

Abstract
MECP2 duplication syndrome is a childhood neurological disorder characterized by intellectual disability, autism, motor abnormalities, and epilepsy. The disorder is caused by duplications spanning the gene encoding methyl-CpG-binding protein-2 (MeCP2), a protein involved in the modulation of chromatin and gene expression. MeCP2 is thought to play a role in maintaining the structural integrity of neuronal circuits. Loss of MeCP2 function causes Rett syndrome and results in abnormal dendritic spine morphology and decreased pyramidal dendritic arbor complexity and spine density. The consequences of MeCP2 overexpression on dendritic pathophysiology remain unclear. We used in vivo two-photon microscopy to characterize layer 5 pyramidal neuron spine turnover and dendritic arborization as a function of age in transgenic mice expressing the human MECP2 gene at twice the normal levels of MeCP2 (Tg1; Collins et al., 2004). We found that spine density in terminal dendritic branches is initially higher in young Tg1 mice but falls below control levels after postnatal week 12, approximately correlating with the onset of behavioral symptoms. Spontaneous spine turnover rates remain high in older Tg1 animals compared with controls, reflecting the persistence of an immature state. Both spine gain and loss rates are higher, with a net bias in favor of spine elimination. Apical dendritic arbors in both simple- and complex-tufted layer 5 Tg1 pyramidal neurons have more branches of higher order, indicating that MeCP2 overexpression induces dendritic overgrowth. P70S6K was hyperphosphorylated in Tg1 somatosensory cortex, suggesting that elevated mTOR signaling may underlie the observed increase in spine turnover and dendritic growth.
Introduction
MECP2 duplication syndrome is a severe childhood neurological disorder characterized by autism, intellectual disability, and motor dysfunction (Ramocki et al., 2010), which is caused by increased levels of methyl-CpG-binding protein-2 (MeCP2), a protein involved in modulating chromatin and gene expression. Mice engineered to express MECP2 at twice the normal level via transgenic insertion of the human MECP2 gene (Tg1) develop a progressive neurologic phenotype with repetitive stereotypes, abnormal social behavior and learning, anxiety, spasticity, and seizures (Collins et al., 2004; Luikenhuis et al., 2004; Jugloff et al., 2008; Na et al., 2012; Samaco et al., 2012) providing an important model for the human disorder.
MeCP2 loss of function mutations cause Rett syndrome as well as some cases of autism and other neuropsychiatric disorders (Chahrour and Zoghbi, 2007). Defects in dendritic structure, synaptic plasticity, and synaptic homeostasis are thought to represent the structural basis of circuit malfunction in Rett syndrome (Ramocki and Zoghbi, 2008). Rett pyramidal neurons have simplified dendritic arbors, decreased dendritic spine densities, smaller spine heads, longer spine necks, and decreased spine motility (Kishi and Macklis, 2004; Armstrong, 2005; Chao et al., 2007; Landi et al., 2011; Stuss et al., 2012). Evidence is accumulating that dendritic and synaptic defects also represent an integral manifestation of the MECP2 duplication syndrome, but the natural history of these abnormalities remains poorly characterized. On one hand, mouse neurons overexpressing MeCP2 have increased density of excitatory synapses (Chao et al., 2007) and increased excitatory drive (Na et al., 2012) compared with controls. On the other, overexpressing MECP2 in rat hippocampal slices decreased spine density (Chapleau et al., 2009) and induced lengthening and thinning of spines (Zhou et al., 2006). Similarly, in vitro transient MECP2 overexpression led to increased (Jugloff et al., 2005) or decreased (Zhou et al., 2006; Chapleau et al., 2009) pyramidal dendritic arbor complexity, while in vivo overexpression of MeCP2 in Xenopus tectum (Marshak et al., 2012) or Drosophila motor neurons (Vonhoff et al., 2012) resulted in simplified dendritic arbors.
The Tg1 mouse model recapitulates many features of the human disorder. Therefore, we undertook the in vivo characterization of layer 5 (L5) pyramidal cell morphology and spine turnover in the barrel cortex of Tg1 mice crossed to the thy1-GFP-M line (Feng et al., 2000) to sparsely label L5 pyramidal neurons. We focused on somatosensory cortex because it is implicated in the pathophysiology of autism and MeCP2 disorders (Yoshikawa et al., 1991; Yamanouchi et al., 1993; Glaze, 2005; Pan et al., 2010; Paluszkiewicz et al., 2011; Zhang et al., 2011; Cascio et al., 2012).
Strikingly, we find that dendritic spine density in terminal branches of L5 pyramidal neurons in Tg1 barrel cortex starts initially higher than in littermate controls, but then falls below normal at approximately the same age that neurobehavioral dysfunction emerges. Dendritic spine turnover stays abnormally high well into adulthood in Tg1 animals. Spine gain and loss rates are both higher, with loss slightly outpacing gain. Apical dendritic arbor complexity is increased in Tg1 L5 pyramidal neurons. Higher levels of phosphorylated p70S6K were found in the somatosensory cortex of Tg1 mice suggesting that mTOR signaling is hyperactive. This may provide a putative mechanism for excessive synaptic plasticity and dendritic overgrowth.
Materials and Methods
All experiments were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Mice.
MECP2-duplication (Tg1) mice on FVB background from Dr. Huda Zoghbi's laboratory (Collins et al., 2004) were crossed to C57BL/6 thy1-GFP-M mice obtained from The Jackson Laboratory. F1 generation mice have layer 5 pyramidal neurons sparsely labeled with green fluorescent protein (GFP). A total of 58 male F1 MeCP2-Tg1; thy1-GFP-M mice and 62 male thy1-GFP-M littermate controls (WT) were used in the experiments.
Surgery.
Chronic window surgeries were performed as previously described in (Holtmaat et al., 2009). Briefly, mice were anesthetized deeply with a 1–2% isoflurane-oxygen mixture, and 2% lidocaine was injected subcutaneously under the scalp. Dexamethasone (1.5 μg/g) and carprofen (3.3 μg/g) were administered subcutaneously (Holtmaat et al., 2009). The scalp hair was trimmed, the skin was disinfected with alternating scrubs of povidone-iodine and 70% ethanol solutions, and a central incision was performed. The exposed skull was scraped gently to remove the periosteum and dried. A thin layer of cyanoacrylate-based glue was then applied to the exposed skull surface. A custom titanium washer-headpost (6 mm inner diameter) was cemented to the skull with dental acrylic to immobilize the mouse's head during imaging (Fig. 1A), with the washer's opening centered on the somatosensory barrel cortex (1 mm posterior and 3 mm lateral to the bregma). A 3 mm diameter craniotomy was drilled at slow speeds carefully using an electric drill (Foredom Electric Company; drill bit 100–5860 from Henry Schein) over the right barrel cortex (Fig. 1B). Special care was taken to minimize injury to the dura during drilling and removal of skull fragments. Sterile saline and gel foam were used to clean debris and blood from the dura as well as to keep it moist at all times. A 3 mm sterile glass coverslip was placed into the craniotomy, and the window was sealed carefully with a small amount of cyanoacrylate glue between the edge of the coverslip and the skull. Finally, dental acrylic was applied throughout the skull surface anchoring the window and sealing it from pathogens. The animal was monitored on a heated water blanket as it recovered from anesthesia.
Figure 1.
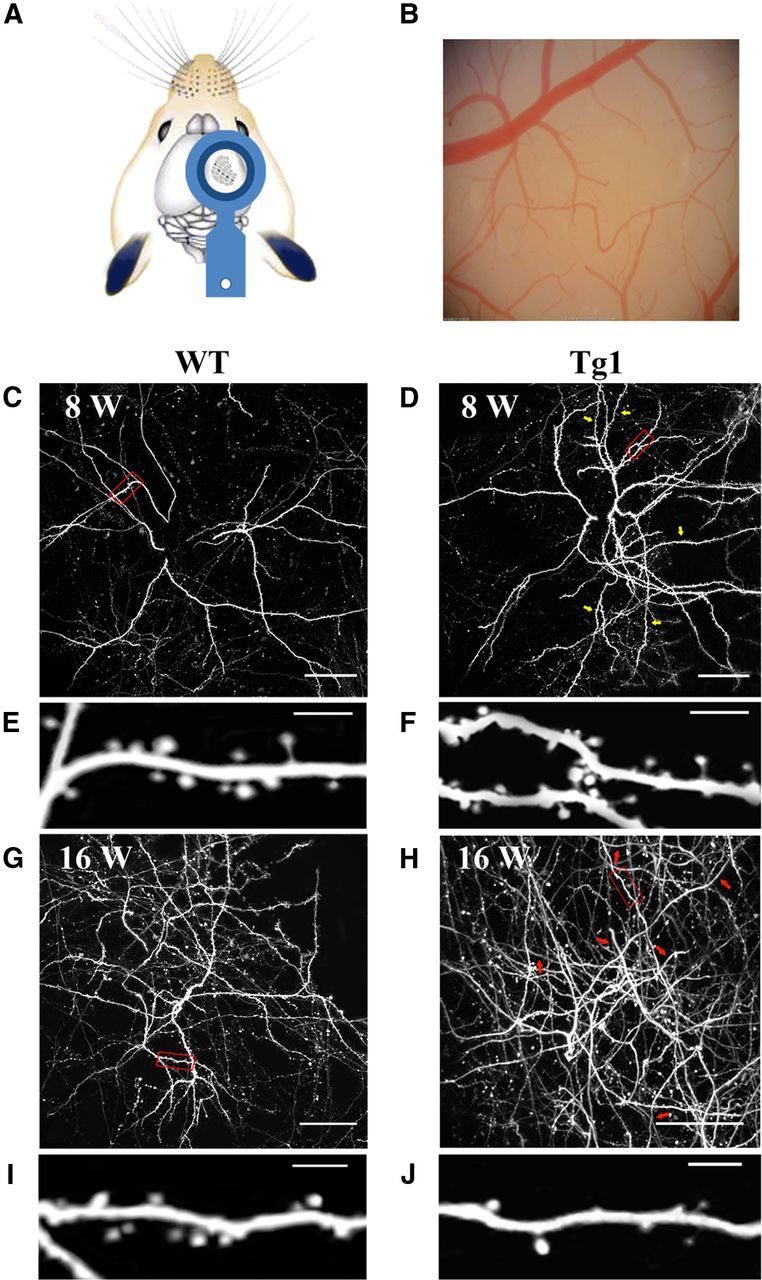
Experimental set up. A, Custom-designed washer-headpost placement above the somatosensory (barrel) cortex. B, Bright-field image of a mouse's chronic window. C,D,G,H, Low-magnification images of apical dendritic fields of L5 pyramidal neurons. Each image is a z-stack projection of 110–150 1-μm-thick two-photon slices taken from the top cortical layers. C, Projection taken from an 8-week-old WT mouse. Dendritic branches and spine density appear evenly distributed. Red outline shows an area seen in high magnification in E. D, Projection taken from an 8-week-old Tg1 mouse. Dendritic branches and spine density appear increased compared with the WT mouse seen in C. Yellow arrows indicate branches of relatively high spine density. Red outline shows an area seen in high magnification in F. G, Projection taken from a 16-week-old WT mouse. Red outline shows an area seen in high magnification in I. H, Projection taken from a 16-week-old Tg1 mouse, with high magnification shown in J. More dendritic branches are visible in the FOV, though spine density appears decreased in several branches compared with the WT mouse seen in G and to the young Tg1 mouse seen in D. Red arrows indicate dendritic branches with relative spine loss. Scale bars: C, D, G, H, 50 μm; E, F, I, J, 5 μm.
In vivo two-photon imaging
Acute.
Following window placement mice were head-fixed to a custom stage, and immobilized under a custom-built two-photon microscope (Prairie Technologies), while anesthesia was maintained with a mixture of ∼1% isoflurane and oxygen. Optical images of the craniotomy were captured at 4× magnification to identify vascular landmarks (Fig. 1B). The dura and superficial neuropil were next visualized by two-photon microscopy using a 20× objective lens (Olympus, 0.95 NA water immersion), excitation provided by a Chameleon titanium:sapphire laser tuned to 890 nm equipped with two Hamamatsu photomultiplier tubes. Prairie View 4.1.1.4 software was used for microscope control and image capture (Prairie Technologies). Nine partially overlapping (620 × 620 μm FOV) stacks, placed 500 μm from each other, tiling a 1500 × 1500 μm field centered in the middle of the window were captured as a rough map to locate GFP-expressing neurons (Fig. 1C,D,G,H). For reconstruction of apical dendritic arbors and measurement of terminal dendrite spine density, two or three GFP-expressing pyramidal neurons per craniotomy were recorded as a stack of images (2 μm section thickness) from the cell body to the dura using a long working distance 40× objective lens (Olympus, 0.8 NA water immersion; Fig. 1E,F,I,J). Fluorescent cell bodies were usually found at ∼600 μm cortical depth consistent with the L5 origin of the vast majority of GFP-positive neurons in the thy1-GFP-M line (Feng et al., 2000). The nominal resolution for each optical section (1024 × 1024 or 2048 × 2048 pixels) was 0.16 or 0.32 μm/pixel. The actual resolution of the 40× objective, as measured by imaging of pollen grains, was 0.32 μm/pixel in the X-Y plane and 2 μm/pixel in the z-axis.
For the time-lapse imaging of spine turnover, stacks were acquired every 10 min over 60 min sessions (1 μm section thickness, 1024 × 1024 pixels or 2048×2048, 0.16 or 0.32 μm/pixel). Apical dendritic branches used for spine analysis were generally imaged between the pia and 100 μm cortical depth. The laser power measured at the sample was strictly limited to below 40 mW (∼20 mW average) at all times and was adjusted to achieve near identical nonsaturating, fluorescence intensities during each imaging session. Tg1 and littermate controls (WT) were imaged in cohorts at 8, 12, 16, 20 and 40 weeks of age, 3–5 mice of each genotype at each time point. Importantly, imaging parameters, corticosteroid administration and anesthesia levels were identically applied for animals in all ages and genotypes.
Chronic.
In addition, chronic time-lapse imaging was performed in five Tg1 mice and five littermate controls at 8, 12, and 16 weeks of age to track changes in protrusion density over time in the same dendritic arbors. The acquisition parameters were as follows: 2048 × 2048 pixels, 60× 1.0 NA Olympus water-immersion lens, 225 × 225 μm FOV, 1-μm-thick optical sections, with an actual resolution of 0.33 μm in the X-Y plane (nominal resolution 0.11 μm/pixel), and 1.8 μm in the z-axis, as measured by imaging of pollen grains. At each time point, arbors were imaged twice, 1 h apart. In all measurements of spine density and spine turnover we limited our analyses to the terminal branches of apical dendritic arbors. Spine densities were expressed in number per micron (that is, spine number in terminal branch/terminal branch length in μm). In some figures (Fig. 1E,F,I,J, Fig. 3A, Fig. 4A,B, Fig. 6A) nonspecific background fluorescence from axons and other processes was masked in Adobe Photoshop for display purposes only.
Figure 3.
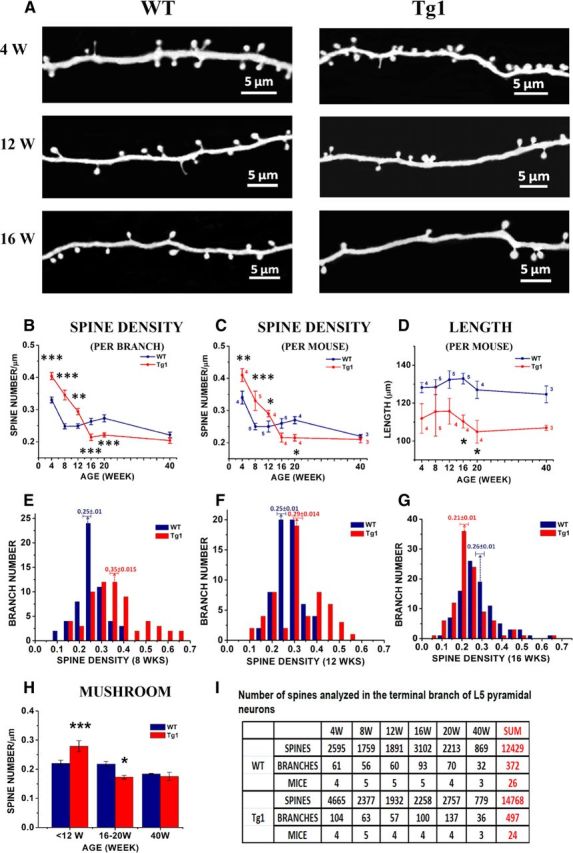
Spine density in terminal branches of Tg1 and WT mice as a function of age. A, High-magnification views of GFP-labeled terminal apical dendrites of L5 somatosensory cortical neurons by two-photon microscopy in vivo from Tg1 mice (right) and littermate controls (WT; left). Representative example images were taken from 4-, 12-, and 16-week-old animals. B, Apical terminal dendrite spine densities at different ages in Tg1 (red) and WT (dark blue) mice, statistical significance computed per analyzed dendritic branch. The spine density time course was significantly different in Tg1 mice (p < 0.001, two-way ANOVA interaction). C, Apical terminal dendrite spine densities at different ages in Tg1 (red) and WT (dark blue) mice, statistical significance computed per animal (number of animals appears next to each time point). D, The average length of terminal dendritic branches measured in 3D, analyzed per mouse (number of animals appears next to each time point). E–G, Histograms displaying the distribution of spine densities (number of spines per micron) across terminal dendritic branches counted for Tg1 mice (red bars) and littermate controls (dark blue bars). E, Histogram of spine densities seen in terminal branches in postnatal week 8. Note the wide distribution of spine densities in Tg1 animals, with some Tg1 branches having more than twice the median density seen in littermate controls. F, Histogram of spine densities seen in terminal branches in postnatal week 12. The distributions still do look different but they appear to converge. G, Histogram of spine densities seen in terminal branches in postnatal week 16, demonstrating a shift toward decreased spine densities in Tg1 animals. H, Densities of mushroom spines in presymptomatic (<12 weeks) versus symptomatic (16–20 and 40 weeks) Tg1 mice, analyzed per mouse. A majority of spines were mushroom like in morphology at all time points in both genotypes. I, Table of spines, dendrites, and mice analyzed per time point in WT and Tg1 mice. ***p < 0.001, **p < 0.01, and *p < 0.05, two-way ANOVA followed by Tukey's multiple-comparison test.
Figure 4.

Time-lapse observations of spine density and dendrite length demonstrate the time course of structural abnormalities in individual dendrites of Tg1 mice. A, B, Example terminal dendrites followed from 8 to 16 weeks of age in Tg1 (B) and WT (A). Red arrows indicate spines lost. Yellow arrows indicate spines gained. C, Spine densities of serially imaged dendrites at 8, 12, and 16 weeks. D, Length of serially imaged dendrites over time. *p < 0.05, **p < 0.01, ***p < 0.001, n = 38 Tg1 branches from five animals, 44 WT branches from five animals, two-way ANOVA followed by Tukey's multiple-comparison test.
Figure 6.
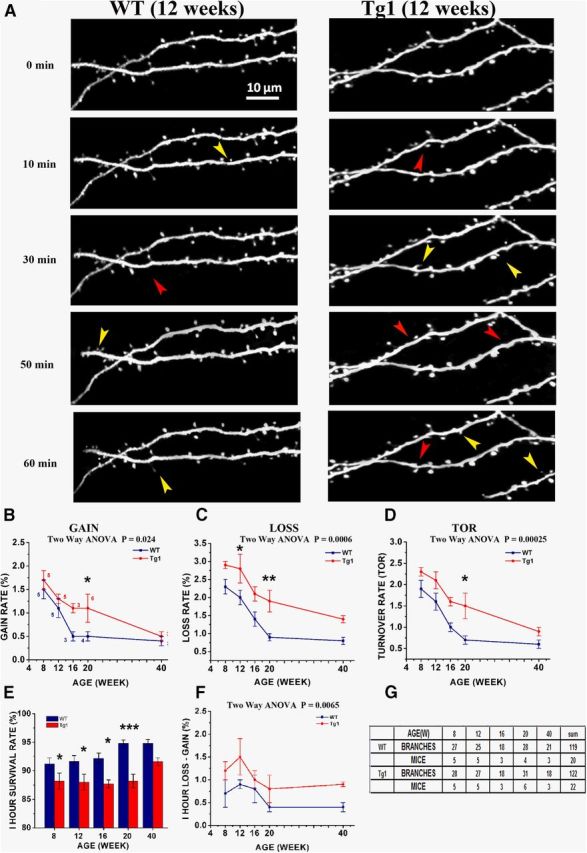
The developmental decrease in spine turnover rate is delayed in Tg1 mice. A, High-magnification time-lapse images of terminal dendritic branches in Tg1 (right) and WT (left). Images were captured every 10 min over 1 h. Only a portion of the analyzed dendrite is displayed at select time points. Yellow arrows indicate spines gained; red arrows spines lost. Scale bar, 10 μm. B–D, Spine gain (B), loss (C), and turnover (D) expressed as percentage of total spines gained/lost/gained-or-lost per mouse over a 10 min interval as a function of age for Tg1 mice (red) and littermate controls (dark blue). Number of animals used to calculate each value is indicated in B. Overall two-way ANOVA significance values are reported above each graph. Significance at individual time points indicated by asterisks. Note that spine gain and loss remain elevated in adult Tg1 animals compared with controls. E, Spine survival calculated as the percentage of spines that survive over 1 h as a function of age in Tg1 mice (red) and littermate controls (dark blue). Note that at all ages spine survival is lower for Tg1 animals, though the gap seems to decrease at older ages (week 40). F, Spine loss minus spine gain (net spine loss) over 10 min as a function of age, averaged per mouse. Note that net spine loss is greater at all ages for Tg1 animals. G, Table providing number of spines, dendrites, and animals analyzed at each time point. *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA followed by Tukey's multiple-comparison test.
Dendritic protrusion classification, counting, and turnover analysis
Spine density and spine turnover in L5 pyramidal neuron apical dendrites from Tg1 mice and littermate controls were analyzed using Neurolucida software (MicroBrightField). Dendritic trees were traced using Neurolucida on a 3D stack of images, and skeletonized. Terminal dendritic branches were identified and selected for protrusion analysis. Dendritic protrusions were carefully identified in individual slices, and their presence or absence was tracked across each time point by scanning through the stacks slice by slice. Only protrusions clearly originating from the analyzed dendrite were tracked. Due to in vivo two-photon microscopy's relatively poor resolving power in the z-axis, only structures protruding laterally along the X-Y plane were included in the analysis, following the standard in this field (Holtmaat et al., 2009). For a protrusion to be selected for analysis it had to project out of the dendritic shaft by at least 4 pixels (>0.64 μm), which corresponds approximately to 2 SDs of the noise blur on either side of the dendritic shaft.
The spine classification system of mushroom, stubby, and thin spines was applied to each protrusion (Peters and Kaiserman-Abramof, 1970; Harris et al., 1992). A protrusion was classified as a stubby spine if it had no discernible neck (diameter of the neck ≈ length of the spine). Of the remaining necked protrusions, if the neck length was shorter than the head diameter, the protrusion was classified as a mushroom spine. Of protrusions whose neck length was longer than the head diameter, mushroom spines had a head diameter >2 times the neck diameter, and thin spines had a head diameter <2 times the neck diameter. Although occasionally long (>4 μm) filopodia-like protrusions with no head were found in young mice (4 weeks old); they were rarely observed at other time points.
To calculate spine formation and elimination rates, spines were first marked along the terminal dendrite branch tracing in the image stack obtained at the initial time point. The tracing file with the marked spines was then superimposed over the second image stack (10 min time point). Dendrites in the new stack were registered with the marked tracing. New spines formed and old spines eliminated were recorded and marked. A cross-check was then made with the prior stack to ensure the fidelity of the process. This procedure was performed recursively on each 10 min time point up to the last imaging time point (60 min). The rate of the fraction of spines gained and lost (Fgain and Flost) were calculated, respectively, based on the spines that appeared and disappeared between two successive images (i.e., over a 10 min interval), relative to the total spine number in the initial stack at time 0. Loss/gain rates were calculated as the average rate across all recorded 10 min intervals at each postnatal age. Turnover rate (TOR) was calculated as the sum of protrusions lost and gained divided by twice the total protrusion number.
Dendritic arbor analysis
To analyze the effect of MECP2 duplication on dendritic arbor complexity, the apical dendrites of 25 adult pyramidal neurons (13 from WT, 12 from Tg1) were traced in Neurolucida starting at the cell body in L5 to the apical tufts on stacks of 2-μm-thick optical sections (345 × 345 μm FOV, 40× Olympus lens, 0.8 NA). Total branch length, total branch number, and max branch order were calculated, and 3D Sholl analysis (Sholl, 1953) and dendrogram analysis were performed (Uylings and van Pelt, 2002). For the Sholl analysis, a series of concentric 3D spheres were drawn centered at the soma with radius increasing at 50 μm intervals, and the number of dendritic intersections with each sphere plotted. Apical dendritic arbors were clustered into simple-tufted and complex-tufted morphologies as described previously (Holtmaat et al., 2006) using k-means cluster analysis along four parameters: (1) total dendrite length, (2) the total number of branch points, (3) the max branch order, and (4) the cortical depth of the first major apical bifurcation.
Spine morphology analysis
Detailed spine morphology analysis was performed in 8- and 16-week-old Tg1 mice and littermate controls. High-resolution stacks of terminal apical dendritic branches (2048 × 2048 pixels, 60× lens, 225 × 225 μm FOV, 0.25- to 1 μm-thick optical sections) were processed by a custom polynomial interpolation method, which performs a weighted average of the intensities of neighboring pixels according to the equations:
 |
and
 |
where, Ic represents the calculated (interpolated) intensity, and Iexp the actual intensity measured in the experiment. The interpolation is calculated for each pixel by the least-squares method using ∼100 pixels within an ∼1.16 × 1.16 μm2 field. As tissue fluorescence is highly correlated locally, while shot noise is not, this polynomial interpolation significantly improves the visibility of small dendritic structures, particularly the spine neck (Fig. 5A). GFP-labeled terminal dendrites in each FOV were randomly selected and the spine head volume, neck length, and neck diameter of each laterally projecting spine were quantified in ImageJ at the best plane of view (Konur et al., 2003; Arellano et al., 2007; Cruz-Martín et al., 2010). Spine head volume was calculated as the sum of the intensities of all the pixels in a manual tracing around the head, normalized by the mean nearby dendritic shaft intensity. Neck length was calculated as the distance along the spine neck from the base of the spine head to the point where the neck joined the dendritic shaft. Neck width was estimated as the maximum intensity at the center of the neck normalized by the mean nearby dendritic shaft intensity.
Figure 5.

Subtle changes of spine morphology in Tg1 mice. A, High-resolution stacks of apical dendrites from Tg1 mice and littermate controls were processed with a polynomial interpolation algorithm to reduce noise (see Materials and Methods), which enhanced the visibility of small dendritic spine components. B, Illustration of the methods used to calculate spine head volume, neck width, and neck length. C–E, Assessments of spine head volume (C), neck width (D), and neck length (E). Left and middle, histograms of measured values at 8 and 16 weeks for Tg1 (red) and WT (black). Right, cumulative distributions of all conditions. Significant differences determined by Mann–Whitney U test are shown in the insets, *p < 0.05; 8 week Tg1, n = 351 spines, two animals; 8 week WT, n = 281 spines, two animals; 16 week Tg1, n = 205 spines, two animals, 16 week WT, n = 153 spines, two animals.
Transcardial perfusion and 4′, 6-diamidino-2-phenylindole staining.
To investigate whether the cell density in different layers of cortex was altered in Tg1 mice compared with WT, three Tg1;thy1-GFP mice and four thy1-GFP controls were transcardially perfused with saline followed by 4% paraformaldehyde (PFA)/5% sucrose. Brains were removed and immersed in 4% PFA/5% sucrose at 4°C overnight. Brains were transferred into 30% sucrose in PBS and incubated another night at 4°C. The 35-μm-thick coronal brain slices were then prepared using a freezing sliding Microtome (Leica, SM2000R). The sections were stained with 4′, 6-diamidino-2-phenylindole (DAPI) and mounted on slides in aqueous medium. Slices containing somatosensory cortex were imaged using an epifluorescence microscope (Zeiss, Axio Imager Z1). Three serial somatosensory cortex slices were analyzed per brain. Bilateral 100 μm wide rectangular boxes were traced in Neurolucida to lie perpendicular to the dura and span all cortical layers centered on a barrel (identified by cell density in L4). Cortical layers were distinguished based on cell size and density. Within the box, the number of DAPI-labeled cells were counted in each layer (Fig. 2A,B). In addition, GFP-positive L5 cell bodies were counted in three consecutive sections of barrel cortex in WT and Tg1 mice (Fig. 2C,D).
Figure 2.
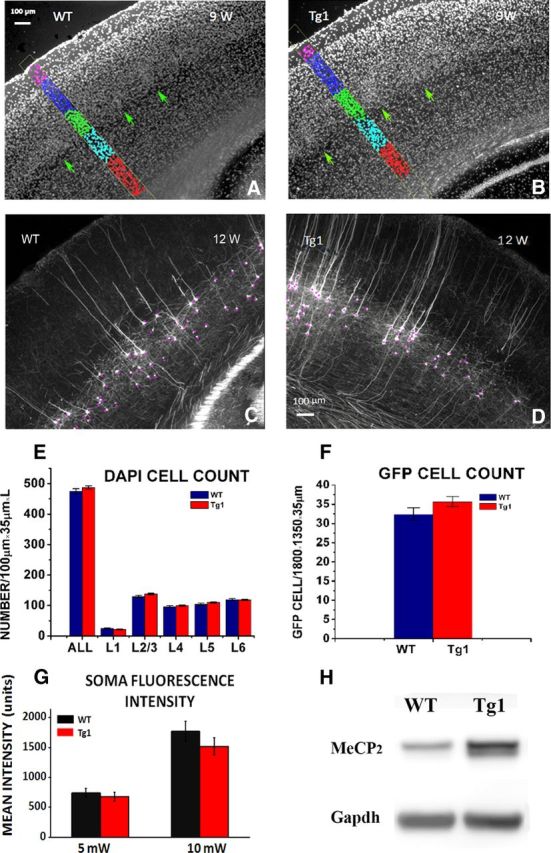
Cell number and GFP protein quantification in barrel cortex. A, B, Demonstration of cell body quantification. Thirty-five micrometer thick fixed barrel cortex sections were stained with DAPI, and cell bodies were counted in each layer within a 100 μm wide box, from three consecutive sections in 9-week-old WT (A) and Tg1 (B) mice. Barrels are indicated by green arrows. C, D, Demonstration of quantification of GFP-labeled cells. GFP-labeled cells were counted in three consecutive sections in layer 5 in WT (C) and Tg1 (D) animals. Scale bar, 100 μm. E, DAPI-labeled cell density per cortical layer in Tg1 (red) and WT (dark blue) animals. No significant difference was noted in overall cell counts at any layer. F, GFP-labeled cell density in layer 5 in Tg1 (red) and WT (dark blue). Note that there is no significant difference in GFP-labeled cell counts suggesting that there is no interaction between probability of GFP expression and Tg1 genotype. G, Average somatic fluorescence intensity at two laser power settings (5 and 10 mW) illustrating that there is no significant difference in mean somatic fluorescence and therefore in mean somatic GFP concentration in L5 neurons of Tg1 animals and controls. H, Western blots of MeCP2 protein extracted from the barrel cortex of Tg1 mice and littermate controls. Note that the MeCP2 protein levels are approximately double in Tg1 animals as expected. Statistical comparisons were made using two-way ANOVA, followed by the Tukey's multiple-comparison test.
Western blotting.
Tg1 mice and littermate controls aged 16, 20, and 40 weeks were killed after anesthesia by isoflurane. Brains were dissected rapidly on a glass plate over ice. In one hemisphere, the somatosensory (barrel) cortex was dissected away from the remaining cortical tissue and these two portions were lysed separately in cold extraction buffer (2% SDS and 50 mm Tris-Cl, pH 7.4) by Dounce homogenization. Insoluble material was pelleted at maximal speed in a table-top centrifuge for 10 min and the supernatant was mixed with Laemmli Buffer. Equal amounts of protein were loaded on to a NuPAGE 4–12% Bis-Tris gel (Invitrogen) and post electrophoresis transferred onto PVDF membranes. For measurement of MeCP2 level, membranes were blocked and incubated with the following primary antibodies: rabbit anti-MeCP2 (homemade; 1:1000) and mouse anti-GAPDH (Advanced ImmunoChemical 6C5; 1:20,000). For assessment of p70S6K phosphorylation, membranes were first probed with rabbit anti-phospho-S6K (#9234; 1:1000) and rabbit anti-H3 (Millipore 06–755; 1:10,000). Blots were then incubated with horseradish peroxidase-conjugated secondary antibody (GE Healthcare) followed by incubation with SuperSignal West Dura Chemiluminescence Substrate (Thermo Scientific) per the manufacturer's instructions. Blots were visualized with an Image Quant LAS 4000. Images were processed and analyzed with ImageJ software. P-S6K blots were then stripped by incubation in Restore Western blot stripping buffer (Thermo Scientific) for 10 min, reprobed with Rabbit S6K (Cell Signaling Technology #9202; 1:1000), and visualized as above. The effectiveness of the stripping protocol was assessed by loss of the loading control (H3) band in the second image acquisition.
Statistics.
Results are presented as mean ± SEM, normalized per terminal dendrite. All statistical analyses were performed with SigmaStat. To determine statistical significance one-way ANOVA or two-ANOVA followed by the Tukey's or Holm–Sidak multiple-comparison tests, Student's t test, and Mann–Whitney U test were used in data analysis. Graphs were designed with Origin, Excel, and MATLAB. Subjects were blinded as to the genotype when performing spine turnover analysis.
Results
Our goal was to study, in vivo, the structural dendritic abnormalities seen in the Tg1 mouse model of MECP2 duplication syndrome to characterize in vivo the synaptic plasticity pathophysiology of this neurodevelopmental disorder. To visualize pyramidal neuron dendritic arbors for analysis, we crossed the Tg1 mouse line to the thy1-GFP-M line (Feng et al., 2000) to brightly label sparse populations of L5 pyramidal neurons with GFP for in vivo imaging. We imaged the apical dendrites of L5 pyramidal neurons in the barrel cortex of male F1 Tg1;thy1-GFP mice and thy1-GFP littermate controls (WT) over 4–40 weeks of age (Fig. 1C–J), spanning the course of the mouse's progressive neurological phenotype from asymptomatic to symptomatic.
Cell densities, GFP expression, and single-cell fluorescence in Tg1 mouse barrel cortex
MeCP2 is known to regulate the expression of hundreds or even thousands of genes (Chahrour et al., 2008; Skene et al., 2010). If MECP2 overexpression significantly altered the expression of the thy1-GFP fluorescent reporter gene, it could potentially alter the visibility of dendritic spines and bias our results. A previous study of thy1-YFP-labeled pyramidal neurons in Rett mice, interestingly, noticed a decrease in the density of fluorescent cells in their mice without a change in single-cell fluorescence (Stuss et al., 2012). To test whether a similar process could occur with MeCP2 gain-of-function, we first confirmed that the relative cell densities across layers were not significantly different in Tg1 compared with controls (Fig. 2A,B,E), and the number of GFP-expressing cells in L5 of barrel cortex was not altered in Tg1 (Fig. 2C,D,F). We next tested whether GFP fluorescence per cell body was different in Tg1, by imaging L5 pyramidal neuron somata at 5 and 10 mW laser powers by two-photon imaging in neocortical slices. Although the fluorescence concentrations varied from cell soma to cell soma, in line with thy1-GFP's variable expression profile, the fluorescence distribution across cells was essentially identical for the two genotypes (Fig. 2G). These findings, in conjunction with our protocol for standardizing imaging conditions, nullifies the risk that our results could be the result of differential levels of fluorescence expressed in the two genotypes. We also confirmed by immunoblot that MeCP2 was overexpressed in experimental Tg1 animals (Fig. 2H; n = 3 per genotype).
Spine density profiles in terminal branches of layer 5 pyramidal neurons are abnormally increased in juvenile and decreased in adult Tg1 mouse barrel cortex
We first measured the density of dendritic protrusions (spines, see Materials and Methods) in terminal apical dendritic branches of L5 pyramidal neurons at different ages (Fig. 3). Spine density decreases with age, as has been reported previously in normal animals (Holtmaat et al., 2005; Zuo et al., 2005b), but the profile of spine density as a function of age differs between Tg1 and controls (Fig. 3B). At 4, 8, and 12 weeks of age terminal dendritic branches of Tg1 mice have significantly higher spine density compared with littermate controls (Tg1: 0.4 ± 0.011/μm, WT: 0.33 ± 0.009/μm, p < 0.001 at 4 weeks; Tg1: 0.35 ± 0.015/μm, WT: 0.250 ± 0.008/μm, p < 0.001 at 8 weeks; Tg1: 0.29 ± 0.005/μm, WT: 0.25 ± 0.007/μm, p < 0.01 at 12 weeks, two-way ANOVA followed by Tukey's test; Fig. 3B). While spine density was relatively stable in WT past 8 weeks, Tg1 spine densities continued to fall between 12 and 16 weeks. From 16 to 20 weeks, Tg1 spine density plateaued at a lower level than WT (Tg1: 0.21 ± 0.01/μm, WT: 0.26 ± 0.01/μm, p < 0.001 at 16 weeks; Tg1: 0.22 ± 0.006/μm, WT: 0.26 ± 0.01/μm, p < 0.01 at 20 weeks; two-way ANOVA followed by Tukey's test; Fig. 3B). After 20 weeks, spine densities in WT mice appear to decrease gradually such that by 40 weeks (the last time point examined) they are commensurate with those of Tg1 mutant mice (Tg1: 0.20 ± 0.01/μm, WT: 0.22 ± 0.01/μm, p > 0.05, two-way ANOVA followed by Tukey's test). The time course of change in spine density was significantly different in Tg1 mice (p < 0.001, two-way ANOVA genotype × age interaction). To control for interanimal variability we also analyzed the data per animal after pooling dendrites from each animal and found that the significance of these results was maintained (Tg1: 0.41 ± 0.02/μm, WT: 0.34 ± 0.02/μm, p <0.05 at 4 weeks; Tg1: 0.33 ± 0.03/μm, WT: 0.250 ± 0.007/μm, p <0.05 at 8 weeks; Tg1: 0.29 ± 0.009/μm, WT: 0.25 ± 0.02/μm, p <0.05 at 12 weeks; Tg1: 0.22 ± 0.01/μm, WT: 0.26 ± 0.01/μm, p = 0.08 at 16 weeks; Tg1: 0.22 ± 0.01/μm, WT: 0.27 ± 0.02/μm, p < 0.05 at 20 weeks; ANOVA interaction: p < 0.001; n = 4–5 animals per sample, two-way ANOVA followed by Tukey's test; Fig. 3C). Note that the observed difference in spine densities cannot be due to the fact that Tg1 terminal branches are on average of higher branching order than terminal branches in WT animals, because spine density did not depend significantly on the branching order of analyzed terminal dendritic branches in either Tg1 or WT animals (data not shown). Interestingly, the age at which Tg1 mice first become symptomatic, i.e., 12–16 weeks of age (Collins et al., 2004), approximately coincides with the time that dendritic spine densities fall below normal.
To better illustrate how spine densities vary across the population of dendritic branches as a function of age for each genotype, we plotted histograms of spine densities per terminal dendritic branch in presymptomatic (8 weeks; Fig. 3E), mildly symptomatic (12 weeks; Fig. 3F), and symptomatic (16 weeks; Fig. 3G) Tg1 animals (red) compared with controls (black). Note that at 8 weeks of age, the distribution of spine densities in Tg1 terminal dendritic branches is skewed toward high densities and has both a higher mean and a higher variance than that of controls, with a subset of Tg1 dendrites having spine densities twice as high as controls (Fig. 3E). Over time, the spine density distribution of Tg1 terminal dendritic branches narrows and moves to the left (toward lower densities) of the distribution derived from controls (Fig. 3F,G). It is interesting to note that there is increased variability in dendritic spine density counts across terminal L5 pyramidal branches in young Tg1, with some branches carrying essentially normal or supranormal spine densities, while others appearing denuded of spines.
We next classified dendritic protrusions using standard criteria into mushroom spines, stubby spines, and thin spines (see Materials and Methods; Peters and Kaiserman-Abramof, 1970) and plotted their densities separately. Mushroom spines were the large majority of dendritic protrusions observed in L5 dendritic branches in barrel cortex at the ages examined. The density of mushroom spines was increased in Tg1 mice at 4–12 weeks (Tg1: 0.28 ± 0.02/μm, WT:0.22 ± 0.011/μm, p < 0.001, n = 13–14 animals per genotype) and decreased versus WT at 16–20 weeks (Tg1:0.17 ± 0.006/μm, WT:0.22 ± 0.009/μm, p < 0.05, n = 8–9 animals per genotype; Fig. 3H). Thin spine and stubby spine densities were not significantly different versus WT (data not shown). Therefore the abnormal fall in spine density over time in Tg1 animals is primarily mediated by mushroom spines, which are believed to represent the most mature excitatory synapse morphology. We note that filopodia were rare at the ages examined and are not illustrated here. A summary of the number of spines, dendrites, and animals analyzed across conditions is provided in Figure 3I.
An increase in the length of terminal dendritic branches could result in decreased spine density per unit length, if spine density is regulated such that a fixed number of presynaptic contacts are made per terminal branch. To determine whether spine density changes are related to changes in dendritic branch length, we measured the length of terminal L5 dendritic branches, in vivo, in the two genotypes. We found that Tg1 terminal branch length was 13–23 μm shorter than WT at all time points (p < 0.001, n = 26 WT, 24 Tg1 animals, two-way ANOVA; Fig. 3D), and this effect reached significance at individual time points at 16 and 20 weeks (p < 0.05, n = 4–5 animals per genotype, two-way ANOVA followed by Tukey's test). This ∼10–18% decrease in total terminal dendrite branch length indicates that the abnormal decrease in spine density observed in Tg1 is not a homeostatic mechanism to maintain a fixed synapse number on a lengthening dendrite, but rather suggests that MECP2 duplication also influences dendritic morphology in addition to its effect on dendritic spines (Chapleau et al., 2009; Vonhoff et al., 2012).
Chronic time-lapse imaging of apical dendrites confirms that spine density decreases over time in Tg1 mice
In a subset of mice, apical dendrites were followed over time to see how the net decrease in spine density with age in Tg1 animals (Fig. 4) emerges at the level of individual dendrites. We imaged apical tufts of L5 pyramidal neurons in somatosensory cortex every 4 weeks from postnatal week 8 to 12 (Tg1: 38 branches from five animals; WT: 44 branches from five animals). As observed in the animal cohorts (Fig. 3), time-lapse imaging of the same dendrites confirmed that Tg1 spine density is elevated at 8 weeks compared with controls (Tg1: 0.27 ± 0.01/μm; WT: 0.23 ± 0.007/μm; p < 0.01, two-way ANOVA followed by Tukey's test), but subsequently decreases so that by postnatal week 16 it is significantly lower, on average, than in controls (Tg1: 0.18 ± 0.008/μm; WT: 0.21 ± 0.007/μm; p < 0.05; Fig. 4A–C). Furthermore, while chronically imaged WT dendritic branches did not demonstrate a significant change in spine density between 8 and 16 weeks, Tg1 dendritic branches showed a significant decrease in spine density over that period (8–12 weeks, p < 0.01; 12–16 weeks, p < 0.001, two-way ANOVA followed by Tukey's test). Again, L5 terminal dendrites were found to be shorter on average in Tg1 compared with controls (p < 0.05, two-way ANOVA followed by Tukey's test; Fig. 4D). This reduction in length may be partially explained by the decrease in spine density, as recent data suggest that terminal dendrites retract to remaining spines when the most distal spines are eliminated (Schubert et al., 2011).
Subtle, if any, differences in dendritic spine morphology in Tg1 mice
Dendritic spines are highly specialized structures, and the high morphological diversity of spines allows neurons to achieve a high functional diversity (Yuste, 2011). Spine head volume correlates directly with the size of the postsynaptic density and the strength of the synapse, while spine neck length and neck width together regulate the biochemical and electrical isolation of each synapse. As disorders of the nervous system often lead to abnormal spine morphology (Fiala et al., 2002; Dierssen and Ramakers, 2006), including in Rett syndrome (Armstrong, 2005; Belichenko et al., 2009), we reasoned that MECP2 duplication may alter spine morphology in L5 pyramid apical dendrites. High-resolution stacks of apical dendrite spines were captured at postnatal week 8 and 16 and processed by a polynomial interpolation method to enhance image quality (Fig. 5A; see Materials and Methods). Protrusion heads and necks were traced and head volume, neck length, and neck width quantified (Fig. 5B; see Materials and Methods). We compared distributions for each morphological parameter from two Tg1 animals (281 spines) and two controls (351 spines) at postnatal weeks 8 (presymptomatic, left) and 16 (postsymptomatic, middle; Fig. 5C–E). The right side shows cumulative distributions of the results for all ages and genotypes.
Spine head volumes decreased on average in both Tg1 mice and controls between 8 and 16 weeks (Fig. 5C, right; p < 0.01, Mann–Whitney U test). Spine head volumes were slightly larger in Tg1 animals compared with controls at 16 weeks (Fig. 5C; p < 0.01, Mann–Whitney U test), while this difference was not significant at 8 weeks. Spine necks were slightly thinner and longer in Tg1 mice compared with controls at 8 weeks (Fig. 5D, neck width; p < 0.01; Fig. 5E, neck length; p < 0.05, Mann–Whitney U test), but by 16 weeks this difference was not significant. Together, differences in spine head volume, neck length, and neck width between Tg1 animals and controls were quite subtle, if present.
Dendritic spine turnover remains high into adulthood in Tg1 mice
It has been shown recently that L5 apical dendritic spines continuously form and retract throughout the life of an animal (Holtmaat and Svoboda, 2009), and this structural plasticity can serve as a substrate for learning and procedural memory (Xu et al., 2009; Yang et al., 2009). In vivo imaging experiments have identified abnormal dendritic spine structural plasticity in Mecp2-null mice (Landi et al., 2011) supporting the hypothesis that MeCP2 may play an important role in synaptic homeostasis (Ramocki and Zoghbi, 2008). Tg1 mice have initially enhanced capacity for learning and memory (Collins et al., 2004) suggesting an enhanced state of plasticity.
To determine whether structural plasticity of dendritic spines is altered in Tg1, we measured the formation and elimination of dendritic spines in L5 pyramidal neuron apical dendrites at 10 min intervals over 1 h as described previously 0(Cruz-Martín et al., 2010) at postnatal weeks 8, 12, 16, 20, and 40 (Fig. 6A; see Materials and Methods). As reported previously (Zuo et al., 2005b; Holtmaat et al., 2006), spine gain, loss, and turnover (sum of spine gain and loss divided by twice the spine count, see Materials and Methods) decreases with age in controls (Fig. 6B–D, blue curve). The natural course of deceleration in the rate of spine gain was strikingly disrupted in Tg1 animals (Fig. 6B), such that the rate of spine gain in 20-week-old Tg1 mice (1.1 ± 0.3%) remained approximately equal to that seen at week 12 in controls (1.1 ± 0.2%), whereas the rate of spine gain in 20-week-old control animals was significantly lower at 0.5 ± 0.1% (Fig. 6B, n = 4–6 animals per genotype, p < 0.05, two-way ANOVA with Tukey's post hoc test). The rate of spine gain per unit time (10 min) compared between the two genotypes across ages was significantly higher in Tg1 animals (two-way ANOVA, p = 0.024). The rate of spine loss was also increased at all time points examined in Tg1 mice (Fig. 6C). Comparison between genotypes was highly significant (p = 0.0006, two-way ANOVA), and single time-point significance was reached for week 12 (Tg1: 2.8 ± 0.4%, WT: 2.0 ± 0.2%, p < 0.05, n = 5 animals per genotype, two-way ANOVA followed by Tukey's test) and week 20 (Tg1: 1.9 ± 0.3%, WT: 0.9 ± 0.1%, p < 0.01, n = 4 WT and 6 Tg1, two-way ANOVA followed by Tukey's test). Spine turnover (average between spine gain and loss) is also higher in Tg1 mice compared with controls (p= 0.0025, two-way ANOVA; Fig. 3D), and this effect reached single time-point significance at week 20 (Tg1: 1.5 ± 0.3%, WT: 0.7 ± 0.1%, p <0.05, two-way ANOVA followed by Tukey's test).
Plotting the percentage of spines surviving after 1 h of observation revealed a gradual increase in spine survival with age in both genotypes, with Tg1 spine survival being lower than those of controls at every age examined (Fig. 6E) The decrease in the percentage of spines surviving over 1 h in Tg1 mice compared with controls was significant at postnatal weeks 8–20. For weeks 8, 12, and 16 survival was 88.2 ± 1.2%, 88.0 ± 1.4%, and 87.7 ± 0.7% for Tg1 mice, respectively, versus 91.2 ± 1.1%, 91.7 ± 1.0%, and 92.2 ± 0.9% for WT mice (p < 0.05; two-way ANOVA followed by Tukey's test). At 20 weeks percentage spine survival for Tg1 mice was 88.2 ± 1.2% versus 94.8 ± 0.7% for WT controls (p < 0.001; two-way ANOVA followed by Tukey's test; Fig. 6E). The rate of spine loss slightly exceeded the rate of spine gain in both Tg1 and controls (Fig. 6F), as reported previously in WT mice at 3–6 months of age (Mostany et al., 2010). The difference between spine loss and gain was significantly higher in Tg1 mice compared with WT (p = 0.0065, n = 20–22 animals per genotype, two-way ANOVA), suggesting an imbalance between the formation and elimination of synapses in favor of spine loss (Fig. 6F). Total numbers of spines, dendrites, and animals analyzed at each time point are provided in Figure 6G.
Apical dendritic arbors are more highly branched in Tg1 mice
A greater number of GFP-labeled dendritic branches were visible in superficial cortical layers in Tg1, suggesting alterations in dendritic arbor complexity. To determine whether MeCP2 overexpression alters dendritic arbor complexity, we imaged the apical dendrites of 25 adult pyramidal neurons (12 from Tg1, 13 from WT) from the apical tufts to the soma and reconstructed them in Neurolucida (Fig. 7A–D). Reconstructed neurons were clustered into simple-tufted (Fig. 7A,B) and complex-tufted (Fig. 7C,D) subtypes using established criteria (see Materials and Methods; Holtmaat et al., 2006). In somatosensory cortex, simple-tufted cells correspond to L5A pyramids projecting to the striatum and contralateral cortex, and complex-tufted cells correspond to L5B pyramids projecting to the thalamus, superior colliculus, and pons (Groh et al., 2010).
Figure 7.

Increased dendritic arbor complexity in Tg1 L5 pyramidal neurons. Apical dendritic arbors of simple- and complex-tufted L5 pyramidal neurons were captured and reconstructed in 3D with Neurolucida software. A–D, Left, 3D projection of an imaged dendritic tree. Middle, Neurolucida tracing of the imaged tree. Right, The same dendritic tracing viewed from above. Scale bar, 50 μm. A, WT simple-tufted cell. B, Tg1 simple-tufted cell. C, WT complex-tufted cell. D, Tg1 complex-tufted cell. E, G, Total branch number per apical pyramidal dendritic tree after the major bifurcation in simple-tufted (E) and complex-tufted (G) cells of Tg1 (red) and littermate controls (dark blue) respectively. F, H, Total apical dendritic tree length after the major bifurcation in simple-tufted (F) and complex-tufted (H) cells of Tg1 (red) and littermate controls (blue), respectively. Note that the apical dendritic trees of Tg1 L5 pyramidal neurons have more branches and higher overall length than littermate controls. *p < 0.05, **p < 0.01, two-tailed t test. Simple-tufted: Tg1, n = 5 trees; WT, n = 6 trees. Complex-tufted: Tg1, n = 7 trees; WT, n = 7 trees. Images were captured from seven animals per genotype. Note that all animals whose dendrites were imaged were adult (≥4 weeks old), and that there was no correlation between dendritic arborization parameters and age in our cohort (data not shown).
The total number of branches per apical dendrite in both simple-tufted (Tg1: 19 ± 2 branches; WT: 11 ± 1 branches, p < 0.01, two-tailed t test; Fig. 7E) and complex-tufted (Tg1: 26 ± 2 branches; WT: 18.1 ± 0.8 branches, p < 0.01, two-tailed t test; Fig. 7G) Tg1 cells was significantly increased compared with littermate controls. Total apical dendrite length of both cell types was also increased in Tg1 animals (Tg1: simple-tufted 2466.1 ± 232 μm, complex-tufted 3177 ± 176.1 μm; WT: simple-tufted 1903.1 ± 81.6 μm, complex-tufted 2612.5 ± 171.1 μm; p < 0.05, two-tailed t test; Fig. 7F,H). Similar numbers of cells were clustered into simple-tufted and complex-tufted in both genotypes, and the increase in dendrite length and branch number caused by MeCP2 duplication appeared to affect both cell types equally.
Sholl analysis of reconstructed dendrites revealed that starting at ∼500–550 μm from the soma, i.e., in the superficial cortical layers within 100 μm of the pia, the arbor complexity (number of Sholl intersections per concentric sphere) increased in Tg1 compared with controls (p < 0.001 at 550 μm, p < 0.01 at 500 μm, two-way ANOVA followed by Tukey's test; Fig. 8B). These results suggest that farther than 500 μm from the soma (approximately in layer I) Tg1 neurons are more likely to bifurcate and extend laterally, leading to a denser coverage of superficial cortical territory. We note that these changes could not be explained by differences in soma depth (Tg1: 612.9 ± 9.03 μm, WT: 586.9 ± 12.8 μm), the area of the contour traced by the XY projection of the dendritic arbor (data not shown), or the depth of the first major apical dendritic bifurcation (Tg1: 269.8 ± 33.8 μm, WT: 275.2 ± 32.3 μm), which were not significantly different between Tg1 and WT. Apical dendritic arbors were acquired in adulthood, and there was no correlation between total branch number or branch length and age in either genotype (data not shown).
Figure 8.
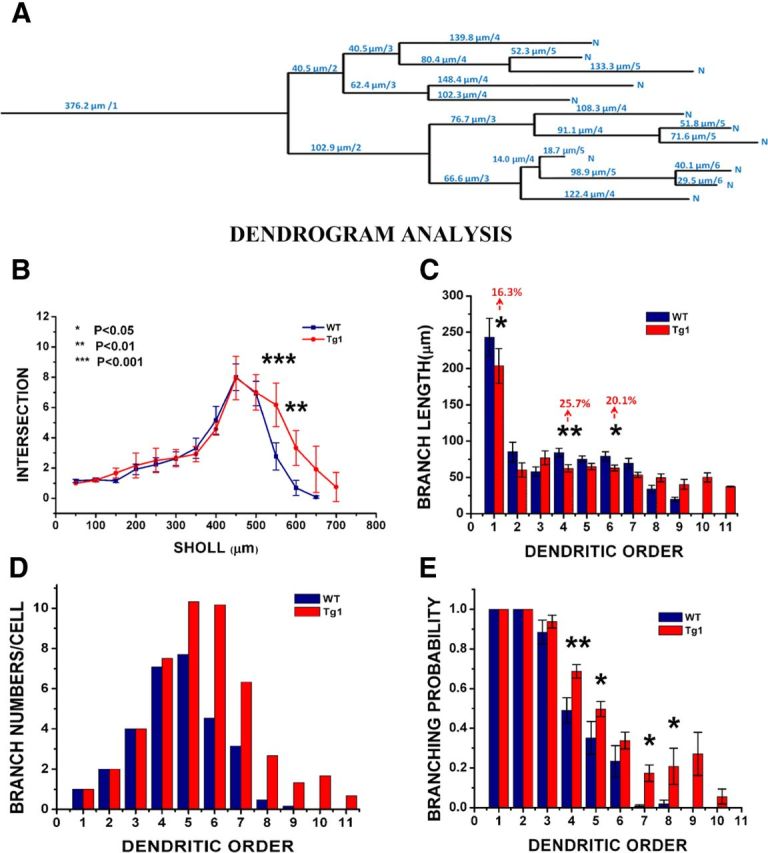
Dendrogram analysis of apical tree branching properties. A, Example dendrogram of a L5 pyramidal neuron. The dendrogram conceptualizes the apical dendritic arbor as a topological tree of branch lengths and branch points. Every branch point increases the order of distal dendritic segments by one. The major bifurcation of the primary apical dendrite was considered the first branch point (occasional minor oblique branches emanating from the main apical dendritic stem before its first major bifurcation were ignored in the analysis). B, 3D Sholl analysis of dendritic intersections with concentric spheres. C, Average branch length by order. D, Total number of branches per cell counted at each order. E, Probability of branching by order. Tg1: n = 12 arbors from seven animals. WT: n = 13 arbors from seven animals. *p < 0.05, **p < 0.01, and ***p < 0.001, two-way ANOVA with Tukey's multiple-comparison test.
To better characterize the Tg1 dendritic arborization abnormality, we conceptualized the imaged dendritic arbors as topological trees, or dendrograms (Fig. 8A; Uylings and van Pelt, 2002). We analyzed branching abnormalities by dendritic order, which would detect changes as a function of the number of branch points between a given segment and the soma. Tg1 pyramidal neurons exhibit more branches of higher order, starting approximately at order 5 (Fig. 8D). The vast majority of branches of order 8–11 belong to Tg1 neurons. Average dendritic branch length was generally similar between Tg1 and controls for the first 7 orders (Fig. 8C), with Tg1 exhibiting slightly shorter branches than controls (this trend reached significance for orders 1, 4, and 6, with a relative difference in length from 15 to 26%; two-way ANOVA with Tukey's test). For higher orders the comparison had little meaning because of the low number of control dendritic branches of that order.
Within a dendritic tree, each dendritic segment can be conceptualized as having two options as it ramifies away from the soma: it can branch with probability p(B), or it can terminate with probability p(T) = 1 − p(B). The increase in branch number at fifth and higher branch points away from the soma in Tg1 mice suggests that Tg1 dendritic segments have a higher probability of branching at high dendritic orders compared with controls. We plotted the probability of branching as a function of dendritic order (Fig. 8E), calculated as the number of dendrites of a given order that bifurcate divided by the total number of branches at that order. As expected, low-order dendrites (1–3) were very likely to branch in both Tg1 and controls. Interestingly, after the third order the probability of branching drops less in Tg1 mice than controls. Thus fourth-order dendritic branches bifurcate with probability ∼0.5 (WT) versus ∼0.7 (Tg1), while higher (>sixth) order branches bifurcate with probability ∼0.02 in controls as compared with ∼0.2 in Tg1. These measurements reveal a dramatic difference in the dendritic arborization pattern of Tg1 L5 apical pyramidal tufts compared with controls.
Increased protein p70S6K phosphorylation in Tg1 neocortex
The increased dendritic arborization and accelerated dendritic spine plasticity we observed in our mice are reminiscent of changes observed with increased BDNF-mTOR signaling (Horch et al., 1999; Horch and Katz, 2002; Kumar et al., 2005). The mTOR signaling pathway consists of a complex of proteins that regulate protein translation locally within dendrites, is enriched in autism-associated genes, and has been previously implicated in MeCP2 disorder pathophysiology (Kelleher and Bear, 2008; Ricciardi et al., 2011). Several genes in the BDNF-mTOR pathway are dysregulated in MeCP2-overexpressing mice in a direction that would predict increased mTOR signaling (Chahrour et al., 2008; Ben-Shachar et al., 2009; Fig. 9A). Although mTOR signaling has been shown to be decreased in Mecp2-null mice (Ricciardi et al., 2011) and Rett syndrome patient-derived stem cells (Li et al., 2013), this pathway has not been directly tested, to the best of our knowledge, in MeCP2-overexpressing mice. To test whether activation of the mTOR pathway differs between Tg1 mice and controls, we quantified the relative amount of phosphorylated protein p70S6K, a downstream effector of the mTOR pathway, versus total S6K. Somatosensory cortical tissue was harvested from six symptomatic (∼20-week-old) Tg1 mice and six littermate controls, and quantitative immunoblots were performed for phosphorylated S6K and total S6K (Fig. 9B); H3 level was measured as a loading control. The intensity of the p-S6K band normalized to the total S6K band was significantly higher in Tg1 mice compared with WT controls (Fig. 9C; p < 0.05, n = 6 samples per genotype, two-tailed t test). Total protein levels of S6K were not significantly altered in Tg1 mice (data not shown). These data indicate that the mTOR pathway is hyperactivated in the somatosensory cortex of Tg1 mice, providing a putative pathway that may play a role in mediating the structural plasticity we report here. We note that the antibody used probes a phosphorylation site solely targeted by mTOR complex 1 (mTOR-C1); mTOR-C2 may have a similar or divergent pattern of activation.
Figure 9.

Biochemical evidence for increased mTOR signaling in Tg1 mice. A, Schematic of the mTOR pathway in neurons (adapted from Hoeffer and Klann, 2010). Genes that are upregulated in Tg1 mice are bolded and italicized, based on Chahrour and Zoghbi, 2007. Note that several genes upstream to mTOR, including Akt, BDNF, NMDAR, and mGluR, have increased expression in Tg1 mice. B, Representative bands from an immunoblot of Tg1 and WT littermate somatosensory cortex tissue, probed for phospho-S6K (top) and total S6K (middle). Histone H3 (bottom) was used as a loading control. C, Quantification of band intensities reveals increased phosphorylation of S6K in Tg1 tissue (*p < 0.05, n = 6 animals per genotype, two-tailed t test).
Discussion
Autism and MECP2-associated developmental abnormalities could arise from pathological changes in cortical circuit homeostasis (Ramocki and Zoghbi, 2008), but this hypothesis has yet to be evaluated in vivo. We studied dendritic arborization and spine turnover over several stages of disease progression in the Tg1 mouse model of MECP2 duplication syndrome (Fig. 10). We found that MeCP2 overexpression leads to exuberant dendritic arborization and dendritic spine formation during early postnatal development. This is followed by a period of elevated spine turnover and synapse loss, persisting well into young adulthood. P70S6 kinase was hyperphosphorylated in Tg1 somatosensory cortex indicative of elevated mTOR-C1 signaling. These abnormalities of dendritic and synaptic homeostasis may contribute in part to the pathophysiology of the MECP2 duplication syndrome.
Figure 10.

Summary of disease progression and experimental results. Disease phenotype progression (adapted from Collins et al., 2004) and incorporating the time course of our observations here.
Increased dendritic arborization and spine density in young postnatal Tg1 mice
Postnatal development of L5 pyramidal neurons is characterized by dendritic arborization within the first two postnatal weeks, significant dendritic pruning in the second week, and marked synaptogenesis in the first 3–4 weeks of postnatal life (Miller, 1981; White et al., 1997; Lendvai et al., 2000; Cruz-Martín et al., 2010; Romand et al., 2011). By postnatal week 3–4, WT L5 apical dendrites have reached mature segment length, branching structure, and synapse density (Romand et al., 2011). Tg1 apical dendrites demonstrate significantly increased total branch length and branching complexity (Figs. 7, 8) as well as significantly increased terminal branch dendritic spine density at postnatal week 4–12 (Fig. 3), indicating that MECP2 overexpression leads to excessive postnatal dendritic and synaptic growth.
Tg1 apical dendrites have more branches of higher order leading to an ∼30% increase of total dendritic tree length on average, which occurs even though branches of a given order tend to be slightly (∼15–26%) shorter in Tg1 mice. Simple- and complex-tufted cells (Holtmaat et al., 2006), which generally correspond to corticostriatal and corticopontine neurons (Groh et al., 2010), respectively, have equally increased arborization in animals with MECP2 overexpression. Increased complexity of dendritic trees could result from increased branching or decreased pruning during dendrite development, and could be affected by the presence (or absence) of layer-specific presynaptic partners (Wong and Ghosh, 2002; Niell et al., 2004). This enhanced dendritic arborization is opposite to the simpler dendritic arbors observed in Mecp2-null mouse cortical pyramidal neurons (Kishi and Macklis, 2004; Stuss et al., 2012), as may be anticipated by the opposite transcriptional effects of MeCP2 gain versus loss (Chahrour et al., 2008). Notably, the effect that MECP2 overexpression has on dendritic arborization depends on the experimental model: In vivo MECP2 overexpression in Xenopus tectal neurons (Marshak et al., 2012) and in Drosophila motor neurons (Vonhoff et al., 2012) leads to simplified as opposed to complex dendritic arborization patterns, in contrast to our results here.
In addition to increased dendritic arborization, ∼24% more spines per dendritic length are found in terminal dendritic branches of layer 5 Tg1 pyramidal neurons at postnatal week 4. This suggests that MECP2 overexpression promotes more robust synaptogenesis in early postnatal development and agrees with in vitro electrophysiological and immunostaining data (Chao et al., 2007; Na et al., 2012).
Accelerated Tg1 spine turnover with a bias toward loss
Once L5 pyramidal neurons reach their maximum dendritic and synaptic extent at 3–4 weeks of life (Romand et al., 2011), a period of synaptic pruning begins and is sustained into adulthood at a steadily decreasing rate (Zuo et al., 2005a; Grutzendler et al., 2007; Yang et al., 2009; but see Romand et al., 2011). Accordingly, in control animals, we observed a sharp decline in the spine density of terminal L5 dendritic branches from postnatal week 4 to 8 reaching a plateau thereafter. Interestingly, the time course of this profile differs markedly in Tg1 animals: spine density, which is initially significantly higher than controls, decreases over a longer period extending to postnatal week 16 to eventually reach a lower plateau (Fig. 3B). This characteristic profile is robust and was observed both by population analysis (Fig. 3) and by chronic monitoring of individual dendritic branches (Fig. 4). The prolonged decrease in synaptic density we observe in Tg1 animals agrees with a trend toward synapse loss previously seen in juvenile Tg1 mice (Chao et al., 2007) and in an in vitro MECP2-overexpression study (Chapleau et al., 2009).
Excess synaptic pruning appeared to be spine-subtype specific, affecting mushroom spines while relatively sparing thin and stubby spines (Fig. 3H). Mushroom spines represent the most mature and stable spine morphologies (Fiala et al., 2002), while thin spines are thought to be more transient loci of plasticity (Holtmaat et al., 2005; Zuo et al., 2005b). Nonetheless, overall changes in spine head volume, spine neck width, and spine neck length, if present, are quite subtle in Tg1 mice (Fig. 5). This is in contrast to the markedly abnormal spine morphology observed in other developmental brain disorders (Dierssen and Ramakers, 2006; Penzes et al., 2011), and in particular in Rett syndrome (Armstrong, 2005; Belichenko et al., 2009).
The period of net synaptic pruning we observed in Tg1 mice occurs in association with a sustained period of elevated spine turnover rate (Fig. 6). In WT mice spine formation and elimination rates decrease with age reflecting a shift in favor of stability in adult neural circuits (Grutzendler et al., 2002; Holtmaat et al., 2005; Zuo et al., 2005b). This shift is markedly delayed in Tg1 animals. Spine gain, loss, and turnover rates in Tg1 animals are initially comparable to controls (Fig. 6). By week 12, spine loss has decreased in controls but remains abnormally high in Tg1 animals. At 20 weeks of age, both spine gain and loss remain at an abnormally elevated rate. Spine loss is slightly higher than spine gain at all ages in both controls (Zuo et al., 2005a; Mostany et al., 2010) and Tg1 but net loss appears to be greater in Tg1 (Fig. 6F). One hour spine survival rates are lower in Tg1 than in controls for all ages tested (Fig. 6E), reaching significance for postnatal weeks 8–20. An analogous increase in spine turnover has been reported in the barrel cortex of Fragile X mice and is thought to reflect a more immature state (Cruz-Martín et al., 2010; Pan et al., 2010).
Potential mechanisms
Although multiple pathways affecting dendritic and synaptic plasticity are known to be modulated by MeCP2 levels (Chahrour et al., 2008; Ben-Shachar et al., 2009), several lines of evidence argue that deranged mTOR signaling could underlie the initial dendritic and synaptic overgrowth, the accelerated spine turnover, and other behavioral and electrophysiological phenotypes observed in Tg1 mice (Chen et al., 2003; Collins et al., 2004; Chang et al., 2006; Larimore et al., 2009; Tropea et al., 2009; Kline et al., 2010; Samaco et al., 2012; Li et al., 2013). Several components of the mTOR signaling pathways, including BDNF, Akt, Tiam1, and Rac, are overexpressed in Tg1 mice and underexpressed in Mecp2-null mice (Chahrour et al., 2008). Increased BDNF-mTOR signaling accelerates apical dendritic growth and branching (McAllister et al., 1995; Horch et al., 1999; Horch and Katz, 2002; Jaworski et al., 2005; Kumar et al., 2005; Kwon et al., 2006; Fraser et al., 2008; Chow et al., 2009; Luikart et al., 2011), increases spine density (Shimada et al., 1998; Alonso et al., 2004; Tavazoie et al., 2005; Napolioni et al., 2009; Chapleau and Pozzo-Miller, 2012), and accelerates dendritic spine turnover (Horch et al., 1999), all features observed in our study (Fig. 10). We confirmed biochemically that mTOR-C1 signaling is upregulated in 20-week-old Tg1 neocortex (Fig. 9). We note that mTOR can be activated through multiple parallel pathways, particularly through the BDNF-TrkB signaling pathway and the glutamate-NMDA/mGluR signaling pathway (Fig. 9A), both of which show increased gene expression in Tg1 mice (Chahrour et al., 2008). Future experiments studying the role of hyperactive mTOR signaling in MECP2 duplication syndrome pathophysiology, including testing the effects of rapamycin and other mTOR pathway modulators, may be warranted given our findings.
This initially accelerated dendritic and spine growth may place the system into an abnormal state of local hyperconnectivity analogous to that observed in autism spectrum disorders (Happé and Frith, 2006; Anagnostou and Taylor, 2011). Assuming that spine densities are relatively homogeneous throughout the apical tuft (Parnavelas et al., 1973), we infer that, at postnatal week 4, apical dendritic tufts of Tg1 L5 pyramidal neurons are both ∼30% longer and harbor ∼24% more spines per dendritic length compared with controls, resulting in ∼60% more total synapses. Tg1 neurons, however, maintain a high spine turnover rate (Fig. 6D) with spine loss predominating (Figs. 3B, 4C), leading over time to a relative correction of the synaptic spine excess. By postnatal week 16, Tg1 terminal dendritic branches lose ∼50% of their spines compared with ∼30% loss in controls. Tg1 spine density plateaus at a lower level compared with WT but Tg1 dendritic arbors are longer, and a rough calculation suggests that the total apical tuft synapse number in Tg1 animals is brought down to within 10% of controls. These approximate estimates suggest that homeostatic mechanisms attempting to preserve total excitatory synapse number may in part account for the prolonged Tg1 spine density decline. However, homeostatic mechanisms are unlikely to explain high spine turnover, which persists beyond the stabilization of spine density in adult Tg1 animals. This accelerated spine turnover likely represents a pathological imbalance between structural stability and plasticity of Tg1 dendritic spines that temporally correlates with and may, in part, contribute to behavioral dysfunction.
Footnotes
We thank the March of Dimes Foundation and the Simons Foundation for support (S.S.). B.S. was in part funded through CHRCDA (NIH, Kl2 HD4164S). We thank Yu Dong, John Dani, Joseph G. Duman, Peyman Golshani, Emmanouil Froudarakis, Joanna L. Jankowsky, Ji-yoen Kim, Tang-Cheng Lee, Ricardo Mostany, Masataka Nishimura, Sanyong Niu, Carlos Portera-Cailliau, Matt N. Rasband, John Swann, Andreas Tolias, Kimberly R. Tolias, Kechun Yang, and Chuansheng Zhang for their help with histological methods and comments on this manuscript.
The authors declare no competing financial interests.
References
- Alonso M, Medina JH, Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostou E, Taylor MJ. Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Mol Autism. 2011;2:4. doi: 10.1186/2040-2392-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arellano JI, Benavides-Piccione R, Defelipe J, Yuste R. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci. 2007;1:131–143. doi: 10.3389/neuro.01.1.1.010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. Widespread changes in dendritic and axonal morphology in <I>Mecp2</I>-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009;514:240–258. doi: 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet. 2009;18:2431–2442. doi: 10.1093/hmg/ddp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascio CJ, Moana-Filho EJ, Guest S, Nebel MB, Weisner J, Baranek GT, Essick GK. Perceptual and neural response to affective tactile texture stimulation in adults with autism spectrum disorders. Autism Res. 2012;5:231–244. doi: 10.1002/aur.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett Syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 Controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapleau CA, Pozzo-Miller L. Divergent roles of p75NTR and Trk receptors in BDNF's effects on dendritic spine density and morphology. Neural Plast. 2012;2012:578057. doi: 10.1155/2012/578057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, Kudo S, Armstrong DL, Percy AK, Pozzo-Miller L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2009;35:219–233. doi: 10.1016/j.nbd.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Chow DK, Groszer M, Pribadi M, Machniki M, Carmichael ST, Liu X, Trachtenberg JT. Laminar and compartmental regulation of dendritic growth in mature cortex. Nat Neurosci. 2009;12:116–118. doi: 10.1038/nn.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Cruz-Martín A, Crespo M, Portera-Cailliau C. Delayed stabilization of dendritic spines in Fragile X mice. J Neurosci. 2010;30:7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M, Ramakers GJ. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5:48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev. 2002;39:29–54. doi: 10.1016/S0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaze DG. Neurophysiology of Rett syndrome. J Child Neurol. 2005;20:740–746. doi: 10.1177/08830738050200090801. [DOI] [PubMed] [Google Scholar]
- Groh A, Meyer HS, Schmidt EF, Heintz N, Sakmann B, Krieger P. Cell-type specific properties of pyramidal neurons in neocortex underlying a layout that is modifiable depending on the cortical area. Cereb Cortex. 2010;20:826–836. doi: 10.1093/cercor/bhp152. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Helmin K, Tsai J, Gan WB. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer's disease. Ann N Y Acad Sci. 2007;1097:30–39. doi: 10.1196/annals.1379.003. [DOI] [PubMed] [Google Scholar]
- Happé F, Frith U. The weak coherence account: detail-focused cognitive style in autism spectrum disorders. J Autism Dev Disord. 2006;36:5–25. doi: 10.1007/s10803-005-0039-0. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation [published erratum appears in J Neurosci 1992 Aug;12:following table of contents] J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5:1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Horch HW, Krüttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/S0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19:18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet. 2008;17:1386–1396. doi: 10.1093/hmg/ddn026. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konur S, Rabinowitz D, Fenstermaker VL, Yuste R. Systematic regulation of spine sizes and densities in pyramidal neurons. J Neurobiol. 2003;56:95–112. doi: 10.1002/neu.10229. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi S, Putignano E, Boggio EM, Giustetto M, Pizzorusso T, Ratto GM. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci Rep. 2011;1:45. doi: 10.1038/srep00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimore JL, Chapleau CA, Kudo S, Theibert A, Percy AK, Pozzo-Miller L. BDNF overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol Dis. 2009;34:199–211. doi: 10.1016/j.nbd.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K. Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature. 2000;404:876–881. doi: 10.1038/35009107. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, Kwok SM, Feldman DA, Bateup HS, Gao Q, Hockemeyer D, Mitalipova M, Lewis CA, Vander Heiden MG, Sur M, Young RA, Jaenisch R. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett Syndrome neurons. Cell Stem Cell. 2013;13:446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. 2011;31:4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak S, Meynard MM, De Vries YA, Kidane AH, Cohen-Cory S. Cell-autonomous alterations in dendritic arbor morphology and connectivity induced by overexpression of MeCP2 in Xenopus central neurons in vivo. PLoS One. 2012;7:e33153. doi: 10.1371/journal.pone.0033153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-X. [DOI] [PubMed] [Google Scholar]
- Miller M. Maturation of rat visual cortex. I. A quantitative study of Golgi-impregnated pyramidal neurons. J Neurocytol. 1981;10:859–878. doi: 10.1007/BF01262658. [DOI] [PubMed] [Google Scholar]
- Mostany R, Chowdhury TG, Johnston DG, Portonovo SA, Carmichael ST, Portera-Cailliau C. Local hemodynamics dictate long-term dendritic plasticity in peri-infarct cortex. J Neurosci. 2010;30:14116–14126. doi: 10.1523/JNEUROSCI.3908-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Adachi M, Autry AE, Mahgoub MA, Kavalali ET, Monteggia LM. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci. 2012;32:3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolioni V, Moavero R, Curatolo P. Recent advances in neurobiology of tuberous sclerosis complex. Brain Dev. 2009;31:104–113. doi: 10.1016/j.braindev.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Niell CM, Meyer MP, Smith SJ. In vivo imaging of synapse formation on a growing dendritic arbor. Nat Neurosci. 2004;7:254–260. doi: 10.1038/nn1191. [DOI] [PubMed] [Google Scholar]
- Paluszkiewicz SM, Olmos-Serrano JL, Corbin JG, Huntsman MM. Impaired inhibitory control of cortical synchronization in fragile X syndrome. J Neurophysiol. 2011;106:2264–2272. doi: 10.1152/jn.00421.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Aldridge GM, Greenough WT, Gan WB. Dendritic spine instability and insensitivity to modulation by sensory experience in a mouse model of fragile X syndrome. Proc Natl Acad Sci U S A. 2010;107:17768–17773. doi: 10.1073/pnas.1012496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG, Globus A, Kaups P. Continuous illumination from birth affects spine density of neurons in the visual cortex of the rat. Exp Neurol. 1973;40:742–747. doi: 10.1016/0014-4886(73)90108-8. [DOI] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, Pizzorusso T, Giustetto M, Broccoli V. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- Romand S, Wang Y, Toledo-Rodriguez M, Markram H. Morphological development of thick-tufted layer V pyramidal cells in the rat somatosensory cortex. Front Neuroanat. 2011;5:5. doi: 10.3389/fnana.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Mandel-Brehm C, McGraw CM, Shaw CA, McGill BE, Zoghbi HY. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet. 2012;44:206–211. doi: 10.1038/ng.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert V, Lebrecht D, Holtmaat A. Structural stability and plasticity of pyramidal cell dendrites and axons in the adult mouse barrel cortex after whisker amputations. Soc Neurosci Abstr. 2011;37 74.12/LL14. [Google Scholar]
- Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–8570. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuss DP, Boyd JD, Levin DB, Delaney KR. MeCP2 mutation results in compartment-specific reductions in dendritic branching and spine density in layer 5 motor cortical neurons of YFP-H mice. PLoS One. 2012;7:e31896. doi: 10.1371/journal.pone.0031896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uylings HB, van Pelt J. Measures for quantifying dendritic arborizations. Network. 2002;13:397–414. doi: 10.1088/0954-898X/13/3/309. [DOI] [PubMed] [Google Scholar]
- Vonhoff F, Williams A, Ryglewski S, Duch C. Drosophila as a model for MECP2 gain of function in neurons. PLoS One. 2012;7:e31835. doi: 10.1371/journal.pone.0031835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EL, Weinfeld L, Lev DL. A survey of morphogenesis during the early postnatal period in PMBSF barrels of mouse SmI cortex with emphasis on barrel D4. Somatosens Mot Res. 1997;14:34–55. doi: 10.1080/08990229771204. [DOI] [PubMed] [Google Scholar]
- Wong RO, Ghosh A. Activity-dependent regulation of dendritic growth and patterning. Nat Rev Neurosci. 2002;3:803–812. doi: 10.1038/nrn941. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanouchi H, Kaga M, Arima M. Abnormal cortical excitability in Rett syndrome. Pediatr Neurol. 1993;9:202–206. doi: 10.1016/0887-8994(93)90085-Q. [DOI] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa H, Kaga M, Suzuki H, Sakuragawa N, Arima M. Giant somatosensory evoked potentials in the Rett syndrome. Brain Dev. 1991;13:36–39. doi: 10.1016/S0387-7604(12)80295-6. [DOI] [PubMed] [Google Scholar]
- Yuste R. Dendritic spines and distributed circuits. Neuron. 2011;71:772–781. doi: 10.1016/j.neuron.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Sun QQ. The balance between excitation and inhibition and functional sensory processing in the somatosensory cortex. Int Rev Neurobiol. 2011;97:305–333. doi: 10.1016/B978-0-12-385198-7.00012-6. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent BDNF transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005a;436:261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005b;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]