

Abstract
Tissue-specific overexpression of the human systemic amyloid precursor transthyretin (TTR) ameliorates Alzheimer's disease (AD) phenotypes in APP23 mice. TTR–β-amyloid (Aβ) complexes have been isolated from APP23 and some human AD brains. We now show that substoichiometric concentrations of TTR tetramers suppress Aβ aggregation in vitro via an interaction between the thyroxine binding pocket of the TTR tetramer and Aβ residues 18–21 (nuclear magnetic resonance and epitope mapping). The KD is micromolar, and the stoichiometry is <1 for the interaction (isothermal titration calorimetry). Similar experiments show that engineered monomeric TTR, the best inhibitor of Aβ fibril formation in vitro, did not bind Aβ monomers in liquid phase, suggesting that inhibition of fibrillogenesis is mediated by TTR tetramer binding to Aβ monomer and both tetramer and monomer binding of Aβ oligomers. The thousand-fold greater concentration of tetramer relative to monomer in vivo makes it the likely suppressor of Aβ aggregation and disease in the APP23 mice.
Introduction
The first suggestion of a salutary functional interaction between transthyretin (TTR) and β-amyloid (Aβ) peptides in vivo came from experiments in which coexpression of wild-type (WT) human TTR (huTTR) and Aβ1–42 in Caenorhabditis elegans muscle cells resulted in normalization of the abnormal motility seen when the Aβ construct was expressed alone (Link, 1995). A later study demonstrated that unilateral cerebral injection of anti-TTR antibody in Tg2576 Aβ transgenic mice enhanced Aβ-associated pathology on the side of the injection relative to the non-injected hemisphere presumably by lowering the free TTR concentration on the injected side (Stein et al., 2004). More recently, controlled studies from our laboratory showed that genetically programmed overexpression of a WT human TTR transgene suppressed both the neuropathologic and behavioral abnormalities seen in the well validated APP23 transgenic mouse model of human Aβ deposition and that silencing the endogenous Ttr gene accelerated the appearance of Aβ-associated neuropathology, a finding made independently in APPswe/PS1ΔE9 transgenic mice (Choi et al., 2007; Buxbaum et al., 2008). Collectively, these experiments indicate that the early in vitro studies leading to the proposal that TTR “sequestered” Aβ, protecting the brain from the effects of Alzheimer's disease (AD), might be correct in concept, if not in detail (Schwarzman et al., 1994).
Experiments exploring the mechanism underlying the apparent beneficial effect of TTR on AD showed that most (70%) hippocampal and cortical neurons from human AD brains stain with an antibody for TTR as do all such neurons in APP23 and Tg2576 mice (Stein and Johnson, 2002; Li et al., 2011). Endogenous Ttr transcription is increased in both adult APP23 brains and primary neurons cultured from 14- to 16-d-old APP23 embryos (Li et al., 2011). TTR–Aβ complexes can be coimmunoprecipitated from APP23 cortical lysates and from similar preparations of some human AD brains (Li et al., 2011). Studies in which Aβ1–40 or Aβ1–42 are preincubated with TTR have demonstrated TTR inhibition of Aβ-induced cytotoxicity in a variety of assay systems (Mazur-Kolecka et al., 1995; Giunta et al., 2005; Costa et al., 2008b; Li et al., 2011).
In vitro surface plasmon resonance (SPR) experiments showed that both TTR monomer and tetramer bound to immobilized Aβ monomers and fibrils, whereas ELISA filter-binding-based assays indicated that TTR monomer is the major binder of Aβ monomer in vitro (Buxbaum et al., 2008; Du and Murphy, 2010). In an effort to further understand the biochemical mechanism underlying the ameliorative effect of TTR on Aβ aggregation-associated toxicity in vivo, we used TTRs with varying thermodynamic and kinetic stabilities to examine the relationship between TTR tetramer stability, binding to Aβ, and inhibition of Aβ aggregation in vitro with liquid-phase binding assays, including isothermal titration calorimetry (ITC) and nuclear magnetic resonance (NMR) spectroscopy, SPR, and solid-phase assays of binding of TTR or Aβ conformers bound to nitrocellulose or plastic.
Materials and Methods
Recombinant TTR and synthetic Aβ preparation.
Recombinant TTR [WT huTTR, T119M, K15A, V30M, V122I, mouse TTR (muTTR), and human monomeric TTR (M-TTR; WT TTR with the substitutions F87M/L110M that does not tetramerize)], 15N,2H-labeled huTTR, 15N,2H-labeled TTRV30M, and 15N-labeled M-TTR were prepared in an Escherichia coli system and purified using fast protein liquid chromatography and gel filtration as described previously (Reixach et al., 2008). Aβ1–40 and Aβ1–42 were synthesized and purified by HPLC, and their identity was confirmed by mass spectrometry as described previously (Du et al., 2011).
Aβ monomerization.
Lyophilized Aβ powder was monomerized as described previously (Du et al., 2011; Li et al., 2011) if not indicated. Briefly, Aβ was dissolved in 2 mm NaOH at 2.5 mg/ml and pH was adjusted to 10.5 before being sonicated in cold water bath for 30 min. The solution was filtered through 10 kDa cutoff centricon filter (Millipore), and its concentration was determined by UV absorbance at 280 nm (ε = 1280 m−1 cm−1).
Aβ aggregation assay.
Initially monomeric synthetic Aβ1–40 (10 μm) and 20 μm thioflavin T (ThT) in NaPi (50 mm sodium phosphate, 150 mm NaCl at pH 7.4) were mixed, and 100 μl of the mixture was added to a 96-well plate (Costar) and sealed with a microplate cover. Fluorescence intensity was recorded every 10 min at 37°C after agitation of 5 s with excitation/emission wavelengths of 420/485 nm (Tecan Safire II; Tecan). The half-maximal fluorescence time point (t50), calculated as the time ThT fluorescence reached the midpoint between lag phase and post-aggregation plateau, was used to describe the aggregation properties of Aβ. At least three replicates were measured for each treatment (Cohen et al., 2006).
To test the effect of TTR variants on Aβ aggregation, buffer or TTR variants (muTTR, K15A, T119M, huTTR, V30M, V122I, or M-TTR) were added to the Aβ1–40 aggregation assays to a final concentration of 0.25, 0.5, 1, and 2 μm. The experiment was repeated, and a representative experiment was graphed. Incubations of TTR alone under these conditions did not result in an increase in ThT fluorescence and were not graphed.
Aβ-derived diffusible ligands.
Lyophilized Aβ1–42 was dissolved in hexafluoro-2-propanol (HFIP; Sigma-Aldrich), evaporated under nitrogen stream at room temperature. During usage, Aβ was resuspended in anhydrous DMSO (Sigma-Aldrich) to 5 mm, vortexed for 10 s, and then sonicated for 10 min. Aβ was then diluted with ice-cold phenol red-free Ham's F-12 (Caissonlabs) to a final concentration of 60 μm either with or without 6 μm TTR (huTTR, muTTR, and M-TTR) and incubated at 4°C for 2 d (Lambert et al., 1998).
Western blots.
Samples were mixed with Tris-tricine sample buffer and separated on 15% Tris-tricine SDS-PAGE. The proteins were transferred onto a PVDF (Bio-Rad) membrane, and the blots were blocked with 5% dry milk, incubated in primary antibody (Covance 6E10 anti-Aβ or Dako anti-TTR) for 1 h and then secondary antibody (IRDye secondary antibody), imaged, and quantified using an Odyssey system (LI-COR Biosciences).
Light scattering.
huTTR (10 μm in NaPi) was mixed with Aβ1–40 monomer (10 μm) in a 20 ml disposable scintillation vial at room temperature quiescently for 1 d and then loaded into a Dawn EOS light-scattering photometer (Wyatt Technology). Light-scattering intensity data at 90° were recorded every 2 s over the course of 120 min at 37°C.
8-Anilino-1-naphthalene sulfonate monitored Aβ1–42 aggregation.
Aβ1–42 (25 μm) aggregation alone or in the presence of 2.5 μm huTTR, muTTR, or M-TTR at pH 7.4 with agitation was monitored by 8-anilino-1-naphthalene sulfonate (ANS; Sigma-Aldrich) binding (which increased fluorescence intensity with Aβ aggregation). When ANS fluorescence was monitored (λ excitation of 380 nm), the emission wavelength increased from day 0 through day 2, corresponding to the formation of oligomers on days 1 and 2.
Transmission electron microscopy.
Transmission electron microscopy (TEM) analysis of the inhibition of Aβ aggregation by huTTR, muTTR, and M-TTR was performed by incubation of 25 μm Aβ1–40 alone or with 25 μm TTRs (100 μm M-TTR to achieve the same Aβ/TTR monomer ratios) in NaPi at 37°C in a 96-well plate for 5 d. The TEM grids were prepared by adding 5 μl of preformed fibrils onto copper grids, covered with Formvar and carbon films, and counterstained with 2% (w/v) uranyl acetate, using the droplet technique. Specimens were examined with a Zeiss 900 electron microscope operated at an acceleration voltage of 80 kV. Magnification of 30,000× was used for imaging.
Atomic force microscopy imaging TTR inhibition of Aβ1–42 aggregation.
huTTR, muTTR, or M-TTR at 2.5 μm was added to 25 μm Aβ1–42 aggregation in 0.1× PBS buffer. The mixture was shaken at 300 rpm at 25°C. Samples were spotted on mica and absorbed for 30 min before being washed and dried. Images were taken using an Asylum Research MFP 3D atomic force microscopy (AFM) system as described previously (Ladiwala et al., 2012). Representative images (3 × 3 μm) were presented for samples taken at days 0, 1, 2, and 5.
SPR.
Sensor chip CM5 (GE Healthcare) was preconditioned by running 20 μl of 0.1% HCl, 50 mm NaOH, 0.1% SDS, and 0.085% H3PO4 at a speed of 20 μl/min using the BIACORE3000 (GE Healthcare). Monomeric Aβ1–40 peptide was immobilized using an amine coupling kit (GE Healthcare) per the recommendation of the manufacturer. NaOH at 2 mm and 150 mm NaCl were used as a regenerant. To compare the relative binding among TTRs to Aβ, 20 μm TTR was allowed to flow through the cell for 3 min at 40 μl/min. Each experiment was performed on a new chip double referenced with buffer before and after sample injection, and response units (RUs) were normalized to the initial plateau. Two thousand two hundred to 2600 RUs of Aβ monomer was immobilized.
ITC.
Direct interactions between TTRs and Aβ1–40 were studied on a Microcal iTC200 system (GE healthcare). TTRs were dialyzed overnight at 4°C against 25 mm HEPES buffer, pH 7.0, obtained by diluting 1 m HEPES buffer, pH 8.0 (Mediatech), with MilliQ water. Lyophilized Aβ1–40 powder was dissolved in HFIP for 1 h and evaporated in a fume hood (Wang et al., 2010). HEPES buffer at 25 mm (Brockhaus et al., 2007) was then added to the tubes, vortexed, and centrifuged at 13,000 × g for 10 min at 4°C to pellet insoluble Aβ. For the titration experiment, TTRs were present in the cell and Aβ in the syringe. Injections were performed at 37°C. Control titrations included Aβ1–40 to buffer and buffer to TTRs (data not shown). Control titrations of Aβ1–40 into buffer were subtracted from all the experiments. In each experiment, 16 injections of 2.5 μl Aβ with 180 s intervals were made with syringe stirring speed of 1000 rpm. The experimental titrations were performed using at least three different concentrations of both TTRs (19, 25, 30, 35 μm) and Aβ (190, 250, 300, 350 μm) while maintaining a 10:1 ratio. All gave similar results. Stoichiometry (N), binding constant (KD), enthalpy (ΔH) and entropy (ΔS) were calculated from the fitting curves using Origin 7.0 from Microcal.
NMR spectroscopy.
NMR spectra were recorded using Bruker DRX 600 and Avance 800 MHz spectrometers. Assignments for TTR, V30M, and M-TTR were made using standard multidimensional triple-resonance NMR experiments (Cavanagh et al., 2007). Two-dimensional 1H-15N transverse relaxation optimized spectroscopy (TROSY)-heteronuclear single quantum coherence (HSQC) spectra of 100 μm huTTR and V30M with or without 80 μm Aβ1–40 were recorded at 25°C in buffer containing 50 mm potassium phosphate, pH 7.6, 50 mm KCl, and 1 mm EDTA. Two-dimensional 1H-15N HSQC spectra of 20 μm M-TTR with or without 80 μm Aβ1–40 were recorded at 37°C in buffer containing 50 mm potassium phosphate, pH 7.2, 50 mm KCl, and 1 mm EDTA.
Effect of TTR stabilizers on the inhibition by TTRs of Aβ aggregation.
Diflunisal or Tafamidis solubilized in DMSO was incubated with TTR at a 2:1 molar ratio for 1 d at 37°C. Incubation with DMSO alone served as control. TTRV122I or M-TTR at 0.25 μm or 0.5 μm HuTTR (WT) with Diflunisal, Tafamidis, or DMSO were added to 10 μm Aβ. Fibril formation was monitored by ThT fluorescence.
SDS Tris-tricine PAGE analysis of TTR and Aβ incubation.
Synthetic Aβ1–40 and Aβ1–42 were first monomerized as described above (see Aβ monomerization). Monomeric Aβ1–40 or Aβ1–42 at 80 μm with or without 20 μm M-TTR, huTTR, or TTRK15A was incubated with agitation at 280 rpm in NaPi for 3 d at 37°C. The mixtures were analyzed on 15% native Tris-tricine SDS PAGE gels, transferred to PVDF membranes, and probed with anti-Aβ (6E10) and anti-TTR (Dako) antibodies. Images were obtained using the Odyssey system (LI-COR; Fig. 6, Aβ, green; TTR, red; left panels, Aβ alone; right panels, merge of Aβ and TTR). Under these conditions, Aβ (alone) aggregation results in a heterogeneous smear and large aggregates trapped at the top of the gels. No smear or trapped material is seen in the presence of M-TTR (Fig. 6, red arrows). The major Aβ species in the M-TTR lanes had mobilities consistent with an Aβ pentamer and decamer (Fig. 6, yellow arrows). The distribution of Aβ aggregates in the lanes showing interaction with huTTR tetramer was intermediate between M-TTR and TTRK15A.
Figure 6.

huTTR inhibition of Aβ1–40/42 aggregation. Synthetic monomeric Aβ1–40 (A) or Aβ1–42 (B) at 80 μm with or without 20 μm M-TTR, huTTR, or TTRK15A was incubated with agitation in NaPi for 3 d at 37°C. The mixtures were analyzed on 15% native Tris-tricine SDS PAGE gels and probed with anti-Aβ (6E10, green) and anti-TTR (Dako, red) antibodies. Under these conditions, Aβ (alone) aggregation results in a heterogeneous smear and large aggregates trapped at the top of the gels. No smear or trapped material is seen in the presence of M-TTR (red arrows). The major Aβ species in the M-TTR lanes had mobilities consistent with an Aβ pentamer and decamer (yellow arrows). The distribution of Aβ aggregates in the lanes showing interaction with huTTR tetramer was intermediate between M-TTR and TTRK15A.
Dot-blot epitope mapping TTR–Aβ1–42 binding site.
Aβ1–42 (American Peptide) was dissolved in an aqueous 50% acetonitrile solution (1 mg/ml). The peptide was aliquoted, lyophilized, and then stored at −20°C. Aβ oligomers were prepared by dissolving the peptide in 100% HFIP (Fluka). The solvent was evaporated, and Aβ was dissolved in 50 mm NaOH (1 mg/ml Aβ), sonicated (30 s), and diluted in PBS (25 μm Aβ). The Aβ peptide was then centrifuged (22,000 × g for 30 min), and the pelleted fraction (5% of starting volume) was discarded. The supernatant was incubated at 25°C for 1–3 d without agitation to form prefibrillar oligomers (A11-positive) or 4–6 d to form A11-negative oligomers. Fibrils were formed in the same way except that monomers were mixed with preexisting fibrils (10–20 weight percent seed) without agitation for 24 h at 25°C. To identify the TTR binding site on Aβ, 220 ng of Aβ1–42 conformers were spotted on nitrocellulose membranes (Hybond ECL; GE Healthcare) and then incubated with sequence specific antibodies for epitopes present on Aβ, including the following: N-terminal Aβ3–10, 6E10 (Sigma-Aldrich); middle Aβ18–22, 4G8 (Covance); Aβ16–21, A3356; C-terminal, Aβ30–35 (Sigma-Aldrich); and Aβ35–39, 9F1 (Santa Cruz Biotechnology) at 25°C for 2 h in blocking buffer. The blot was washed with PBST (PBS with 0.05% Tween 20) and incubated with 10 μm biotinylated TTR at 4°C overnight. The blot was washed again and incubated with streptavidin-conjugated HRP and developed.
Quantification of binding strength of TTR and Aβ1–42 conformers.
The TTR was biotinylated with Sulfo-NHS-Biotin (Pierce). Approximately 2 μg of Aβ fibrils, oligomers (A11 positive), or monomers (prepared as in the previous section) were immobilized in 96-well plates (Nunc Maxisorb; Thermo Fisher Scientific), and biotinylated TTR was added at various concentrations. The plate were washed (three times, PBST) and developed using avidin–HRP. The IC50 values were calculated using SigmaPlot.
Brain lysate ELISA.
Brain tissue (cortices, 60 mg) of APP23 mice of either sex overexpressing huTTR without endogenous muTTR (APP23/TTR+/Ttr−) was homogenized in 800 μl of Tris buffer [50 mm Tris, 150 mm NaCl, and 3 mm EDTA with protease inhibitors (Roche), pH 7.5] on ice. The lysate was centrifuged at 21,000 × g at 4°C for 4 min, and the supernatant was collected as representing the extracellular fraction. The pellet was rehomogenized in Triton buffer (Tris buffer with 0.5% Triton X-100) and centrifuged at 21,000 × g, and the supernatant was collected as Triton extracts (intracellular fraction). The pellet was dissolved in 2% SDS and spun again at room temperature, and the samples were stored at −80°C. ELISA plates (Immulon 4 HBX; Thermo Fisher Scientific) were coated with primary antibody (Dako or 6E10) in 50 mm carbonate buffer, pH 9.6, at 4°C overnight. The plate was washed with TBST (Tris buffered saline with 0.05% Tween 20) and blocked with 5% non-fat milk in TBST. The samples and standard (recombinant huTTR or synthetic Aβ1–40) in blocking buffer were incubated on the plate for 2 h at 37°C. The alkaline phosphatase (Dojindo)-labeled goat anti-human preablumin (Meridian Life Science) or biotin-4G8 (Covance) was incubated for 1 h at 37°C. For TTR detection, 0.5 mg/ml p-nitrophenylphosphate (NPP; Sigma-Aldrich) in NPP buffer (10 mm diethanolamine, 0.5 mm MgCl2) was added, and the plate was read by SpectroMAX (Molecular Devices) at 405 nm. For Aβ detection, the plate was incubated with 0.04% streptavidin–HRP (Invitrogen) for 1 h at room temperature before being developed using QnataBlue kit (Thermo Fisher Scientific) and read at excitation/emission at 340/400 nm (Tecan Safire II; Tecan).
TTRs did not disrupt preformed Aβ aggregates.
The disaggregation assay was designed to investigate whether TTR disaggregates or digests mature Aβ fibrils. Monomeric Aβ1–40 (10 μm) was aggregated into fibrils in a black 96-well plate in NaPi buffer for 5 d. ThT at 20 μm and 1 μm TTRs (huTTR, muTTR, and M-TTR) were added to 100 μl of fibril mixture, and fluorescent intensity was recorded for 5 d. The ThT fluorescence intensity was normalized against that of the day 0 when TTR was added to the fibrils.
TTRs (10 μm) incubated quiescently with preformed Aβ1–42 oligomers and fibrils (25 μm) for 24 h, and AFM analysis of the incubation showed that the quantity and conformation of Aβ aggregates did not change in the presence of TTR over this period.
Statistical analyses.
Statistical analysis was performed in Minitab 13 (Minitab). Post hoc analysis was performed when the ANOVA result was significant. For aggregation assays, post hoc Dunnett's simultaneous tests were performed. The t50 values were compared only with the samples on the same plate, and representative data were plotted. For quantification of inhibition of oligomer formation on Western blot, the Bonferroni's correction was used for t tests. For aggregation assays, one-way ANOVA was used, followed by post hoc t tests with Bonferroni's corrections. Means ± SD were graphed for all experiments. For each treatment, the replicates were more than three, and each experiment was repeated at least twice. Significance is shown at *p < 0.05 and **p < 0.01.
Results
TTRs inhibit Aβ fibril and oligomer formation
Using the ThT binding assay, we successfully reproduced results from other laboratories showing that huTTR inhibits Aβ fibril formation in vitro (Schwarzman et al., 1994, 2004; Liu and Murphy, 2006; Costa et al., 2008b; Du and Murphy, 2010). We then compared the inhibitory activities of three forms of TTR, huTTR (WT), muTTR, which is 80% homologous to huTTR but kinetically much more stable (Reixach et al., 2008), and M-TTR, an engineered human TTR monomer that contains mutations (F87M/L110M) that alter the monomer–monomer interface preventing tetramer formation under physiologic conditions (Jiang et al., 2001). We measured the t50 values of Aβ aggregation (time required to reach half-maximal ThT fluorescence) in the presence of each of the TTRs (Fig. 1A). The rank order of inhibitory capacity is M-TTR > huTTR > muTTR, and it is concentration dependent for all forms of TTR. TEM confirmed that no fibrils were formed when Aβ1–40 was coincubated with any of the TTRs at high concentrations (25 μm TTR/25 μm Aβ1–40; Fig. 1B).
Figure 1.
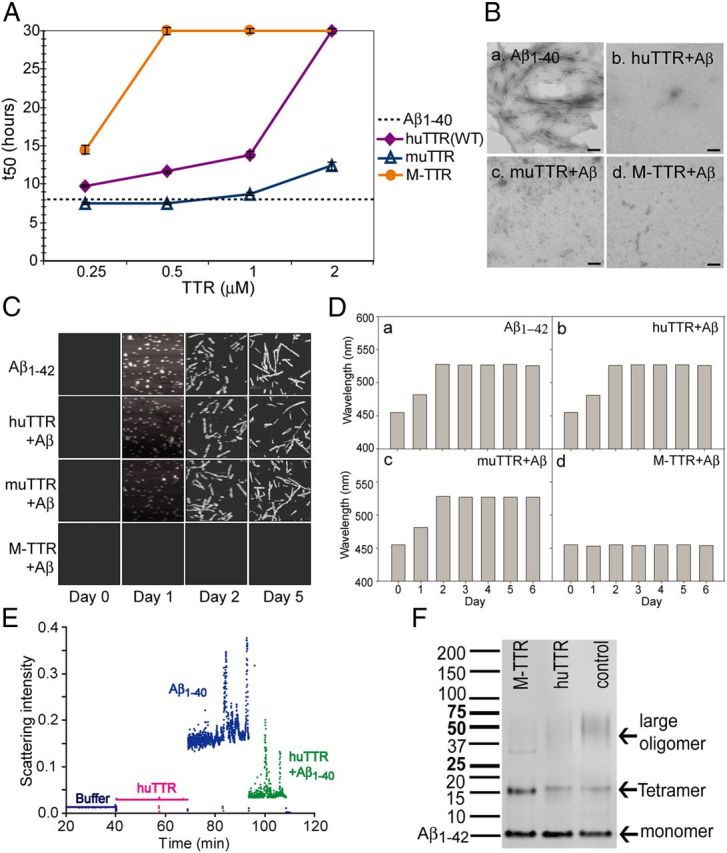
TTRs inhibit Aβ oligomer and fibril formation. A, WT huTTR, muTTR, and M-TTR each increased the aggregation t50 (i.e., decreased fibril formation) of 10 μm solution of monomeric Aβ1–40 monitored by ThT fluorescence in a concentration-dependent manner within concentration ranges found in human CSF (0.3–0.5 μm) or serum (3–5 μm). The dotted line shows the t50 of Aβ aggregation under the same conditions without TTR. B, TEM images of Aβ1–40 alone (a) or with huTTR (b), muTTR (c), and M-TTR (d) show that TTRs inhibited Aβ fibril formation. Aβ1–40 at 25 μm and 25 μm TTR tetramers (100 μm M-TTR to achieve the same Aβ/TTR monomer ratios) in NaPi were incubated at 37°C for 5 d. Scale bars, 200 nm. C, AFM showed M-TTR and huTTR, but muTTR (2.5 μm) inhibited Aβ1–42 (25 μm) fibril formation with agitation in diluted PBS. Each panel equals 3 × 3 μm. D, Aβ1–42 at 25 μm aggregation alone (a) or with 2.5 μm huTTR (b), muTTR (c), or M-TTR (d) at pH 7.4 with agitation monitored by ANS binding suggested that M-TTR inhibited Aβ conformation change (i.e., aggregation). E, Light scattering shows that huTTR and Aβ1–40 coincubation resulted in smaller aggregates compared with Aβ1–40 incubated alone, demonstrating that huTTR inhibited Aβ oligomer formation. F, M-TTR or huTTR were added to Aβ1–42 (1:10 molar ratio) during ADDLs preparation. The products were analyzed by Western blot probed with 6E10 against Aβ. M-TTR and huTTR both inhibited large oligomer formation by Aβ1–42.
AFM studies of mixtures of TTR (2.5 μm) and Aβ1–42 (25 μm) incubated for 5 d with agitation show that M-TTR completely inhibited Aβ aggregation. huTTR reduced the quantity of Aβ fibrils formed, and muTTR had no detectable effect (Fig. 1C). The findings were similar when Aβ1–42 aggregation was monitored by ANS, which shifts its maximum fluorescence emission spectrum on Aβ aggregation. The shift was inhibited by M-TTR but not huTTR or muTTR (Fig. 1D). These analyses suggest that the inhibitory capacities of various TTRs varied depending on the assay (as well as whether aggregation of Aβ1–40 or Aβ1–42 is being analyzed); however, in most assays, the order of inhibitory capacity was M-TTR > huTTR > muTTR.
It is thought that Aβ oligomers (rather than fibrils) are the toxic species in AD. They have been shown to increase Ca2+ influx, induce the generation of reactive oxygen species, and decrease the viability of primary cultured neurons and a variety of cultured cell lines (Walsh et al., 2002; De Felice et al., 2007; Wang et al., 2009; Li et al., 2011). To study the effect of TTR on Aβ oligomer formation, Aβ1–40 monomer was incubated in the presence or absence of huTTR at room temperature without agitation, conditions known to result in oligomer but not fibril formation (Sarroukh et al., 2011). Light-scattering analysis showed that the oligomeric Aβ aggregates formed in the presence of huTTR were smaller than those formed in the absence of TTR (Fig. 1E). When tetrameric huTTR or M-TTR was added during the preparation of toxic Aβ1–42 oligomers [Aβ-derived diffusible ligands (ADDLs)], the proportion of large Aβ oligomers (37–75 kDa) present was greatly reduced (t test, n = 4, Bonferroni's correction, p < 0.05), with M-TTR being more effective than huTTR in reducing large oligomer formation (Fig. 1F). In addition, the fraction of Aβ remaining as monomer was greater in the presence of both M-TTR and huTTR. The results suggest that TTR inhibits Aβ fibril formation by reducing oligomer formation and/or suppressing the conversion of Aβ oligomers to larger oligomers, which can serve as fibril seeds that can initiate/accelerate Aβ fibrillogenesis in vitro.
To examine the mechanism of inhibition in greater detail, we compared the abilities of recombinant TTRs of different kinetic and thermodynamic stabilities to inhibit Aβ1–40 fibril formation. We reasoned that, because monomeric TTR was the best inhibitor of in vitro fibrillogenesis and is generated by tetramer dissociation, perhaps inhibition of fibril formation required a dissociation step. We used WT huTTR tetramer, several destabilized naturally occurring huTTR mutant tetramers, naturally occurring and engineered kinetically stable tetramers and M-TTR (Fig. 2A). By adding identical molar concentrations of the different TTRs to the aggregation reaction (containing the same amount of Aβ1–40) at time 0 and calculating the t50 value, we could compare the relative abilities of various TTRs to inhibit fibril formation. At 0.25 μm, the approximate TTR concentration in human CSF, huTTR, TTRV122I, and M-TTR significantly suppressed aggregation of a 10 μm Aβ solution (an Aβ concentration far in excess of that found in vivo; Dunnett's test, p < 0.05; Figs. 1A, 2B). At 0.5 μm, TTRT119M and TTRV30M (TTR tetramers with increased kinetic stability) also inhibited Aβ1–40 fibril formation (Dunnett's test, p < 0.05). At higher concentrations (up to 2 μm, a 1:5 molar ratio of TTR/Aβ), all TTRs tested, regardless of their thermodynamic or kinetic stability, significantly delayed Aβ fibril formation. The results demonstrated a concentration-dependent effect on the inhibition of Aβ1–40 fibril formation. It is interesting to note that, under these conditions, the most kinetically unstable tetramer, i.e., TTRV122I, was a better inhibitor of aggregation than the thermodynamically unstable TTRV30M mutant. However, even the rare highly kinetically stable variant TTRT119M could suppress Aβ aggregation at TTR concentrations within the range found in human serum (3–5 μm), indicating that in vitro, although M-TTR was the best inhibitor of aggregation, TTR tetramer dissociation to monomer was not required for suppression.
Figure 2.

TTR capacity of inhibition Aβ fibril formation. A, Thermodynamic and kinetic properties of the TTR variants used in experiments. B, TTRV122I, huTTR (WT), TTRV30M, TTRT119M, and TTRK15A each increased the t50 (i.e., decreased fibril formation) of 10 μm monomeric Aβ1–40 aggregation monitored by ThT fluorescence in a concentration-dependent manner within concentration ranges found in human CSF (0.3–0.5 μm) or serum (3–5 μm).
We assumed that the extent of inhibition of Aβ fibril formation would be related to the affinities of the various TTRs for the Aβ monomer because the monomer is required for the oligomerization that forms the nucleus apparently required for fibrillogenesis. This appeared to be true when the relative association between the various TTRs and Aβ1–40 monomer were measured by SPR (Fig. 3A), with Aβ immobilized to a dextran matrix and the interaction measured in a relatively rapid timeframe. The rank order of association was M-TTR > TTRV30M ≥ huTTR > TTRV122I = TTRT119M > TTRK15A. When TTR binding was compared in terms of the molar concentration of monomer, M-TTR bound Aβ1–40 monomer best. Of the TTR tetramers tested, TTRV30M and WT were the strongest binders to Aβ1–40, and TTRK15A (a highly kinetically stable engineered huTTR variant) was the weakest. Thus, the relative association of various TTRs with Aβ monomers, as measured by SPR, may be related to their stabilities when the correlation is based on experimentally determined “combined stability scores” (Sekijima et al., 2005). However, the relationship is not statistically significant (p > 0.05).
Figure 3.

SPR and ITC analyses of TTR binding to Aβ1–40 monomer. A, TTRs bound to Aβ1–40 monomer immobilized on chips in SPR (Biacore). RUs were normalized to the initial flat phase (baseline), and response curves were from multiple chips without regeneration. B–D, ITC binding isotherms for Aβ1–40 monomer to huTTR (B), muTTR (C), and M-TTR (D). The top rows in B–D show raw data after correction for background; the bottom rows show the binding isotherms by plotting integrated peak areas against the molar ratio of Aβ1–40/TTR. E, The table compares the thermodynamic parameters of binding of Aβ to human and murine TTR tetramers as measured by ITC.
It is known that TTR, as a systemic amyloidogenic precursor, aggregates in vitro. However, consistent with published data (Hammarström et al., 2002), TTR incubated alone under the conditions of our experiments (NaPi buffer at neutral pH) did not aggregate as monitored by either ThT fluorescence or AFM/TEM microscopy (data not shown).
To determine the concentrations of TTR and Aβ present in vivo in circumstances in which TTR had been shown to inhibit the behavioral and neuropathologic manifestations of Aβ deposition, we performed TTR and Aβ specific ELISAs on Tris extracts of the cerebral cortices of APP23 mice overexpressing WT huTTR. The TTR concentration was 1.4 ± 0.4 μm, whereas the soluble Aβ concentration was 0.7 ± 0.5 nm (n = 12).
ITC of TTR–Aβ interaction
ITC analysis of huTTR, muTTR, and M-TTR binding to Aβ1–40 monomer showed that, despite their great differences in inhibiting Aβ fibril formation, human and mouse tetrameric TTRs bound Aβ1–40 with similar affinities, with KD values in the micromolar range (Fig. 3B,C). The huTTR–Aβ and muTTR–Aβ interactions were enthalpy driven (ΔH < 0), with muTTR appearing to have a higher change in enthalpy than huTTR (Fig. 3B,C,E). It is unclear whether the entropy factor is favorable or unfavorable for the huTTR–Aβ interaction, whereas it appeared to be strongly unfavorable (−TΔS > 0) in the muTTR–Aβ interaction. Surprisingly, despite M-TTR being the most effective inhibitor of Aβ aggregation, the M-TTR–Aβ interaction did not exhibit an exothermic isotherm under the same ITC conditions used for TTR tetramers (Fig. 3D), indicating that, under these conditions, it did not bind monomeric Aβ or that the interaction did not result in a detectable heat change.
TTRs bind Aβ in the thyroxine site of TTR
To further elucidate the structural basis of huTTR binding to Aβ monomer, we acquired two-dimensional 1H-15N HSQC NMR spectra to identify the Aβ monomer binding sites on tetrameric WT huTTR and TTRV30M. We recorded the TROSY-HSQC spectra of huTTR or TTRV30M tetramers (each at 25 μm) in the absence and presence of Aβ1–40 (80 μm; Fig. 4A,B). The majority of HSQC cross-peaks remain unchanged or only marginally shifted on Aβ addition, suggesting that the native TTR tetramer structure was not affected by binding Aβ monomer. However, significant changes were observed for several cross-peaks corresponding to amino acids that could be involved in the TTR–Aβ interaction.
Figure 4.
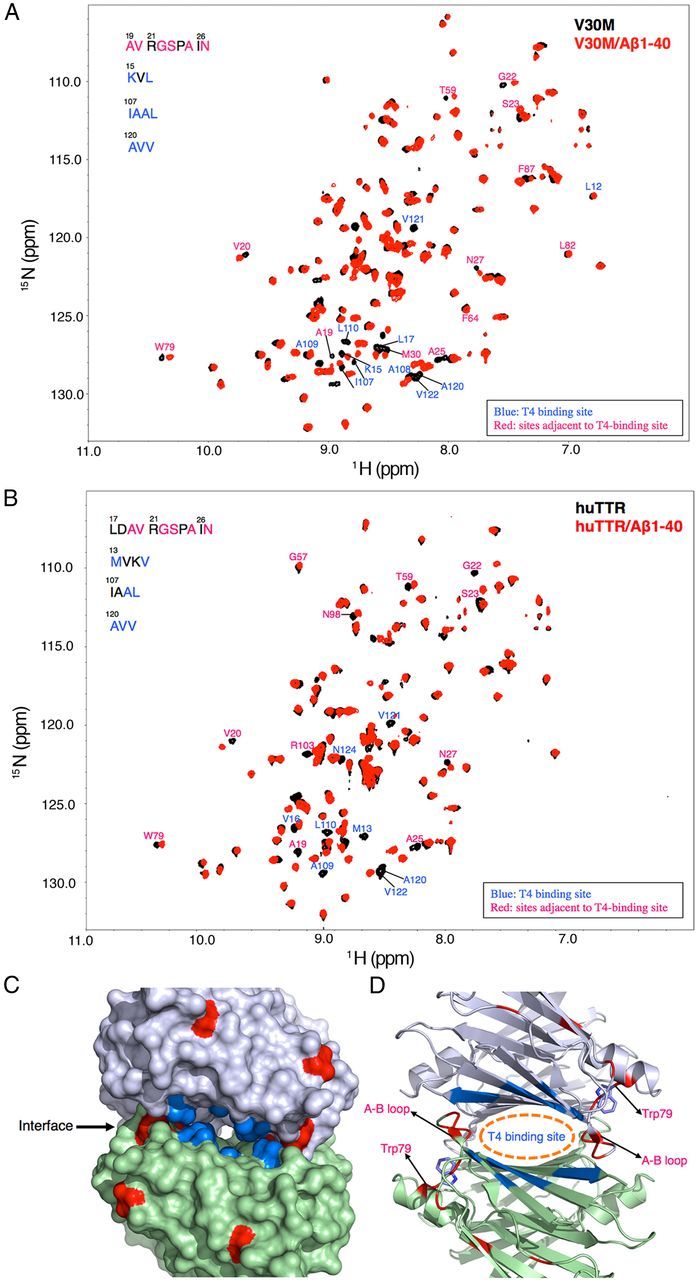
NMR analysis of TTRV30M and WT-TTR binding to Aβ1–40. A, B, Aβ1–40 binding to TTRV30M (A) and WT-TTR (B) causes significant changes in the amide-proton chemical shifts of TTRs as shown by the two-dimensional TROSY-HSQC spectra. C, D, Residues showing changes in their chemical shifts are mapped onto the TTRV30M x-ray structure (3GKS) (C) and ribbon diagram (D). These residues are primarily distributed around the T4-binding site (blue) and its adjacent regions (red).
Residues in or around the thyroxine (T4) binding site (i.e., L17, T106, A108, L110, and V121; Hamilton and Benson, 2001) exhibited the greatest changes in chemical shift during binding to Aβ, i.e., Lys15, Leu17, Ile107, Ala108, Ala109, Leu110, Ala120, Val121, and Val122 (Fig. 4C,D, blue patches). Similar shifts in residues close to the T4 binding site (Met13, Val16, Ala109, Leu110, Ala120, Val121, and Val122) were also observed in WT huTTR (Fig. 4B). The findings suggest a crucial role of this region in the binding of Aβ monomer by TTR. In addition, residues in the A-B loop/helix, which were adjacent to the T4 binding site, i.e., Ala19, Val20, Gly22, Ser23, Ala25, and Asp27 (Fig. 4C,D, red), exhibited shifted resonances. Although these residues showed significant chemical shift changes, they might not reflect direct interaction with Aβ but rather report on conformational perturbations induced by the interaction between Aβ and the T4 binding pocket. Similarly, the chemical shift change of Trp79 was likely to be related to the fact that its side chain pointed toward the T4 binding site and the A-B loop (Fig. 4D). The relevance of the chemical shift perturbations near the T4 binding site were reinforced by studies in which small molecules known to occupy the T4 binding site of the tetramer (Johnson et al., 2012) diminished the capacity of TTR to inhibit Aβ fibril formation (Fig. 5A). No chemical shift changes were observed in the regions known to be involved in TTR–retinol binding protein interaction (Monaco et al., 1995).
Figure 5.
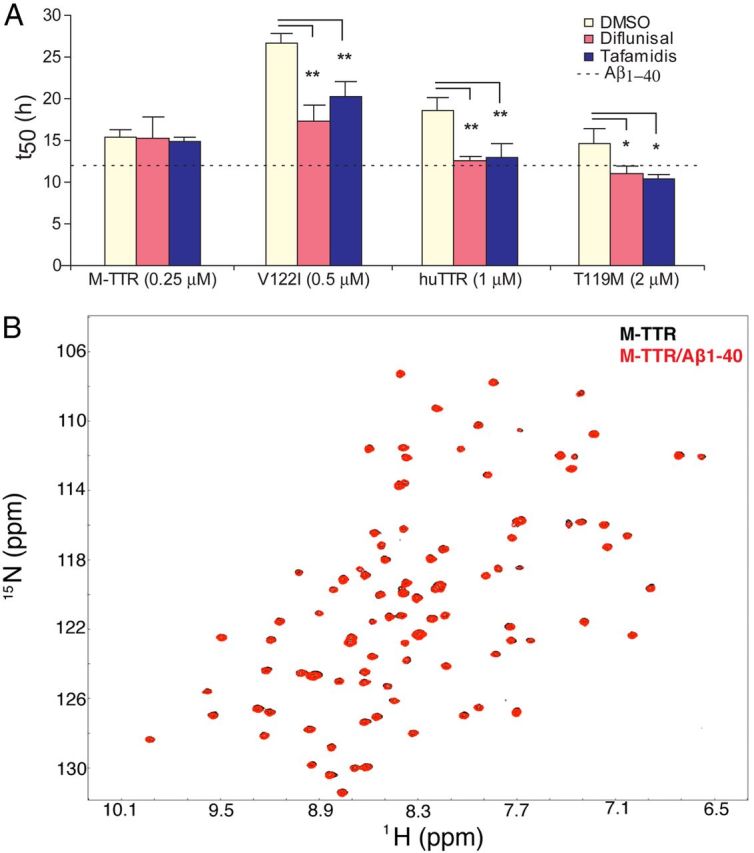
Effect of occupied T4 pocket on inhibition of Aβ aggregation and NMR analysis of M-TTR binding to Aβ1–40. A, TTR stabilizers bind to T4 and abolish the capacity of tetrameric TTRs to inhibit Aβ aggregation. M-TTR, TTRV122I, huTTR (WT), and TTRT119M incubated with Diflunisal, Tafamidis, or DMSO (1:2 molar ratio for 24 h at 37°C) or buffer (dotted line) was added to 10 μm Aβ aggregation. Different concentrations of TTRs were selected to achieve t50 values <30 h (**p < 0.01, n > 3 for each incubation). B, Aβ1–40 binding to M-TTR produced no changes in the amide-proton chemical shifts as shown by the two-dimensional TROSY-HSQC spectra.
Intriguingly, despite its potent inhibition of Aβ fibril formation, we observed no shifted cross-peaks in M-TTR after incubation with Aβ monomer (Fig. 5B). The absence of such changes is consistent with the lack of a detectable signal in the ITC experiments in which M-TTR was titrated with Aβ monomer. Thus, our results demonstrate that M-TTR is a highly efficient inhibitor of Aβ fibrillogenesis in vitro, although it does not appear to bind Aβ monomer as measured by ITC or examined in the NMR experiments (Figs. 1A, 3D, 5B). These results suggest that the inhibitory activity of the monomer resides in its capacity to bind relatively small Aβ oligomers, formed over a longer time than measured by ITC, which prevents additional assembly beyond tetra/pentamers or nono/decamers (Fig. 6A,B, lanes of Aβ+M-TTR).
TTR binding to Aβ involves Aβ amino acids 18–21
To determine the TTR binding site on Aβ, Aβ1–42 monomer and aggregates were immobilized on nitrocellulose and probed with antibodies specific for epitopes formed by different regions of the peptide (Kayed et al., 2003; Ladiwala et al., 2011). The antibodies interacting with the central hydrophobic region of Aβ, i.e., residues 18VFFAE22 (4G8) and residues 16KLVFFA21 (A3356), abolished the TTR/Aβ1–42 interaction as monitored by the biotin–TTR–streptavidin–HRP (conjugated horseradish peroxidase) system (Fig. 7A). The results are consistent for both tetrameric and monomeric TTR and for all Aβ conformers that were assayed.
Figure 7.

Dot-blot epitope mapping of Aβ1–42 conformers bound by TTR and IC50 values of binding. A, TTR-bound Aβ1–42 conformers at the hydrophobic central region was demonstrated by epitope mapping dot blot. Aβ conformers (25 μm, 2 μl) were spotted on nitrocellulose membrane and incubated with antibody specific to different regions (N terminal Aβ3–10; middle, Aβ18–22, Aβ16–21; C terminal, Aβ30–35, Aβ35–39). The second spot on each strip is spotted with oligomers that were A11 (oligomer-specific) antibody positive, whereas the third spot is not. B, Quantification of binding (IC50) of biotinylated TTRs to Aβ1–42 conformers immobilized on Nunc Maxisorb plates.
The binding capacities (IC50 values) of TTR (huTTR, muTTR, and M-TTR) to Aβ conformers to immobilized TTR (huTTR, muTTR, and M-TTR) on 96-well plates, measured as IC50, were also compared. In this assay, all TTRs bound to Aβ oligomers better than to Aβ fibrils (Fig. 7B). M-TTR bound to all forms of Aβ aggregates and showed the strongest binding (the smallest IC50) among three TTRs. M-TTR bound to all forms of Aβ aggregates better than huTTR. Both tetrameric TTRs bound to Aβ oligomers significantly better than to Aβ fibrils. In this assay, neither tetramer bound to Aβ monomers, whereas the strength of M-TTR binding to Aβ monomers was weak relative to its binding to Aβ aggregates under these conditions.
Discussion
Many observations have suggested an interaction between the systemic amyloid precursor TTR and the AD amyloid peptide Aβ (Riisøen, 1988; Hatterer et al., 1993; Schwarzman et al., 1994; Serot et al., 1997; Castano et al., 2006; Brettschneider et al., 2010; Schultz et al., 2010). However, the specificity of the interaction was questioned in the absence of detailed characterization of the binding (for review, see Li and Buxbaum, 2011). We used ITC to determine the KD and stoichiometry of the Aβ monomer–TTR tetramer interaction and NMR to characterize the Aβ binding site on TTR in vitro in solution. Our estimation of the KD differs substantially from previously estimated binding parameters of KS at 2300 m−1 (Liu and Murphy, 2006) by tryptophan fluorescence quenching and KD of 28 nm using competition binding (Costa et al., 2008b). Our NMR data showed that the Aβ binding site on TTR involves amino acids in and around the T4 binding site of the tetramer, partially confirming and extending the recently reported involvement of TTR residues L17, L110, and L82 (but S85) (Du et al., 2012; Yang et al., 2013).
huTTR tetramers of varying stabilities and M-TTR inhibit Aβ oligomerization and fibril formation at substoichiometric concentrations, which is consistent with previous reports that TTRs inhibit Aβ fibril formation (Schwarzman et al., 2004; Liu and Murphy, 2006; Costa et al., 2008b). However, when liquid-phase binding interactions are measured by ITC, M-TTR, the best inhibitor of Aβ aggregation, yields no heat signature and presumably does not bind monomeric Aβ under these conditions, whereas human and murine tetramers, which differ considerably in their inhibitory capacities, have similar binding characteristics.
The ITC data are reinforced by the NMR results in which binding of Aβ produces clear shifts in resonances of amino acids comprising the T4 binding site of the human TTR tetramer. Experiments showing reduced inhibition of Aβ aggregation when the T4 site is occupied by small molecules confirm its involvement in Aβ binding. These results are consistent with, but more definitive than, previous data from cross-linking experiments, alanine scanning mutagenesis, and peptide inhibition of TTR–Aβ interaction, which suggested major roles for residues L110 in strand G and L82 in the EF helix/loop in binding Aβ (Du et al., 2012; Yang et al., 2013). Our data suggest that L82, rather than serving as an Aβ oligomer sensor (Yang et al., 2013), may influence the orientation of the side chain of W79, which usually points to the T4 binding site.
The relatively unstable TTRV30M tetramer shows resonance shifts of the same residues greater than those seen with WT human TTR, indicating greater access of Aβ to the hydrophobic T4 binding site of the tetramer, perhaps accounting for its greater capacity to inhibit fibrillogenesis. There were no shifts when M-TTR was incubated with monomeric Aβ, despite its substantial capacity to inhibit Aβ aggregation but consistent with its lack of binding in the ITC experiments. The fact that M-TTR does not show interaction with Aβ monomer detectable by ITC or NMR, yet efficiently inhibits Aβ oligomer and fibril formation, strongly suggests that M-TTR binds species of Aβ other than monomer, most likely some form of Aβ oligomer. The data in Figure 6 are consistent with that hypothesis.
The data from solid-phase binding assays are not completely consistent with the characteristics of binding or inhibition of fibril formation in liquid phase, i.e., huTTR and muTTR, did not bind to Aβ monomer in dot blots or on the plate, whereas M-TTR bound very well, suggesting that the molecular surfaces available in liquid phase for interaction may not be equally accessible when various forms of Aβ are coupled to solid matrices or tethered to the dextran matrix of the Biacore chip. When Aβ was immobilized, we observed preferential binding of M-TTR to Aβ monomer, which is consistent with a previous report, although we found that this varies with the methods used (Du and Murphy, 2010). Interestingly, in dot-blot assays, regardless of the form of TTR used or the aggregation state of Aβ, specific binding for Aβ epitopes (18VFFA21) encompassing amino acids 16–21, the region required for the formation of on-pathway fibrillar aggregates (Ahmed et al., 2010), is blocked, suggesting that the site is available to interact, presumably with either the hydrophobic T4 binding site or the hydrophobic surface of M-TTR (Fig. 8), confirming the previously reported involvement of residues 17–24 (Du and Murphy, 2010).
Figure 8.

Mechanisms of TTR inhibition of Aβ aggregation in vitro. The majority of Aβ monomers aggregate into oligomers and fibrils and ultimately reach equilibrium in vitro. TTR tetramers bind the central hydrophobic region of Aβ monomer (at residues 18VFFA21) in the T4 binding pocket and sequester the monomer from the oligomer forming pool and abolish Aβ form β-sheet structure. A small fraction (Hurshman Babbes et al., 2008) of WT TTR tetramers dissociate into monomers, which have the capacity to efficiently bind small Aβ oligomers, removing them from the fibril forming pathway and inhibiting additional Aβ aggregation. The excess of TTR tetramer relative to monomer in vivo suggests that the salutary effect of TTR on Aβ deposition is likely to take place via TTR tetramer and Aβ monomer association (dotted line circled area). Other alternatives include TTR tetramer interaction with Aβ oligomers and large fibrils, the destabilization of TTR tetramer by the binding of Aβ monomer in the T4 pocket to yield free Aβ-binding monomers (broken arrows), an effect that would be opposite to that of T4 binding (which stabilizes the tetramer), or interaction of Aβ oligomers with TTR monomer in a cellular compartment before the formation of mature TTR tetramers. The precise anatomic site of interaction in vivo is unknown.
In vitro, Aβ monomers adopt many conformations (Tomaselli et al., 2006), creating an ensemble of aggregation intermediates among which are β-sheet-rich oligomers that seed on-pathway aggregation. The formation of such nuclei is the rate-limiting step in primary fibril formation (for review, see Dasilva et al., 2010). In our experiments, the interaction between TTR tetramer (which does not form fibrils under the conditions of the experiments) and Aβ monomers reduced Aβ aggregation, presumably by removing Aβ from the pool of monomers with the exposed central hydrophobic region critical for β-sheet-rich oligomer formation, i.e., TTR tetramers inhibited Aβ seed formation at substoichiometric concentrations. In contrast, the hydrophobic surface of M-TTR (which does not tetramerize or aggregate under the same experimental conditions) can interact with Aβ oligomers to slow large oligomer formation. Given that TTR equilibrium greatly favors tetramer and evidence suggesting that Aβ does not destabilize the TTR tetramer (data not shown), tetrameric TTR is presumably the major player in inhibition of Aβ aggregation in vivo (Fig. 8).
Recently, it was proposed that interaction of Aβ monomer or small oligomers with TTR tetramer leads to tetramer dissociation and subsequent binding of oligomers by the dissociated monomers (Yang et al., 2013). Our experiments indicate that tetramer dissociation is not required for inhibition of Aβ fibrillogenesis by various TTRs. We were also unable to demonstrate destabilization of TTR tetramers by incubation with Aβ1–40 (data not shown).
What are the implications of these data for the apparent protective activity of TTR in transgenic murine models of Aβ deposition or in the context of human AD? The concentration of the Aβ monomer in human brains (and those of APP23 mice) in vivo is in the high picomolar to low nanomolar range (Cirrito et al., 2003; Buxbaum et al., 2008; Moore et al., 2012). The concentration of TTR tetramers in vivo is much higher (0.25–0.5 μm in human CSF, 3–5 μm in human serum). The predicted concentration of monomeric TTR, based on the WT TTR Kassociation of 1.1 × 1024 m−3 (Hurshman Babbes et al., 2008), is much lower (∼25 nm in CSF, ∼46 nm in serum) and consistent with the observed human serum TTR monomer concentration (5–10 nm; Sekijima et al., 2001). In soluble extracts of brains of APP23 mice in which the neuropathologic and behavioral abnormalities are suppressed by overexpressing huTTR, the concentration of huTTR is 2000 times than that of Aβ (1.4 μm vs 0.7 nm). Thus, in vivo, whereas TTR monomer may be present in excess relative to Aβ, the absolute amount of tetrameric TTR is 1000-fold greater. Assuming that there is no anatomic or cellular compartment in which TTR monomers are preferentially increased in the presence of Aβ oligomers, it appears that TTR tetramer binding to Aβ monomers and perhaps Aβ oligomers is the major mechanism of inhibition of Aβ fibrillogenesis in the brains of APP23 mice overexpressing huTTR (Fig. 8).
Studies in hemizygous Igf1r knock-out mice carrying a human AD gene indicated that the mice formed large non-toxic oligomers (Cohen et al., 2009). The present experiments did not show that TTR drives Aβ aggregation to create large non-toxic, non-fibrillar aggregates when various forms of TTR were added to Aβ monomers at the start of the reaction. However, recent studies examining the interaction of TTR conformers with preformed toxic Aβ oligomers suggest that this may be the case (Cascella et al., 2013). In contrast to another report (Costa et al., 2008a), we could not detect disruption of preformed Aβ oligomers and fibrils by any of the TTRs tested (Fig. 9).
Figure 9.

TTRs did not disaggregate Aβ aggregates. A, ThT monitored 1 μm TTRs (huTTR, muTTR, and M-TTR) incubation with preformed Aβ1–40 (10 μm) fibrils. B, AFM analysis of TTRs (10 μm) incubated quiescently with preformed Aβ1–42 oligomers and fibrils (25 μm) for 24 h. Each panel equals 3 × 3 μm.
TTR is present in the interstitial fluid of the brain because the choroid plexus secretes substantial amounts of the protein into the CSF. We also showed increased TTR synthesis by primary neurons cultured from the hippocampus and cortex of APP23 AD model mice (Li et al., 2011). Hence, TTR is available to interact with Aβ either intracellularly or extracellularly. We believe that the results of the current experiments, coupled with those of others, describe the nature of the chemical interactions that may be involved. They are consistent with previously published data that neuronal TTR transcription is enhanced in human and mouse AD models in response to as yet undetermined signals (Stein et al., 2004). It is unclear whether neuronal TTR production delays the onset of human AD or represents a failed natural defense against the toxic effects of Aβ. In any case, the data we present here describe, to the greatest extent so far, the biophysical constraints governing what appears to be a biologically, and perhaps clinically, important protein–protein interaction that may be therapeutically exploitable.
Footnotes
The authors declare no competing financial interests.
The work was supported by National Institutes of Health Grant AG R01 030027 (J.N.B.) and the W.M. Keck Foundation (J.N.B., X.L.). X.Z. is a Howard Hughes Medical Institute Fellow of the Helen Hay Whitney Foundation. We thank Michael Saure for synthesizing Aβ peptide, Dr. Jean-Philippe Julien for his assistance with the ITC analysis and discussion, Prof. Jane Dyson for suggestions regarding experimental design, Prof. Hun Lim and Drs. Lei Zhao, Goran Pljevaljcic, Amber Murray, Jiyong Lee, and Kanchan Garai for complementary experiments (not included), Prof. Ian Wilson for access to the MicroCal instrument, and Gerard Koon for technical assistance with the NMR experiments. We also thank Drs. Evan Powers, John Hulleman, Francesca Cattaneo, and Natalia Reixach for insightful discussions. Prof. Fabrizio Chiti allowed us access to his unpublished data.
References
- Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Lehmensiek V, Mogel H, Pfeifle M, Dorst J, Hendrich C, Ludolph AC, Tumani H. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS) Neurosci Lett. 2010;468:23–27. doi: 10.1016/j.neulet.2009.10.053. [DOI] [PubMed] [Google Scholar]
- Brockhaus M, Ganz P, Huber W, Bohrmann B, Loetscher HR, Seelig J. Thermodynamic studies on the interaction of antibodies with beta-amyloid peptide. J Phys Chem B. 2007;111:1238–1243. doi: 10.1021/jp0664059. [DOI] [PubMed] [Google Scholar]
- Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of A beta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella R, Conti S, Mannini B, Li X, Buxbaum JN, Tiribilli B, Chiti F, Cecchi C. Transthyretin suppresses the toxicity of oligomers formed by misfolded proteins. Biochim Biophys Acta. 2013;1832:2302–2314. doi: 10.1016/j.bbadis.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Castaño EM, Roher AE, Esh CL, Kokjohn TA, Beach T. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer's disease and non-demented elderly subjects. Neurol Res. 2006;28:155–163. doi: 10.1179/016164106X98035. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, III, Rance M, Skelton NJ. Protein NMR spectroscopy: principles and practice. Burlington, MA: Elsevier Academic; 2007. [Google Scholar]
- Choi SH, Leight SN, Lee VMY, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS. Accelerated A beta deposition in APPswe/PS1 Delta E9 mice with hemizygous deletions of TTR (transthyretin) J Neurosci. 2007;27:7006–7010. doi: 10.1523/JNEUROSCI.1919-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Ferreira-da-Silva F, Saraiva MJ, Cardoso I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the Kunitz protease inhibitor. Plos One. 2008a;3:e2899. doi: 10.1371/journal.pone.0002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Gonçalves A, Saraiva MJ, Cardoso I. Transthyretin binding to A-Beta peptide: impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008b;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Dasilva KA, Shaw JE, McLaurin J. Amyloid-beta fibrillogenesis: structural insight and therapeutic intervention. Exp Neurol. 2010;223:311–321. doi: 10.1016/j.expneurol.2009.08.032. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. A beta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Du D, Murray AN, Cohen E, Kim HE, Simkovsky R, Dillin A, Kelly JW. A kinetic aggregation assay allowing selective and sensitive amyloid-beta quantification in cells and tissues. Biochemistry. 2011;50:1607–1617. doi: 10.1021/bi1013744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Murphy RM. Characterization of the interaction of beta-amyloid with transthyretin monomers and tetramers. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Cho PY, Yang DT, Murphy RM. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng Des Sel. 2012;25:337–345. doi: 10.1093/protein/gzs026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S, Valli MB, Galeazzi R, Fattoretti P, Corder EH, Galeazzi L. Transthyretin inhibition of amyloid beta aggregation and toxicity. Clin Biochem. 2005;38:1112–1119. doi: 10.1016/j.clinbiochem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Hamilton JA, Benson MD. Transthyretin: a review from a structural perspective. Cell Mol Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström P, Jiang X, Deechongkit S, Kelly JW. Anion shielding of electrostatic repulsions in transthyretin modulates stability and amyloidosis: insight into the chaotrope unfolding dichotomy. Biochemistry. 2001;40:11453–11459. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatterer JA, Herbert J, Hidaka C, Roose SP, Gorman JM. CSF transthyretin in patients with depression. Am J Psychiatry. 1993;150:813–815. doi: 10.1176/ajp.150.5.813. [DOI] [PubMed] [Google Scholar]
- Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–6984. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Smith CS, Petrassi HM, Hammarström P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Ladiwala AR, Dordick JS, Tessier PM. Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. J Biol Chem. 2011;286:3209–3218. doi: 10.1074/jbc.M110.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladiwala AR, Bhattacharya M, Perchiacca JM, Cao P, Raleigh DP, Abedini A, Schmidt AM, Varkey J, Langen R, Tessier PM. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc Natl Acad Sci U S A. 2012;109:19965–19970. doi: 10.1073/pnas.1208797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from A beta(1–42) are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Buxbaum JN. Transthyretin and the brain re-visited: is neuronal synthesis of transthyretin protective in Alzheimer's disease? Mol Neurodegener. 2011;6:79. doi: 10.1186/1750-1326-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Masliah E, Reixach N, Buxbaum JN. Neuronal production of transthyretin in human and murine Alzheimer's disease: is it protective? J Neurosci. 2011;31:12483–12490. doi: 10.1523/JNEUROSCI.2417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Murphy RM. Kinetics of inhibition of beta-amyloid aggregation by transthyretin. Biochemistry. 2006;45:15702–15709. doi: 10.1021/bi0618520. [DOI] [PubMed] [Google Scholar]
- Mazur-Kolecka B, Frackowiak J, Wiśniewski HM. Apolipoproteins E3 and E4 induce, and transthyretin prevents accumulation of the Alzheimer's β-amyloid peptide in cultured vascular smooth muscle cells. Brain Res. 1995;698:217–222. doi: 10.1016/0006-8993(95)00873-O. [DOI] [PubMed] [Google Scholar]
- Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- Moore BD, Chakrabarty P, Levites Y, Kukar TL, Baine AM, Moroni T, Ladd TB, Das P, Dickson DW, Golde TE. Overlapping profiles of Abeta peptides in the Alzheimer's disease and pathological aging brains. Alzheimer Res Ther. 2012;4:18. doi: 10.1186/alzrt121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N, Foss TR, Santelli E, Pascual J, Kelly JW, Buxbaum JN. Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic: a lesson in the generation of transgenic models of diseases involving oligomeric proteins. J Biol Chem. 2008;283:2098–2107. doi: 10.1074/jbc.M708028200. [DOI] [PubMed] [Google Scholar]
- Riisøen H. Reduced prealbumin (transthyretin) in CSF of severely demented patients with Alzheimer's disease. Acta Neurol Scand. 1988;78:455–459. doi: 10.1111/j.1600-0404.1988.tb03687.x. [DOI] [PubMed] [Google Scholar]
- Sarroukh R, Cerf E, Derclaye S, Dufrêne YF, Goormaghtigh E, Ruysschaert JM, Raussens V. Transformation of amyloid beta(1–40) oligomers into fibrils is characterized by a major change in secondary structure. Cell Mol Life Sci. 2011;68:1429–1438. doi: 10.1007/s00018-010-0529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz K, Nilsson K, Nielsen JE, Lindquist SG, Hjermind LE, Andersen BB, Wallin A, Nilsson C, Petersén A. Transthyretin as a potential CSF biomarker for Alzheimer's disease and dementia with Lewy bodies: effects of treatment with cholinesterase inhibitors. Eur J Neurol. 2010;17:456–460. doi: 10.1111/j.1468-1331.2009.02841.x. [DOI] [PubMed] [Google Scholar]
- Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D. Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid. 2004;11:1–9. doi: 10.1080/13506120410001667458. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Tokuda T, Kametani F, Tanaka K, Maruyama K, Ikeda S. Serum transthyretin monomer in patients with familial amyloid polyneuropathy. Amyloid. 2001;8:257–262. doi: 10.3109/13506120108993822. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Serot JM, Christmann D, Dubost T, Couturier M. Cerebrospinal fluid transthyretin: aging and late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1997;63:506–508. doi: 10.1136/jnnp.63.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APP(Sw) mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli S, Esposito V, Vangone P, van Nuland NA, Bonvin AM, Guerrini R, Tancredi T, Temussi PA, Picone D. The alpha-to-beta conformational transition of Alzheimer's Abeta-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang SH, Liu FF, Dong XY, Sun Y. Thermodynamic analysis of the molecular interactions between amyloid beta-peptide 42 and (−)-epigallocatechin-3-gallate. J Phys Chem B. 2010;114:11576–11583. doi: 10.1021/jp1001435. [DOI] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DT, Joshi G, Cho PY, Johnson JA, Murphy RM. Transthyretin as both a sensor and a scavenger of beta-amyloid oligomers. Biochemistry. 2013;52:2849–2861. doi: 10.1021/bi4001613. [DOI] [PMC free article] [PubMed] [Google Scholar]