

Abstract
Despite the critical importance of voltage-gated ion channels in neurons, very little is known about their functional properties in Fragile X syndrome: the most common form of inherited cognitive impairment. Using three complementary approaches, we investigated the physiological role of A-type K+ currents (IKA) in hippocampal CA1 pyramidal neurons from fmr1-/y mice. Direct measurement of IKA using cell-attached patch-clamp recordings revealed that there was significantly less IKA in the dendrites of CA1 neurons from fmr1-/y mice. Interestingly, the midpoint of activation for A-type K+ channels was hyperpolarized for fmr1-/y neurons compared with wild-type, which might partially compensate for the lower current density. Because of the rapid time course for recovery from steady-state inactivation, the dendritic A-type K+ current in CA1 neurons from both wild-type and fmr1-/y mice is likely mediated by KV4 containing channels. The net effect of the differences in IKA was that back-propagating action potentials had larger amplitudes producing greater calcium influx in the distal dendrites of fmr1-/y neurons. Furthermore, CA1 pyramidal neurons from fmr1-/y mice had a lower threshold for LTP induction. These data suggest that loss of IKA in hippocampal neurons may contribute to dendritic pathophysiology in Fragile X syndrome.
Introduction
Fragile X syndrome (FXS) is the most common form of inherited mental delay, affecting 1 in 4000 males and 1 in 6000 females. Patients with FXS display a variety of phenotypes related to the CNS, including lower cognitive function, impairments of both working and episodic memory, and placement along the autistic spectrum (Van Dam et al., 2000; Jin and Warren, 2003; Bernardet and Crusio, 2006; Dölen et al., 2007; Bassell and Warren, 2008). At the neuronal level, FXS has generally been considered a synaptic disorder. This is due to the large body of evidence showing alterations in synaptic structures and protein expression (Comery et al., 1997; Nimchinsky et al., 2001; Hou et al., 2006). However, synaptic elements are only part of the process that is involved in the regulation of neuronal signaling. Once a signal crosses the synapse, it is processed and integrated by neuronal dendrites, in particular, by the myriad of voltage-gated channels expressed in dendrites and spines (Magee, 2000; London and Häusser, 2005; Johnston and Narayanan, 2008; Sjöström et al., 2008). Investigations into the impact of FXS on voltage-gated channels and the processing ability of dendrites are surprisingly lacking. This is particularly noteworthy because FMRP, the mRNA-binding protein that is lacking in FXS, binds to the mRNAs encoding a number of ion channel proteins (Bell et al., 1991; Darnell et al., 2011).
One channel that is highly enriched in the distal dendrites of hippocampal CA1 pyramidal neurons and influences both synaptic inputs and back-propagating action potentials (b-APs) is the A-type K+ channel (IKA) (Hoffman et al., 1997). This channel is thought to be from the shal or KV4 family of voltage-gated K+ channels (Serôdio et al., 1994), and is critically important for normal dendritic function and excitability (Hoffman et al., 1997; Ramakers and Storm, 2002; Johnston et al., 2003). The rapid kinetics and quick recovery from inactivation allow IKA to influence a variety of neuronal functions. Under normal conditions, IKA regulates dendritic excitability by limiting the amplitude of both locally generated dendritic spikes and b-APs. The high density of IKA can also reduce the magnitude of EPSPs in the distal dendrites and is subject to activity-dependent regulation with certain forms of synaptic plasticity (Frick et al., 2004; Kim et al., 2007; Lin et al., 2011). Two recently published reports using biochemical approaches arrived at conflicting conclusions as to whether expression of KV4.2, the putative pore-forming subunit of hippocampal A-type K+ channels, is elevated (Lee et al., 2011) or reduced (Gross et al., 2011) in hippocampal neurons from the fmr1-/y mouse model of FXS.
Here we demonstrate that dendritic, but not somatic, IKA is lower in CA1 pyramidal neurons from fmr1-/y mice. In addition to the reduced maximum dendritic IKA, the voltage dependence of activation of A-type K+ channels is hyperpolarized in fmr1-/y neurons. Based upon the time course of recovery from inactivation, the IKA present in fmr1-/y dendrites is likely mediated by KV4 containing channels. This overall reduction in IKA results in larger b-APs, more Ca2+ influx in the distal dendrites, and a lowered threshold for LTP induction in fmr1-/y neurons. We thus provide crucial evidence that demonstrates that reduced A-type K+ channel function alters hippocampal dendritic physiology in fmr1-/y mice. These deficits in hippocampal neuron function may, in part, be responsible for the cognitive impairments associated with FXS.
Materials and Methods
Preparation of acute hippocampal slices.
All experiments were conducted in accordance with the University's Institutional Animal Care and Use Committee. Hippocampal slices (300 μm) were prepared from 2- to 4-month-old male wild-type (C57BL/6) and fmr1-/y mice as described in Brager et al. (2012). Briefly, animals were deeply anesthetized with a lethal dose of ketamine and xylazine and intracardially perfused with ice-cold modified ACSF containing the following (in mm): 210 sucrose, 2.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7.0 MgCl2, and 7.0 dextrose bubbled with 95% O2/5% CO2. The brain was removed and bisected along the midline, an oblique cut was made to promote the planar orientation of the dendrites, the brain was mounted to the stage of a Vibratome, and sections were made from the middle hippocampus. Slices were placed in a holding chamber with ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.0 CaCl2, 2.0 MgCl2, and 21 dextrose, pH 7.4, bubbled with 95% O2/5% CO2 at 35°C for 45–60 min and then kept at room temperature.
Electrophysiology.
Slices were placed individually, as needed, into a submerged recording chamber and continuously perfused with oxygenated extracellular saline containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.0 CaCl2, 1.0 MgCl2, and 21 dextrose, pH 7.4, at 32–34°C. Slices were viewed with a Zeiss AxioExaminer D microscope fitted with a 60× water-immersion objective and Dodt contrast optics. Patch pipettes were pulled from borosilicate glass and wrapped with Parafilm to reduce capacitance.
Cell-attached patch recordings.
For cell-attached recordings, pipettes contained the following (in mm): 125 NaCl, 3 KCl, 10 HEPES, 2.0 CaCl2, 1.0 MgCl2, and 0.001 TTX, pH 7.3 with KOH. Membrane currents were recorded using an Axopatch 200B amplifier (Molecular Devices), sampled at 10 kHz, analog filtered at 2 kHz, and digitized by an ITC-18 interface connected to a computer running Axograph X. To separate IKA from total IK, the sustained component was isolated using a depolarized holding potential (−30 mV) in which IKA is inactivated and subtracting that from the total IK obtained from a hyperpolarized holding potential (−100 mV). Activation curves were constructed by using depolarizing voltage commands (−90 to 50 mV in 10 mV steps) to activate IKA from a holding potential of −100 mV. Steady-state inactivation was studied using depolarizing test pulses to a fixed voltage (50 mV) preceded by a series of prepulse conditioning potentials ranging from −100 to −30 mV. Activation and inactivation data were fit to single Boltzmann functions using a least-squares program. Recovery from inactivation was determined by measuring the IKA in response to two steps from −100 to 50 mV. The time between the two steps was varied from 5 to 300 ms. A single exponential fit the recovery from inactivation of IKA. Linear leakage and capacitive currents were digitally subtracted by scaling traces at smaller command voltages in which no voltage-dependent current was activated.
Whole-cell dendritic recording.
The ACSF was the same as for cell-attached experiments except 20 μm DNQX, 50 μm d-AP5, 5 μm gabazine, and 5 μm CGP55845 were included to block synaptic transmission. For whole-cell dendritic recording, pipettes were filled with the following (in mm): 120 K-gluconate, 20 KCl, 10 HEPES, 4 NaCl, 4.0 Mg-ATP, 0.3 Na-GTP, and 14 K2-phosphocreatine, pH 7.3 with KOH. Whole-cell recordings were made using a Dagan BVC-700A in current-clamp mode. Data were sampled at 40 kHz, analog filtered at 5 kHz, and digitized by an ITC-18 interface connected to a computer running Axograph X. Series resistance (RS) was monitored throughout the recording and the experiment was discarded if RS exceeded 30 MΩ or varied by >20%. Back-propagating action potentials were elicited by antidromic extracellular stimulation in the stratum oriens or the alveus. The liquid junction potential was 13 mV and was not corrected.
Ca2+ imaging.
For Ca2+-imaging experiments, slices were viewed using a Zeiss Axioskop FS2 fitted with a 60× objective and differential interference contrast optics. The extracellular saline was the same as for whole-cell dendritic recording except that 20 μm BaCl2 was included to block inwardly rectifying K+ channels. For Ca2+-imaging experiments, the pipette solution was the same as for whole-cell recording except for the inclusion of the calcium indicator bis-fura-2 (100 μm). Neurons were allowed to fill with the Ca2+ indicator for 20 min before imaging. Voltage signals were recorded using a Dagan BVC-700A amplifier, sampled at 20 kHz, analog filtered at 3 kHz, and digitized by an ITC-18 interface connected to a computer running custom written software in IGOR Pro. A CCD camera (Redshirt Imaging) with an 80 × 80 pixel array, 2 kHz frame rate, and single wavelength (380 nm) excitation was used to measure calcium signals in response to back-propagating action potentials (2 nA for 2 ms into the soma). Changes in [Ca2+]i were quantified by calculating ΔF/F0, where F0 is the fluorescence intensity before stimulation (after subtracting autofluorescence) and ΔF is the change in fluorescence during neuronal activity (corrected for bleaching). The autofluorescence of the tissue was measured in a region of equal size but adjacent to the dye-filled neuron in the dendritic field and bleaching was determined by measuring the change in fluorescence at rest (without stimulation). The ΔF/F0 measurements were repeated five times and averaged.
Weak theta-burst pairing LTP experiments.
The methods for the LTP experiments (see Fig. 7) were similar to those described previously (Watanabe et al., 2002; Chen et al., 2006). GABAA- and GABAB-mediated IPSPs were blocked by 2 μm SR95531 and 5 μm CGP55845, respectively. To prevent epileptiform activity, a cut was made between area CA3 and area CA1. Schaffer collateral EPSPs of 2–3 mV were elicited using tungsten-stimulating electrodes placed <20 μm from the apical dendrite ∼180–225 μm from the soma. Back-propagating action potentials were elicited by somatic current injection (1–2 nA from 2 ms). The weak theta-burst pairing (TBP) protocol consisted of a burst of 5 EPSPs at 100 Hz paired with a single somatic current injection (paired with the last EPSP in the burst) and each burst was delivered 10 times at 5 Hz. This train was repeated three times at 10 s intervals. Although there was only a single current injection paired with the burst of EPSPs, the number of elicited action potentials varied from cell to cell. Therefore, the actual number of elicited action potentials was reported for each burst.
Figure 7.
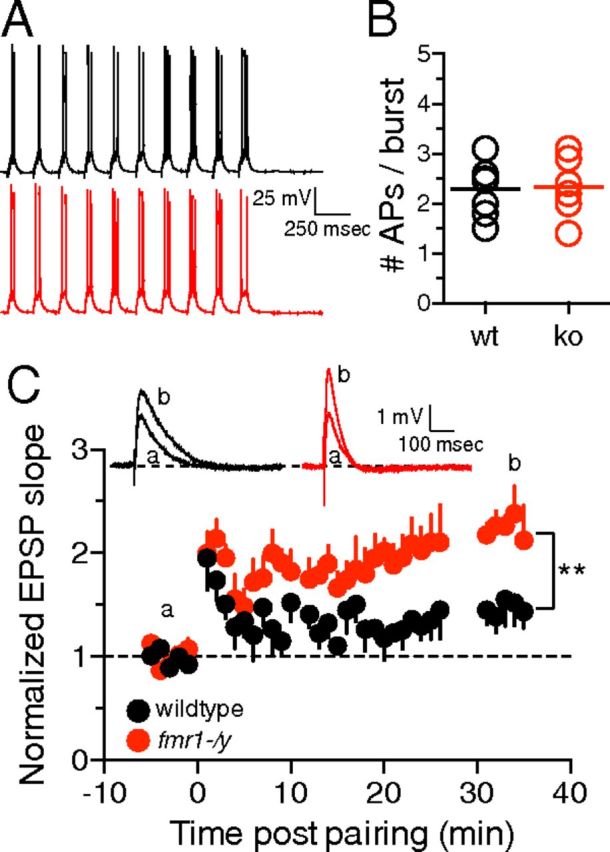
wTBP is more effective at producing LTP in fmr1-/y neurons. A, Representative traces showing the wTBP protocol. B, The mean number of APs per burst is not significantly different between wild-type and fmr1-/y neurons. C, Graph showing the time course of EPSP slope change after a wTBP protocol. Note that the LTP in fmr1-/y neurons is significantly greater than wild-type neurons 30 min after wTBP. Inset, Representative EPSPs during baseline (a) and 30–35 min post wTBP (b). For wild-type: n = 7 cells, 4 mice; fmr1-/y: n = 6 cells, 4 mice **p < 0.01 vs wild-type.
Statistical analyses.
All data were compared using Student's t test, two-way ANOVA, and linear regression where appropriate (see figure legends for specific instances). All fits were performed using Prism software (GraphPad). Data were considered significantly different if p < 0.05. All data are plotted as mean ± SEM.
Results
Fmr1-/y mice lack the distal dendritic enrichment of A-type K+ channels
We made cell-attached patch-clamp recordings from the soma or the distal apical dendrite (range: 175–250 μm from the soma; median: 200 μm) of CA1 pyramidal neurons from wild-type and fmr1-/y mice to measure the current through A-type K+ channels directly. Depolarization to 50 mV from a holding potential of −100 mV resulted in an outward current (total IK) that had both a transient and a sustained component (Fig. 1A). Subtracting the sustained current measured when the patch was held at −30 mV from the total IK isolated the inactivating A-type K+ current (IKA, Fig. 1A,B). Consistent with the previously described increasing gradient of IKA from soma to dendrite, we found that in wild-type neurons, the peak dendritic IKA was significantly larger than the peak somatic IKA (dendritic: 29.0 ± 4.72 pA, n = 10; soma: 14.0 ± 2.55 pA, n = 10; Fig. 1C, black symbols). In contrast, there was no significant difference between the peak somatic and dendritic IKA in fmr1-/y neurons (dendritic: 13.1 ± 2.29 pA, n = 8; soma: 14.1 ± 2.07 pA, n = 7, p > 0.05; Fig. 1C, red symbols). These data suggest that the distal dendritic enrichment of IKA present in wild-type neurons is absent in fmr1-/y mice.
Figure 1.

Maximum dendritic IKA is smaller in fmr1-/y mice. A, Isolation of IKA by subtraction of sustained IK from total IK. B, Representative A-type K+ currents measured at 50 mV in dendritic patches (200 μm from the soma) on wild-type (black) and fmr1-/y (red) neurons. C, Graph of the summary data showing that maximum dendritic IKA is significantly larger than somatic IKA in wild-type neurons, but not in fmr1-/y neurons. For wild-type: dendritic patches, n = 10 from 8 mice; somatic patches, n = 10 from 5 mice. For fmr1-/y: dendritic patches, n = 8 from 6 mice, somatic patches, n = 7 from 4 mice. *p < 0.05 vs soma; p̂ < 0.05 vs wild-type, two-way ANOVA.
A-type K+ channels in fmr1-/y neurons are activated at more negative potentials compared with wild-type neurons
We measured the voltage dependence of A-type K+ channels in somatic and dendritic patches from wild-type and fmr1-/y mice. Outward K+ currents were elicited by depolarizing the patches in 10 mV increments from a holding potential of −100 mV (Fig. 2A). Consistent with previously published results (Hoffman et al., 1997), we found that in wild-type neurons, the activation of dendritic A-type K+ channels was significantly hyperpolarized compared with somatic A-type K+ channels (dendritic V1/2: −0.1 ± 2.87 mV, n = 7; soma V1/2: 9.6 ± 2.9 mV, n = 4, p < 0.05; Fig. 2B). In fmr1-/y neurons, the activation of dendritic A-type K+ channels was similarly hyperpolarized compared with the soma (dendritic V1/2: −10.9 ± 2.43 mV, n = 8; soma V1/2: −2.9 ± 3.6 mV, n = 4, p < 0.05; Fig. 2C). Interestingly, the activation of both somatic and dendritic A-type K+ channels in fmr1-/y neurons was significantly hyperpolarized compared with wild-type (Fig. 2D–F). There was no significant difference in slope factor for either dendritic (kwt: 11.3 ± 0.9; kfmr1-/y: 9.3 ± 1.2, p > 0.05) or somatic A-type K+ channels (kwt: 13.0 ± 2.2 mV; kfmr1-/y: 10.6 ± 1.1; p > 0.05) between wild-type and fmr1-/y neurons. These data suggest that although the IKA current density is lower in the dendrites of fmr1-/y neurons, the channels that are present activate at more hyperpolarized voltages.
Figure 2.

Voltage dependence of IKA activation is hyperpolarized in fmr1-/y neurons. A, Representative IKA traces (top) in response to a family of depolarizing voltage commands from −100 to 50 mV in 10 mV steps (bottom). B, C, Activation curves of transient A-type K+ channels recorded from somatic and dendritic cell-attached patches in wild-type (B) and fmr1-/y (C) neurons. D, E, Activation curves of transient A-type K+ channels recorded from dendritic (D) and somatic (E) cell-attached patches in wild-type and fmr1-/y neurons. F, Group data showing that the V1/2 of activation in dendritic patches is hyperpolarized compared with somatic patches for both wild-type and fmr1-/y neurons. For wild-type: dendritic patches, n = 7 from 7 mice; somatic patches, n = 4 from 3 mice. For fmr1-/y: dendritic patches, n = 8 from 7 mice, somatic patches, n = 4 from 3 mice. *p < 0.05 vs soma; p̂ < 0.05 vs wild-type, two-way ANOVA.
The IKA present in fmr1-/y dendrites is mediated by KV4-containing channels
The inactivation of A-type K+ channels is voltage dependent, with the time constant of inactivation (τinact) increasing with depolarization (Hoffman et al., 1997). In agreement with these results, we found that in wild-type neurons, τinact increased with depolarization (Fig. 3A1,B, black symbols). In contrast, τinact in fmr1-/y neurons was less voltage dependent (Fig. 3A2,B) and the slope of the line fit to the voltage dependence of τinact in fmr1-/y patches was significantly smaller compared with wild-type (wt: 1.0 ± 0.4 ms/10 mV, n = 8; ko: 0.3 ± 0.1 ms/10 mV, n = 6, p < 0.05; Fig. 3C). Due to the reduced voltage dependence, τinact for maximum IKA was significantly slower in wild-type dendrites compared with fmr1-/y dendrites (wt: 19.1 ± 3.6 ms; fmr1-/y: 10.1 ± 2.3 ms, p < 0.05, t test; Fig. 3D).
Figure 3.

The kinetics of recovery from steady-state inactivation suggests that IKA in both wild-type and fmr1-/y neurons is mediated by KV4-containing channels. A, Representative traces showing the slower inactivation of IKA at 50 mV (thick trace) compared with 0 mV (thin trace) in a dendritic patch from a wild-type neuron (A1) and an fmr1-/y neuron (A2). The traces are scaled to peak an on an expanded time scale below. B, τinact of IKA is less voltage dependent in fmr1-/y dendrites compared with wild-type dendrites. C, The slope of the fit to the voltage dependence of τinact for fmr1-/y patches is significantly smaller than wild-type patches. D, The mean τinact for the maximum IKA is significantly faster in fmr1-/y patches (wild-type: n = 8 patches, 8 mice; fmr1-/y: n = 6 patches, 5 mice; *p < 0.05 vs wild-type). E, Steady-state inactivation of dendritic A-type K+ channels is not significantly different between wild-type and fmr1-/y neurons. Inset, Representative traces and voltage protocol used to measure steady-state inactivation (wild-type: n = 4 patches, 3 mice; fmr1-/y: n = 4patches, 4 mice). F, Recovery from steady-state inactivation for dendritic A-type K+ channels from wild-type and fmr1-/y neurons. G, Group data showing the time constant of recovery from steady-state inactivation of dendritic A-type K+ channels for wild-type and fmr1-/y neurons is not significantly different. Note the τrecovery for both wild-type and fmr1-/y neurons is consistent with KV4 containing A-type K+ channels. For wild-type: n = 5 from 5 mice; fmr1-/y: n = 4 from 4 mice.
We next investigated whether there are any differences in steady-state inactivation for A-type K+ channels between dendritic wild-type and fmr1-/y neurons. The patch was depolarized to various prepotentials (−90 to −30 mV) for 500 ms to inactivate any A-type K+ channels present in the patch, which was followed by a test pulse (50 mV) to measure the maximum available IKA (Fig. 3E). There were no significant differences in steady-state IKA inactivation between wild-type and fmr1-/y neurons (V1/2 WT: −63 ± 1.3 mV, n = 4; V1/2 fmr1-/y: −66 ± 2.8 mV, n = 4, p > 0.05; kWT: 7.0 ± 0.7; kfmr1-/y: 7.7 ± 0.5; p > 0.05; Fig. 3E).
A-type K+ channels can be composed of either Kv4 or Kv1 subunits (Coetzee et al., 1999). One method of discriminating between A-type currents mediated by Kv1 and Kv4 potassium channels involves measuring the recovery from steady-state inactivation: KV1 channels recover an order of magnitude slower than KV4 channels (Castellino et al., 1995; Jerng et al., 2004). We found that the time constant of recovery from steady-state inactivation was not different between wild-type and fmr1-/y dendritic A-type channels and was consistent with KV4-containing channels (wt: 12 ± 3 ms, n = 5; fmr1-/y: 18 ± 4 ms, n = 4,p > 0.05; Fig. 3F,G). These data suggest that, similar to wild-type neurons, the dendritic IKA in fmr1-/y neurons is mediated by KV4-containing channels.
Back-propagating action potentials are larger in fmr1-/y dendrites
One physiological role of A-type K+ channels in CA1 pyramidal neurons is to limit the amplitude of b-APs into the distal dendritic arbor (Fig. 4A). Based upon our cell-attached patch recordings and previous data from Kv4.2 knock-out mice (Chen et al., 2006), we predicted that b-AP amplitude would be greater in the distal dendrites of fmr1-/y neurons due to lower IKA. We measured b-APs in distal dendrites (range: 180–225 μm from the soma, median 200 μm), proximal dendrites (range: 75–150 μm from soma, median 100 μm), and in the soma in wild-type and fmr1-/y neurons. In agreement with this prediction, we found that distal dendritic b-APs were significantly larger in fmr1-/y neurons compared with wild-type neurons (wt: 25.5 ± 3.2 mV, n = 5; fmr1-/y: 38.6 ± 2.1 mV, n = 7, p < 0.05; Fig. 4B–D). Consistent with our finding that IKA was not significantly different at the soma between wild-type and fmr1-/y neurons, we found no significant difference in action potential amplitude measured at the soma (wt: 112 ± 1 mV, n = 5; fmr1-/y: 114 ± 1 mV, n = 5, p > 0.05) nor in the proximal dendrite (wt: 83 ± 3.4 mV, n = 6; fmr1-/y: 84 ± 2 mV, n = 6, p > 0.05; Fig. 4D).
Figure 4.
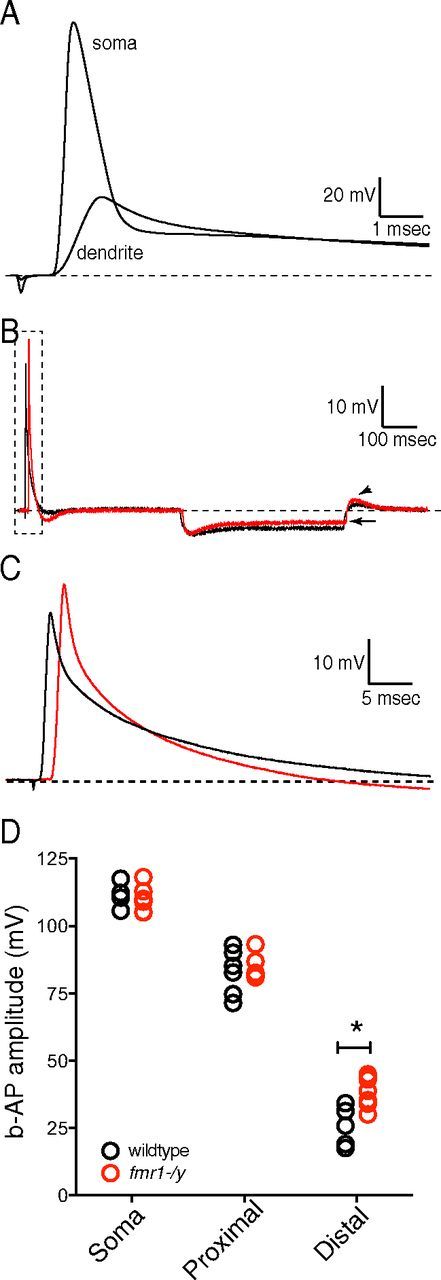
Distal dendritic b-APs are larger in fmr1-/y neurons. A, Representative voltage traces showing the attenuation of b-AP amplitude between the soma and distal dendrite of CA1 pyramidal neurons. B, Recordings of b-APs from the distal dendrites of wild-type (black) and fmr1-/y (red) neurons. Note the lower input resistance (arrow) and higher rebound amplitude (arrowhead) in fmr1-/y dendrites consistent with higher dendritic h-channels in fmr1-/y CA1 pyramidal neuron dendrites. C, The representative b-APs recorded from wild-type (black) and fmr1-/y (red) dendrites (∼ 220 μm from the soma) indicated in the box in B. D, Scatter plot showing the distance-dependent attenuation of bAP amplitude for wild-type and fmr1-/y neurons. Note the larger bAP amplitude in the distal dendrites of fmr1-/y neurons compared with wild-type. For wild-type recordings, n = 5 somatic, 3 mice; n = 6 proximal dendrite, 4 mice; and n = 5 distal dendrite, 4 mice. For fmr1-/y recordings, n = 5 somatic, 2 mice, n = 6 proximal dendrite, 4 mice; and n = 7 distal dendrite, 4 mice. *p < 0.05 vs wild-type, two-way ANOVA.
b-AP-mediated dendritic Ca2+-influx is greater in fmr1-/y neurons
The back-propagation of action potentials leads to increases in dendritic intracellular calcium via the opening of dendritic voltage-gated Ca2+ channels. The lower IKA in fmr1-/y neurons should result in greater dendritic calcium influx in response to b-APs (Chen et al., 2006). We used fluorescent imaging to measure b-AP-mediated Ca2+ influx in the dendrites of CA1 pyramidal neurons (Fig. 5A). Under baseline conditions, and with inwardly rectifying K+ channels blocked by 20 μm Ba2+, b-AP-mediated Ca2+ influx significantly decreased with distance from the soma in wild-type neurons, consistent with an increasing density of A-type K+ channels along the apical dendrite (Fig. 5B1,C, black symbols). Dendritic calcium signals in fmr1-/y neurons also decreased with distance (Fig. 5B2,C, red symbols) from the soma, but were significantly larger than those observed in wild-type dendrites 200 and 250 μm from the soma.
Figure 5.
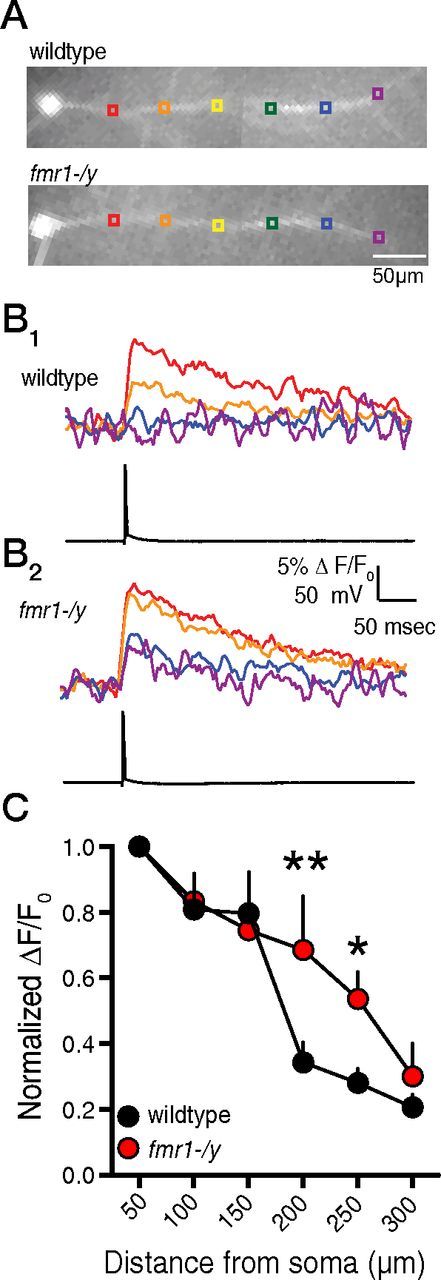
b-AP-mediated Ca2+ influx is larger in the distal dendrites of fmr1-/y neurons. A, CCD images of bis-fura-2-filled CA1 pyramidal neurons from wild-type and fmr1-/y neurons. B, Representative traces showing Ca2+ influx (top) at the indicated regions in A in response to a single action potential elicited by somatic current injection (bottom) for wild-type (B1) and fmr1-/y (B2) neurons. C, Group data showing that distal dendritic b-AP-mediated Ca2+ influx is larger in fmr1-/y neurons (n = 7 cells in 5 mice) compared with wild-type neurons (n = 11 in 8 mice). *p < 0.05 vs wild-type; **p < 0.01 vs wild-type, two-way ANOVA.
To determine whether the difference in b-AP-mediated calcium signals was due to lower IKA in fmr1-/y dendrites, we blocked IKA by raising extracellular Ba2+ to 150 μm (a concentration sufficient to block IKA (Gasparini et al., 2007). Extracellular Ba2+ reversibly increased b-AP-mediated calcium influx in wild-type dendrites (Fig. 6A,B), but not fmr1-/y dendrites (Fig. 6A,C). Furthermore, in the presence of 150 μm Ba2+, there was no difference in the distance-dependent decrease in b-AP-mediated calcium signaling between wild-type and fmr1-/y neurons (Fig. 6D). These data suggest that the lower expression of functional A-type K+ channels in fmr1-/y neurons contributes to the difference in the observed calcium signal.
Figure 6.
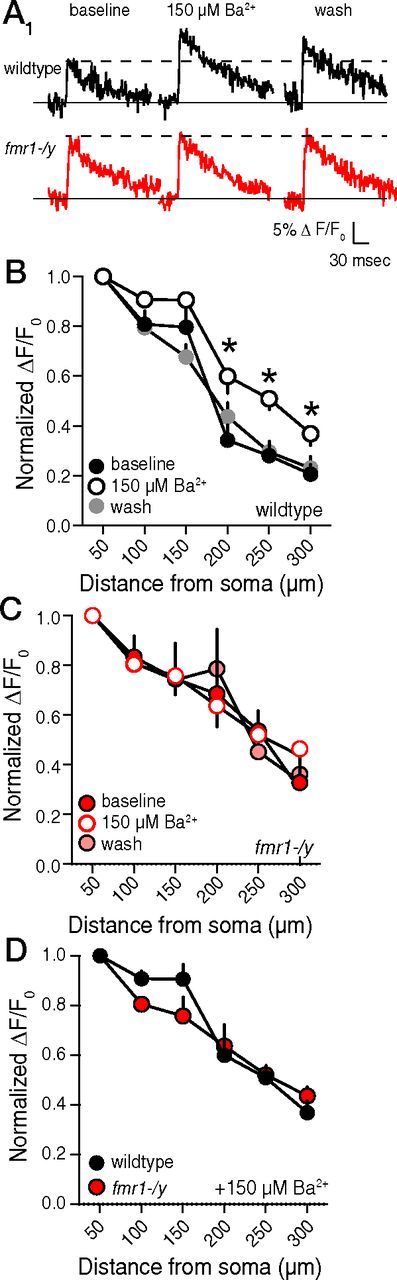
Block of IKA reduces the distance-dependent decrease in b-AP-mediated Ca2+-influx in wild-type, but not fmr1-/y neurons. A, Representative traces showing Ca2+ influx 250 μm from the soma in response to a single action potential during baseline, 150 μm extracellular Ba2+, and washout measured from a wild-type and fmr1-/y neuron. B, 150 μm extracellular Ba2+ increases distal dendritic Ca2+ influx in a reversible manner in the distal dendrites of wild-type neurons. C, 150 μm extracellular Ba2+ has no effect on distal dendritic Ca2+ influx in fmr1-/y neurons. D, Group data showing that distal dendritic b-AP-mediated Ca2+-influx is not different between fmr1-/y and wild-type neurons in the presence of 150 μm extracellular Ba2+. *p < 0.05 vs baseline.
Induction threshold for theta-burst pairing LTP is lower in fmr1-/y neurons
Functional A-type K+ channels and their influence on dendritic b-APs and calcium influx play a role in determining the threshold for TBP-LTP in the hippocampus (Magee and Johnston, 1997; Watanabe et al., 2002). We previously demonstrated that after strong TBP (5 spikes per burst of 5 EPSPs), there was no significant difference in LTP between wild-type and fmr1-/y slices (Brager et al., 2012). Previous work with KV4.2 knock-out mice demonstrated that fewer b-APs were required to induce TBP-LTP in absence of functional A-type K+ channels (Chen et al., 2006). We hypothesized that the larger bAPs and greater calcium influx in fmr1-/y dendrites would permit a weak TBP protocol (wTBP: 1–3 spikes per burst of 5 EPSPs) to induce greater LTP compared with wild-type neurons. We elicited EPSPs using a stimulating electrode placed 180–240 μm from the soma in stratum radiatum. This location corresponded to where we observed the greatest difference in calcium influx between wild-type and fmr1-/y dendrites (Fig. 5C). Although the number of action potentials did not always match the number of current injections, the average number of action potentials elicited per burst of EPSPs was not significantly different between wild-type and fmr1-/y neurons (Fig. 7A,B). Immediately after wTBP, both wild-type and fmr1-/y neurons showed short-term potentiation of EPSP slope (wt: 195 ± 30%, n = 7; ko: 200 ± 21%, n = 6, p > 0.05). In wild-type neurons, EPSP slope decreased over the next 20–30 min, whereas in fmr1-/y neurons, the EPSP slope remained significantly more potentiated (Fig. 7C). The amount of LTP 30 min post-wTBP was significantly greater in fmr1-/y neurons compared with wild-type neurons (wt: 161 ± 9%; ko: 223 ± 10%; p < 0.01).
Discussion
We found that there were significantly less functional A-type K+ channels in the distal dendrites of CA1 pyramidal neurons from the fmr1-/y mouse model of FXS. The maximum dendritic IKA was reduced by 50% in neurons from fmr1-/y mice compared with wild-type. IKA displayed mono-exponential, rapid recovery from inactivation for both wild-type and fmr1-/y dendrites, suggesting that IKA was most likely carried predominantly by KV4-containing channels. Despite smaller maximal IKA, the A-type K+ channels present in fmr1-/y dendrites were more responsive than those in wild-type neurons due to a hyperpolarization of the midpoint of activation. The more negative voltage range for activation for the remaining channels may account for the smaller difference in b-AP amplitude than might be expected for a 50% reduction in dendritic IKA. Nonetheless, the net effect of these changes in A-type K+ channel function was reduced attenuation of b-APs and greater b-AP-mediated calcium influx in fmr1-/y distal dendrites compared with wild-type.
Recently, there have been a few investigations into the pathophysiology of ion channels in FXS. In the auditory brainstem, FMRP interacts directly with and activates the sodium-activated K+ channel Slack-B (Brown et al., 2010), and the tonotopic gradient of KV3.1 in the medial nucleus of the trapezoid body is flattened in fmr1-/y knock-out mice (Strumbos et al., 2010). In a previous study, we provided the first physiological evidence that dendritic function is altered in a model of FXS. We demonstrated that CA1 pyramidal neuron dendrites have elevated Ih due in part to higher expression of the HCN1 subunit of h-channels in neurons from the fmr1-/y mouse compared with wild-type (Brager et al., 2012). In a more recent study, a loss of BK channel function in CA3 pyramidal neurons was thought to underlie increased neurotransmitter release in fmr1-/y mice (Deng et al., 2013).
The KV4.2-fmr1 controversy
Two recently published studies used biochemical and molecular approaches to examine changes in the expression of KV4.2, the putative pore-forming subunit of A-type K+ channels in hippocampal pyramidal neurons, in fmr1-/y and wild-type mice (Gross et al., 2011; Lee et al., 2011). These two studies came to opposite conclusions. Gross et al. (2011) found that there was less KV4.2 protein expressed in the hippocampus of fmr1-/y mice using immunohistochemistry and Western blotting. Furthermore, they demonstrated that there was reduced surface expression of KV4.2, suggesting that there were fewer A-type K+ channels on the neuronal membrane. In contrast, Lee et al. (2011) found that there was more KV4.2 protein and greater surface expression in fmr1-/y neurons. Our results are in agreement with those of Gross et al. However, there are several possible explanations for why our results differ from those of Lee et al.
First, we cannot exclude differences in mouse strains. In particular, it has been suggested that strain differences between 129 and C57BL/6 affect expression of the fmr1-/y phenotype (Paradee et al., 1999). It should be noted that Lee et al. used the 129 mouse, whereas our study and those of Gross et al. used the C57BL/6 mouse. Interestingly, much of the seminal research on the cellular neurobiology of FXS was performed on the C57BL/6 strain of fmr1-/y mice (Huber et al., 2002; Bear et al., 2004; Hou and Klann, 2004; Auerbach et al., 2011). Second, it is possible that hippocampal KV4.2 protein expression is actually greater in fmr1-/y mice, but that there are fewer functional channels expressed on the apical dendrite. For example, there are cases where immunohistochemistry and physiology do not always agree (Hoffman et al., 1997; Kerti et al., 2012). Last, KV4.2 protein and A-type K+ channels are located on individual dendritic spines on CA1 neurons (Kim et al., 2007). It is therefore possible that there is higher expression of KV4.2 at the spines of hippocampal pyramidal neurons and lower expression on the apical dendritic trunk in fmr1-/y mice. If the increase on spines is greater than the decrease on the trunk, then it is possible that by immunohistochemistry and Western blotting, one would observe greater expression of KV4.2 protein while observing less IKA recorded from the apical dendrite. In addition, if there is a greater expression of A-type K+ channels on individual spines, then this may explain the reduced LTP expression observed in fmr1-/y that was rescued by heteropodotoxin (Lee et al., 2011).
Potential mechanisms
How does the loss of FMRP in fmr1-/y mice result in a loss of A-type K+ channel function? The most parsimonious possibility is that, in wild-type mice, FMRP binds to KV4.2 mRNA and promotes translation. The absence of FMRP in fmr1-/y mice would result in a loss of KV4.2 mRNA translation and a subsequent reduction IKA. Alternatively, the loss of FMRP may alter the expression or function of a number of auxiliary A-type K+ channel subunits, including DPP6 and/or KChIP, which could result in lower IKA. The association of KV4 subunits with DPP6 and/or KChIP regulates both the surface expression and biophysical properties of dendritic A-type K+ channels (An et al., 2000; Covarrubias et al., 2008; Sun et al., 2011). We found no significant difference in the recovery from inactivation for IKA from wild-type and fmr1-/y neurons (Fig. 2C,D). The absence of either DPP6 or KChIP should produce a longer recovery from inactivation, suggesting that A-type K+ channels in fmr1-/y dendrites are still in a complex with DPP6 and KChIP. We cannot rule out, however, that an overall reduction in either of these two auxiliary proteins occurs and leads to lower dendritic IKA in fmr1-/y neurons. It is also possible that the loss of A-type K+ channel function is compensatory for other changes in neuronal activity. For example, inactivation of A-type K+ channels occurs in response to some LTP induction paradigms (Frick et al., 2004). It is therefore possible that changes in neuronal activity during development in FXS may lead to compensatory changes in A-type K+ channel function and/or expression.
Impact of reduced IKA on dendritic function
What are the physiological implications of reducing IKA in hippocampal dendrites? As we have demonstrated, there will be greater calcium influx in response to b-APs. b-APs play a crucial role in some forms of associative long-term synaptic plasticity. Large EPSPs (e.g., coincident synaptic events) can inactivate IKA and thereby lead to a temporally restricted window of increased dendritic excitability, during which the amplitude of a single b-AP may be boosted and the threshold for dendritic spike initiation lowered (Magee and Johnston, 1997; Watanabe et al., 2002). By setting up this temporal restriction, IKA can control the narrow time window required for some forms of long-term synaptic plasticity. We previously reported that a strong TBP protocol (five well timed action potentials per burst of EPSPs) produced the same amount of LTP in both wild-type and fmr1-/y neurons (Brager et al., 2012). Furthermore, a train of 10 theta-bursts of EPSPs (TBS, four EPSPs per burst) produces similar increases in field EPSP slope between wild-type and fmr1-/y hippocampal slices (Lauterborn et al., 2007). In this study, we found that a weak-TBP protocol (1–3 action potentials per burst, which were not well timed to the EPSPs) produced greater LTP in fmr1-/y neurons than in wild-type. This suggests that the threshold for TBP-LTP induction, which normally requires precise action potential to EPSP timing, is lower in fmr1-/y neurons, presumably due in part to the greater calcium influx per b-AP (Watanabe et al., 2002; Chen et al., 2006). Previous work demonstrated that a weak TBS (five bursts) produced less LTP of field EPSPs in fmr1-/y mice (Lauterborn et al., 2007; Lee et al., 2011). One major difference between our wTBP-LTP experiments and the weak TBS experiments of Lauterborn et al. and Lee et al. is the requirement of bAPs for TBP-LTP induction (Magee and Johnston, 1997). It is possible that, without bAPs, dendritic EPSPs alone produce insufficient calcium influx to promote LTP induction in fmr1-/y neurons. This hypothesis is consistent with our previous results showing that there is elevated Ih in fmr1-/y dendrites, which could reduce the temporal summation of dendritic EPSPs during the TBS induction train (Brager et al., 2012). The pairing of bAPs with EPSPs during the TBP protocol could overcome this reduced temporal summation and promote calcium influx. In fmr1-/y dendrites, the calcium influx is enhanced due to the lack of function A-type K+ channels, leading to a reduced threshold for LTP induction.
The normalization of calcium signals between the apical trunk and the smaller oblique dendrites occurs in part due to the presence of A-type K+ channels (Frick et al., 2003; Cai et al., 2004). It is possible that, in addition to the greater calcium influx into the distal apical dendrite, the loss of dendritic IKA in fmr1-/y mice will reduce b-AP attenuation into oblique dendrites, leading to greater calcium signals there as well. Because the majority of synaptic inputs are found on oblique dendrites, the elevated calcium levels here would have an even greater influence on synaptic plasticity mechanisms. Furthermore, A-type K+ channels themselves undergo activity-dependent plasticity in response to long-term potentiation—a downregulation of IKA due to a hyperpolarizing shift in inactivation (Frick et al., 2004) and internalization of channels from spines (Kim et al., 2007). It is not known whether IKA plasticity occurs in fmr1-/y mice. However, even if these forms of plasticity persist in fmr1-/y mice, the influence of this shift in A-type K+ channel function would be greatly reduced compared with wild-type neurons simply due to the fact that there is less IKA to begin with.
In addition to the attenuation of b-AP amplitude, the high density of IKA influences the initiation of local dendritic spikes and the magnitude of EPSPs in the distal dendrites (Kim et al., 2007; Losonczy et al., 2008). If our observation of less IKA on the apical dendrite extends to dendritic spines, then miniature EPSPs would be larger in fmr1-/y due to the absence of the inhibitory influence of A-type K+ channels (Kim and Hoffman, 2008). It should be noted that, in addition to these direct consequences of lower IKA, there could be additional compensatory changes (e.g., altered synaptic scaling; Andrásfalvy et al., 2008) that further alter dendritic signal processing in fmr1-/y dendrites.
In summary, we have shown how a loss of A-type K+ channel function in CA1 pyramidal neuron dendrites in the fmr1-/y hippocampus leads to an increase in dendritic b-AP amplitude and resulting calcium influx. We further show that these changes in IKA lower the threshold for TBP-LTP induction. Additional possible ramifications of this loss of A-type K+ channel function include dendritic hyperexcitability, altered intrinsic plasticity, and loss of dendritic compartmentalization. These results could provide mechanistic insight into the cellular mechanisms that may contribute to the cognitive impairments associated with FXS.
Footnotes
This work was supported by the FRAXA Research Foundation (D.H.B.) and the National Institutes of Health (Grant MH048432 to D.J.). We thank Drs. Raymond Chitwood and Richard Gray for help with Ca2+-imaging experiments and Drs. XiXi Chen, Nikolai Dembrow, Kelly Dougherty, and Brian Kalmbach for discussions and comments on the manuscript.
The authors declare no competing financial interests.
References
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Andrásfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knock-out mice. J Physiol. 2008;586:3881–3892. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480:63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60:201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bell MV, Hirst MC, Nakahori Y, MacKinnon RN, Roche A, Flint TJ, Jacobs PA, Tommerup N, Tranebjaerg L, Froster-Iskenius U. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell. 1991;64:861–866. doi: 10.1016/0092-8674(91)90514-Y. [DOI] [PubMed] [Google Scholar]
- Bernardet M, Crusio WE. Fmr1 KO mice as a possible model of autistic features. ScientificWorldJournal. 2006;6:1164–1176. doi: 10.1100/tsw.2006.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, Johnston D. Impaired dendritic expression and plasticity of h-channels in the fmr1(-/y) mouse model of fragile X syndrome. Cell Reports. 2012;1:225–233. doi: 10.1016/j.celrep.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Chen Y, Strumbos JG, Sigworth FJ, Navaratnam D, Kaczmarek LK. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat Neurosci. 2010;13:819–821. doi: 10.1038/nn.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Muralidharan S, Kao JP, Tang CM, Thompson SM. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Castellino RC, Morales MJ, Strauss HC, Rasmusson RL. Time- and voltage-dependent modulation of a Kv1.4 channel by a beta-subunit (Kv beta 3) cloned from ferret ventricle. Am J Physiol. 1995;269:H385–H391. doi: 10.1152/ajpheart.1995.269.1.H385. [DOI] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Bhattacharji A, De Santiago-Castillo JA, Dougherty K, Kaulin YA, Na-Phuket TR, Wang G. The neuronal Kv4 channel complex. Neurochem Res. 2008;33:1558–1567. doi: 10.1007/s11064-008-9650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Rotman Z, Blundon JA, Cho Y, Cui J, Cavalli V, Zakharenko SS, Klyachko VA. FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron. 2013;77:696–711. doi: 10.1016/j.neuron.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Magee J, Koester HJ, Migliore M, Johnston D. Normalization of Ca2+ signals by small oblique dendrites of CA1 pyramidal neurons. J Neurosci. 2003;23:3243–3250. doi: 10.1523/JNEUROSCI.23-08-03243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Losonczy A, Chen X, Johnston D, Magee JC. Associative pairing enhances action potential back-propagation in radial oblique branches of CA1 pyramidal neurons. J Physiol. 2007;580:787–800. doi: 10.1113/jphysiol.2006.121343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 2011;31:5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jin P, Warren ST. New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem Sci. 2003;28:152–158. doi: 10.1016/S0968-0004(03)00033-1. [DOI] [PubMed] [Google Scholar]
- Johnston D, Narayanan R. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 2008;31:309–316. doi: 10.1016/j.tins.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–674. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerti K, Lorincz A, Nusser Z. Unique somato-dendritic distribution pattern of Kv4.2 channels on hippocampal CA1 pyramidal cells. Eur J Neurosci. 2012;35:66–75. doi: 10.1111/j.1460-9568.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hoffman DA. Potassium channels: newly found players in synaptic plasticity. Neuroscientist. 2008;14:276–286. doi: 10.1177/1073858408315041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramár E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007;27:10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011;72:630–642. doi: 10.1016/j.neuron.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Kung F, Dell'Acqua ML, Hoffman DA. AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phospho-regulation of A-type K+ channel trafficking. J Neurosci. 2011;31:1323–1332. doi: 10.1523/JNEUROSCI.5383-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London M, Häusser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Makara JK, Magee JC. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature. 2008;452:436–441. doi: 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMR1 knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradee W, Melikian HE, Rasmussen DL, Kenneson A, Conn PJ, Warren ST. Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience. 1999;94:185–192. doi: 10.1016/S0306-4522(99)00285-7. [DOI] [PubMed] [Google Scholar]
- Ramakers GM, Storm JF. A postsynaptic transient K(+) current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:10144–10149. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serôdio P, Kentros C, Rudy B. Identification of molecular components of A-type channels activating at subthreshold potentials. J Neurophysiol. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Strumbos JG, Brown MR, Kronengold J, Polley DB, Kaczmarek LK. Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J Neurosci. 2010;30:10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Maffie JK, Lin L, Petralia RS, Rudy B, Hoffman DA. DPP6 Establishes the A-type K+ current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron. 2011;71:1102–1115. doi: 10.1016/j.neuron.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Hauben E, Reyniers E, Gantois I, Bakker CE, Oostra BA, Kooy RF, De Deyn PP. Spatial learning, contextual fear conditioning and conditioned emotional response in Fmr1 knockout mice. Behav Brain Res. 2000;117:127–136. doi: 10.1016/S0166-4328(00)00296-5. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]