collected which, as with the Spanish data, differs from the formats InterRett uses for data collection. This might be considered a weakness but by building a specific database compatible with the format of the Spanish data we have been able to overcome this as a problem. In other instances, as with the French data, certain questions were omitted during the translation process. Therefore, data sources vary with respect to the amount of data available for analysis. The issue of missing data is however a common but often unrecognised problem.
Recruitment will continue to increase as the Internet becomes more accessible. Some InterRett families and clinicians chose to submit questionnaires online, a unique feature which has contributed to the large number of countries, 32, represented within the InterRett sample. Whilst the ability to submit questionnaires online is limited by the availability and access to Internet services, internet usage worldwide has increased to nearly 1.5 billion users as of March 2008 (“Internet World Stats: Usage and Population Statistics”) and is increasing daily. It is anticipated that with increasing sample size and greater accessibility to the Internet that the InterRett sample will become more representative in terms of the socio-demographic characteristics investigated in this study.
We have shown that with few exceptions most studies investigating genotype phenotype relationships in Rett syndrome have not been able to demonstrate how their data relate to the underlying population and could have poor external validity. In comparison to our work with the population-based ARSD we had previously been concerned that, although InterRett is a large and statistically powerful dataset, the data might not allow us to generalise our findings, especially in making genotype-phenotype comparisons. However our review of literature in this area has reassured us that very few other studies have been able to take this factor into account. Moreover the results from the comparison with the ARSD give us confidence that conclusions drawn from these international data are comparable to those from a population cohort, especially as the distribution of common mutations is much the same as in the Australian RTT population.
Therefore we would conclude that data analyses using InterRett are no more likely to be biased than any other non-population-based studies.
Furthermore, InterRett with its ongoing recruiting strategies targeting individual families and its linkage with a number of clinical centres throughout the world has now accrued one of the largest collections of phenotype and genotype data on Rett syndrome. Providing a dataset with the power to identify small but real phenotypic differences based on genotype is an important evolution in the epidemiology of Rett syndrome. Using InterRett data we have already replicated genotype and phenotype associations evident within a national population based cohort (ARSD) (Bebbington et al., 2008). This therefore ratifies the value of collecting large amounts of data from disparate sources as a means of increasing understanding of clinical expression in rare disorders. The development of a mechanism for both families and clinicians from across the globe to submit data both on individual cases and in bulk has strengthened research capacity for collaborative worldwide studies and provides a tested model for the investigation of this and other rare disorders.
Acknowledgments
The authors would like to acknowledge the International Rett Syndrome Foundation (IRSF previously IRSA) for their ongoing support of the InterRett project and their continuous encouragement of this international collaboration. We would also like to express our special appreciation to all the families who have participated in the study and all the clinicians who have completed questionnaires. In particular we would like to thank Yael Petel Galil for her submission of clinical data on Israeli cases; Yael Yoshei for her assistance to Israeli families; the Catalan Association of Rett syndrome and Drs Aracil and Naudo in Spain and from France the French Association AFSR, Christiane Roque (past president), Elisabeth Celestin (current president) and Martine Gaudy and Thierry Bienvenu (performed most mutation screening). Finally we'd like to acknowledge those members of the InterRett International Reference panel who helped with the piloting of InterRett. We also acknowledge Neil Leonard and the information technology team at the Telethon Institute for Child Health Research for their information technology expertise and assistance.
Abbreviations
- RTT
Rett syndrome
- MECP2
Methyl CpG binding protein 2
- ARSD
Australian Rett syndrome database
Footnotes
Competing interests: The authors report no conflicts of interest. The funding agencies for this study had no involvement in the study design nor the collection, analysis and interpretation of data.
Authors' contributions: SL designed the study, organized the data, performed the statistical analysis and drafted the manuscript. SF was involved in study conception, participated in its design and made a major contribution to the drafting and revision of manuscript for important intellectual content. AB assisted with the analysis and interpretation of the data and participated in drafting the manuscript. NBB contributed to the provision of data and revised the manuscript critically for important intellectual content. AA made a substantial contribution to the acquisition of data and contributed to the drafting of the manuscript. AP, MP, JA, XW XB and PM contributed data and revised the manuscript for important intellectual content.
HL was involved in study conception and design, had overall responsibility for data collection and made a major contribution to the drafting and revision of manuscript for important intellectual content. All authors have given final approval of the version to be published.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, et al. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Annals of Neurology. 2000;47:670–679. [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, et al. Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. Journal of Medical Genetics. 2007;44:148–152. doi: 10.1136/jmg.2006.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol-Rosseto A, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. American Journal of Human Genetics. 2008;83:89–93. doi: 10.1016/j.ajhg.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australian Bureau of Statistics. Australian and New Zealand Standard Classification of Occupations. 2006. from http://www.abs.gov.au/AUSSTATS/abs@.nsf/mf/1220.0.
- Bebbington A, Anderson A, Ravine D, Fyfe S, Pineda M, de Klerk N, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70:868–875. doi: 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Carrie A, de Roux N, Vinet MC, Jonveaux P, Couvert P, et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Human Molecular Genetics. 2000;9:1377–1384. doi: 10.1093/hmg/9.9.1377. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Philippe C, De Roux N, Raynaud M, Bonnefond JP, Pasquier L, et al. The incidence of Rett syndrome in France. Pediatric Neurology. 2006;34:372–375. doi: 10.1016/j.pediatrneurol.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Chae JH, Hwang H, Hwang YS, Cheong HJ, Kim KJ. Influence of MECP2 gene mutation and X-chromosome inactivation on the Rett syndrome phenotype. Journal of Child Neurology. 2004;19:503–508. doi: 10.1177/08830738040190070501. [DOI] [PubMed] [Google Scholar]
- T, Neilson TC, Mash V, Archer H, Gardiner MT, Knudsen GP, et al. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype-phenotype associations in Rett syndrome. European Journal of Human Genetics. 2005;13:1121–1130. doi: 10.1038/sj.ejhg.5201471. [DOI] [PubMed] [Google Scholar]
- Cheadle JP, Gill H, Fleming N, Maynard J, Kerr A, Leonard H, et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Human Molecular Genetics. 2000;9:1119–1129. doi: 10.1093/hmg/9.7.1119. [DOI] [PubMed] [Google Scholar]
- Colvin L, Leonard H, de Klerk N, Davis M, Weaving L, Williamson S, et al. Refining the phenotype of common mutations in Rett syndrome. Journal of Medical Genetics. 2004;41:25–30. doi: 10.1136/jmg.2003.011130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs J, Young D, de Klerk N, Bebbington A, Baikie G, Leonard H. The impact of scoliosis surgery on activities of daily living in females with Rett syndrome. Journal of Pediatric Orthopedics. 2008 doi: 10.1097/BPO.0b013e3181a53b41. in press. [DOI] [PubMed] [Google Scholar]
- Fyfe S, Cream A, de Klerk N, Christodoulou J, Leonard H. InterRett and RettBASE: International Rett Syndrome Association databases for Rett syndrome. Journal of Child Neurology. 2003;18:709–713. doi: 10.1177/08830738030180100301. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Rett's syndrome: prevalence and impact on progressive severe mental retardation in girls. Acta Paediatrica Scandinavica. 1985;74:405–408. doi: 10.1111/j.1651-2227.1985.tb10993.x. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. European Journal of Paediatric Neurology. 2002;6:293–297. doi: 10.1053/ejpn.2002.0612. [DOI] [PubMed] [Google Scholar]
- Hagberg BA, Skjeldal OH. Rett variants: a suggested model for inclusion criteria. Pediatric Neurology. 1994;11:5–11. doi: 10.1016/0887-8994(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Halldorsson M, Kunst AE, Kohler L, Mackenbach JP. Socioeconomic differences in children's use of physician services in the Nordic countries. Journal of Epidemiology and Community Health. 2002;56:200–204. doi: 10.1136/jech.56.3.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffbuhr K, Devaney JM, LaFleur B, Sirianni N, Scacheri C, Giron J, et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology. 2001;56:1486–1495. doi: 10.1212/wnl.56.11.1486. [DOI] [PubMed] [Google Scholar]
- Huppke P, Held M, Hanefeld F, Engel W, Laccone F. Influence of mutation type and location on phenotype in 123 patients with Rett syndrome. Neuropediatrics. 2002;33:63–68. doi: 10.1055/s-2002-32365. [DOI] [PubMed] [Google Scholar]
- Huppke P, Held M, Laccone F, Hanefeld F. The spectrum of phenotypes in females with Rett Syndrome. Brain & Development. 2003;25:346–351. doi: 10.1016/s0387-7604(03)00018-4. [DOI] [PubMed] [Google Scholar]
- Internet World Stats: Usage and Population Statistics. from http://www.internetworldstats.com/stats.htm.
- Kerr AM, Prescott RJ. Predictive value of the early clinical signs in Rett disorder. Brain & Development. 2005;27(1):S20–S24. doi: 10.1016/j.braindev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kozinetz CA, Skender ML, MacNaughton N, Almes MJ, Schultz RJ, Percy AK, et al. Epidemiology of Rett syndrome: a population-based registry. Pediatrics. 1993;91:445–450. [PubMed] [Google Scholar]
- Kozinetz CA, Skender ML, MacNaughton NL, del Junco DJ, Almes MJ, Schultz RJ, et al. Population-based registries using multidisciplinary reporters: a method for the study of pediatric neurologic disorders. Journal of Clinical Epidemiology. 1995;48:1069–1076. doi: 10.1016/0895-4356(94)00233-g. [DOI] [PubMed] [Google Scholar]
- Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148:347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Leonard H, Bower C, English D. The prevalence and incidence of Rett syndrome in Australia. European Journal of Child & Adolescent Psychiatry. 1997;6(1):8–10. [PubMed] [Google Scholar]
- Leonard H, Colvin L, Christodoulou J, Schiavello T, Williamson S, Davis M, et al. Patients with the R133C mutation: is their phenotype different from patients with Rett syndrome with other mutations? Journal of Medical Genetics. 2003;40:e52. doi: 10.1136/jmg.40.5.e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H, Fyfe S, Leonard S, Msall M. Functional status, medical impairments, and rehabilitation resources in 84 females with Rett syndrome: a snapshot across the world from the parental perspective. Disability & Rehabilitation. 2001;23:107–117. doi: 10.1080/09638280150504180. [DOI] [PubMed] [Google Scholar]
- Leonard H, Moore H, Carey M, Fyfe S, Hall S, Robertson L, et al. Genotype and early development in Rett syndrome: the value of international data. Brain & Development. 2005;27(1):S59–S68. doi: 10.1016/j.braindev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Monros E, Armstrong J, Aibar E, Poo P, Canos I, Pineda M. Rett syndrome in Spain: mutation analysis and clinical correlations. Brain & Development. 2001;23(1):S251–253. doi: 10.1016/s0387-7604(01)00374-6. [DOI] [PubMed] [Google Scholar]
- Moore H, Leonard H, Fyfe S, De Klerk N, Leonard N. InterRett--The application of bioinformatics to International Rett syndrome research. Annals of Human Biology. 2005;32:228–236. doi: 10.1080/03014460500075068. [DOI] [PubMed] [Google Scholar]
- Nectoux J, Bahi-Buisson N, Guellec I, Coste J, De Roux N, Rosas H, et al. The p.Val66Met polymorphism in the BDNF gene protects against early seizures in Rett syndrome. Neurology. 2008;70(22 Pt 2):2145–2151. doi: 10.1212/01.wnl.0000304086.75913.b2. [DOI] [PubMed] [Google Scholar]
- Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70:1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolao P, Carella M, Giometto B, Tavolato B, Cattin R, Giovannucci-Uzielli ML, et al. DHPLC analysis of the MECP2 gene in Italian Rett patients. Human Mutation. 2001;18:132–140. doi: 10.1002/humu.1162. [DOI] [PubMed] [Google Scholar]
- Percy AK, Lane JB, Childers J, Skinner S, Annese F, Barrish J, et al. Rett syndrome: North American database. Journal of Child Neurology. 2007;22:1338–1341. doi: 10.1177/0883073807308715. [DOI] [PubMed] [Google Scholar]
- Renieri A, Mari F, Mencarelli MA, Scala E, Ariani F, Longo I, et al. Diagnostic criteria for the Zappella variant of Rett syndrome (the preserved speech variant) Brain & Development. 2008 doi: 10.1016/j.braindev.2008.04.007. [DOI] [PubMed] [Google Scholar]
- RettBASE: IRSA MECP2 Variation Database. from http://mecp2.chw.edu.au/ [DOI] [PubMed]
- Schanen C, Houwink EJ, Dorrani N, Lane J, Everett R, Feng A, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. American Journal of Medical Genetics A. 2004;126A:129–140. doi: 10.1002/ajmg.a.20571. [DOI] [PubMed] [Google Scholar]
- E, Schollen E, Moog U, Matthijs G, Herbergs J, Smeets H, et al. Rett syndrome in adolescent and adult females: clinical and molecular genetic findings. Americn Journal of Medical Genetics A. 2003;122A:227–233. doi: 10.1002/ajmg.a.20321. [DOI] [PubMed] [Google Scholar]
- StataCorp. Stata Statistical Software: Release 9. TX: StataCorp LP; 2005. [Google Scholar]
- The Central Intelligence Agency. The World Factbook. 2008. from https://www.cia.gov/library/publications/the-world-factbook/index.html.
- Trevathan E. Diagnostic criteria for Rett syndrome. The Rett Syndrome Diagnostic Criteria Work Group. Annals of Neurology. 1988;23:425–428. doi: 10.1002/ana.410230432. [DOI] [PubMed] [Google Scholar]
- Vacca M, Filippini F, Budillon A, Rossi V, Mercadante G, Manzati E, et al. Mutation analysis of the MECP2 gene in British and Italian Rett syndrome females. Journal of Molecular Medicine. 2001;78:648–655. doi: 10.1007/s001090000155. [DOI] [PubMed] [Google Scholar]
- Yaron Y, Ben Zeev B, Shomrat R, Bercovich D, Naiman T, Orr-Urtreger A. MECP2 mutations in Israel: implications for molecular analysis, genetic counseling, and prenatal diagnosis in Rett syndrome. Human Mutation. 2002;20:323–324. doi: 10.1002/humu.9069. [DOI] [PubMed] [Google Scholar]
- Zahorakova D, Rosipal R, Hadac J, Zumrova A, Bzduch V, Misovicova N, et al. Mutation analysis of the MECP2 gene in patients of Slavic origin with Rett syndrome: novel mutations and polymorphisms. Journal of Human Genetics. 2007;52:342–348. doi: 10.1007/s10038-007-0121-x. [DOI] [PubMed] [Google Scholar]