

Abstract
Adverse early-life experiences, such as poor maternal care, program an abnormal stress response that may involve an altered balance between excitatory and inhibitory signals. Here, we explored how early-life stress (ELS) affects excitatory and inhibitory transmission in corticotrophin-releasing factor (CRF)-expressing dorsal-medial (mpd) neurons of the neonatal mouse hypothalamus. We report that ELS associates with enhanced excitatory glutamatergic transmission that is manifested as an increased frequency of synaptic events and increased extrasynaptic conductance, with the latter associated with dysfunctional astrocytic regulation of glutamate levels. The neurosteroid 5α-pregnan-3α-ol-20-one (5α3α-THPROG) is an endogenous, positive modulator of GABAA receptors (GABAARs) that is abundant during brain development and rises rapidly during acute stress, thereby enhancing inhibition to curtail stress-induced activation of the hypothalamic-pituitary-adrenocortical axis. In control mpd neurons, 5α3α-THPROG potently suppressed neuronal discharge, but this action was greatly compromised by prior ELS exposure. This neurosteroid insensitivity did not primarily result from perturbations of GABAergic inhibition, but rather arose functionally from the increased excitatory drive onto mpd neurons. Previous reports indicated that mice (dams) lacking the GABAAR δ subunit (δ0/0) exhibit altered maternal behavior. Intriguingly, δ0/0 offspring showed some hallmarks of abnormal maternal care that were further exacerbated by ELS. Moreover, in common with ELS, mpd neurons of δ0/0 pups exhibited increased synaptic and extrasynaptic glutamatergic transmission and consequently a blunted neurosteroid suppression of neuronal firing. This study reveals that increased synaptic and tonic glutamatergic transmission may be a common maladaptation to ELS, leading to enhanced excitation of CRF-releasing neurons, and identifies neurosteroids as putative early regulators of the stress neurocircuitry.
Keywords: stress, PVN, allopregnanolone, maternal case, glutamate transporter, GABAA receptors
Introduction
Stressful experiences engage a coordinated neuronal and hormonal response that is orchestrated by the hypothalamic-pituitary-adrenocortical (HPA) axis via activation of corticotrophin-releasing factor (CRF)-releasing parvocellular neurons of the hypothalamic paraventricular nucleus (PVN; de Kloet et al., 2005; Joëls and Baram, 2009; Ulrich-Lai and Herman, 2009). Dysfunction of this fundamental survival mechanism is programmed early in life (Bale et al., 2010) and abnormal regulation, for example, as a consequence of adverse early-life experiences, is crucially implicated in various psychiatric disorders including depression (Lupien et al., 2009; Baram et al., 2012; Franklin et al., 2012). Interestingly, perturbations in glutamatergic transmission have been linked to depression in both clinical and animal studies (Popoli et al., 2012; Sanacora et al., 2012). Analysis of molecular markers of glutamatergic transmission suggests that similar adaptations may also underpin HPA axis dysfunction (Flak et al., 2009). Here, we used a mouse model of early-life stress (ELS; Rice et al., 2008) to investigate how negative early-life experiences may affect neurotransmitter function in CRF-expressing (CRF+ve) dorsal-medial (mpd) neurons of the mouse hypothalamus. In a combined electrophysiological and immunohistochemical approach, we show that ELS associates primarily with a significant increase in the excitatory drive (mediated by synaptic and extrasynaptic ionotropic glutamate receptors) onto mpd neurons. Certain endogenously occurring neurosteroids (e.g., 5α-pregnan-3α-ol-20-one [5α3α-THPROG]) abundant early in development (Grobin and Morrow, 2001) potently and selectively enhance GABAA receptor (GABAAR) function at low nanomolar concentrations (Belelli and Lambert, 2005). Intriguingly, neurosteroid levels change dynamically in response to a variety of stimuli, such as after an acute stress challenge (Purdy et al., 1991; Barbaccia et al., 1996; Biggio et al., 2007), suggesting that they may “fine-tune” GABAAR inhibitory function in the PVN (Decavel and Van den Pol, 1990; Boudaba et al., 1996; Miklós and Kovács, 2002, 2012; Di et al., 2005; Verkuyl et al., 2005; Cullinan et al., 2008; Hewitt et al., 2009; Popescu et al., 2010; Wamsteeker et al., 2010) to influence HPA axis activity (Patchev et al., 1994). Here, we show that ELS blunts the potent inhibitory actions of 5α3α-THPROG upon the neuronal discharge of CRF+ve mpd neurons of the neonatal mouse hypothalamus. The loss of neurosteroid efficacy is not caused by a change in GABAergic inhibition, but primarily by the significant increase in the excitatory drive onto mpd neurons, which is sufficient to functionally overcome the suppressant action of the neurosteroid. Dams lacking the GABAAR δ-subunit exhibit a depressive phenotype and altered maternal behavior (Maguire and Mody, 2008). Intriguingly, we find that the offspring of mice lacking the GABAAR δ-subunit (δ0/0) present hallmarks of an abnormal stress response and, importantly, share with ELS mice an increased glutamatergic drive with an associated loss of neurosteroid inhibition. Therefore, this neuronal plasticity may be a common maladaptation of the hypothalamic stress centers to negative early-life experiences. These results provide compelling support for glutamate receptors as an innovative avenue of therapeutic intervention for stress-related pathologies such as depression and raise the exciting prospect that neurosteroids may be an important molecular signal for the maturation of the HPA axis.
Materials and Methods
Breeding of mice
The δ0/0 mice and wild-type (WT) controls were generated on a C57BL/6J-129SvSvJ background at the University of Pittsburgh and bred at the University of Dundee as described previously (Mihalek et al., 1999). Mice were group housed, given free access to standard rodent chow and water, and maintained on a 12 h alternating light-dark cycle with lights on at 07:30.
ELS paradigm
The ELS paradigm was adapted from Rice et al. (2008). Specifically, pregnant dams were housed singly and monitored every 12 h for the birth of pups. The day of birth was termed postnatal day 0 (P0) and both the control and ELS dams were left undisturbed until P2. On P2, litters were adjusted to a maximum of eight pups of both sexes. Control dams were housed in standard sawdust bedding and provided with sufficient nesting material (1 square; DBM Scotland). In the ELS cages, dams were provided with reduced bedding (2/3 square) placed upon a raised, fine-gauge (5 mm) steel mesh platform. The cage floor was covered with a small amount of sawdust to prevent ammonia buildup. All litters were left undisturbed between P2 and P9. On P9, both control and ELS pups were weighed before being returned, with dams, to “control” cages (i.e., with standard bedding and nesting material). Offspring then remained with the dams until weaning at P24.
Assessment and analysis of maternal behavior
The maternal behavior of control (n = 7) and ELS (n = 7) dams was initially evaluated on day of life 3–8 of the pups to validate the previously described model. Briefly, control and ELS dams were observed (by video camera) three times a day during the light (08:30, 15:00) and dark (20:00) phases for 30 min sessions with minimum disturbance to the mice (the experimenter left the room during the recording session). During each 30 min observation period, the number of sorties that the dam made from the nest (i.e., no contact with pups) was recorded. The dam–pup interaction was scored every other minute (resulting in 15 × 1 min epochs) and the position of the dam with regard to the nest (i.e., inside nest, outside nest, or a mixture of both) was recorded for the entire 1 min epoch. The same parameters were used to evaluate the maternal behavior of dams lacking the δ-GABAAR both in control breeding conditions (n = 5) and after ELS (n = 5). The number of sorties and the duration of dam–pup interaction was analyzed for each observation period and a total for both parameters was calculated for every treatment day and averaged across litters. A two-way ANOVA was applied to assess the correlation between treatment group and each postpartum day. Statistical comparisons between the total number of sorties of different treatment groups were made using the unpaired Student's t test.
Electrophysiology
Slice preparation
WT control, WT ELS, and δ0/0 mice of either sex (P18–P26) were killed by cervical dislocation in accordance with Schedule I of the UK Government Animals (Scientific Procedures) Act of 1986. Coronal hypothalamic slices containing the PVN were prepared as described previously (Zaki and Barrett-Jolley, 2002). The brain was rapidly dissected and placed in ice-cold (0–4°C), oxygenated (95% O2) artificial CSF (aCSF) containing the following (in mm): 135 NaCl, 2.5 KCl, 1.25 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, and 10 glucose, 320–335 mOsm, pH ∼7.4. Coronal hypothalamic slices were then cut (300–320 μm) using a Vibratome (Leica) at 0–4°C. Slices were subsequently incubated for at least 1 h at room temperature in a holding chamber containing oxygenated aCSF (as above, but also containing 1 mm ascorbic acid and 3 mm sodium pyruvate) before being transferred to the recording chamber. Recordings were performed on slices perfused (3–6 ml/min) with extracellular solution (ECS) containing the following (in mm): 126 NaCl, 26 NaHCO3, 2.95 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, and 10 glucose, 306–309 mOsm, pH ∼7.4.
Recordings
All recordings were performed using an Axopatch 1D amplifier (Molecular Devices) stored directly to a PC using a NI-DAQmx interface (National Instruments) for analysis offline.
Characterization of parvocellular neurons, current-clamp recordings.
Parvocellular neurons were identified based upon a combination of immunohistochemical, anatomical, and morphological criteria described previously in rats (Luther et al., 2002; Verkuyl and Joëls, 2003) using an Olympus BX50 microscope equipped with IR/DIC optics. For parvocellular neurons, the expression of a Ca2+-dependent low threshold current (LTS) was assessed with a current–clamp protocol as described previously (Luther et al., 2002). Briefly, the input resistance of individual neurons was calculated after a hyperpolarizing current injection of 20–30 pA for a duration of 400 ms. Cells were then tested with a 400 ms hyperpolarizing current injection that lowered the membrane potential of neurons to between −90 and −110 mV, to de-inactivate resident low threshold Ca2+ channels. The size of the current injection was calculated for individual mpd neurons based upon the input resistance of each neuron using Ohms law (I = V/R). For these recordings, patch pipettes with a resistance of 4–6 MΩ were pulled from thick-wall borosilicate glass (1.55 mm outer diameter; 0.95 mm inner diameter; Garner Glass) and filled with an intracellular solution containing the following (in mm): 130 K-gluconate, 10 HEPES, 10 EGTA, 1 NaCl, 1 MgCl2, 1 CaCl2, 2 Mg-ATP, and 0.5 Na-GTP, pH 7.2 with KOH. The osmolarity of the intracellular solution was adjusted to 300–310 mOsm with d-mannitol. A measured liquid junction potential of 11 mV was corrected as described previously (Neher, 1992).
Using such an approach, recordings were made from the mid-rostrocaudal level of the PVN, an area that has a high density of CRF+ve parvocellular neurons (Biag et al., 2012). We found that, in common with the rat (Luther et al., 2002) low threshold Ca2+ spikes (LTS) were absent in the majority (>90%) of mpd neurons of the mouse PVN, whereas neurons located in dorsal and ventral medial parvocellular regions (dp and mpv neurons, respectively) exhibited a prominent LTS (Fig. 2A). Furthermore, post hoc immunohistochemical analysis of biocytin-filled cells (5–10 min recording) confirmed that neurons within the mpd region expressed CRF (see Immunohistochemistry; Visualization of biocytin filled neurons and Fig. 2B). The observed CRF expression is in agreement with the recent immunocytochemical analysis in a C57BL/6J mouse line indicating that CRF+ve parvocellular neurons are concentrated in the dorsal-medial parvocellular region (Alon et al., 2009).
Figure 2.
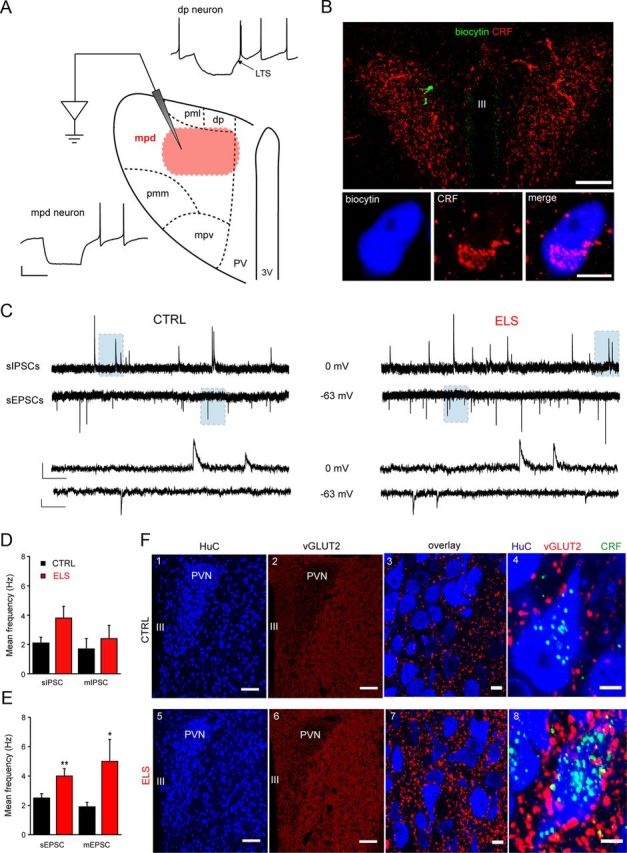
Exposure to ELS greatly increases synaptic glutamatergic drive onto neonatal mpd neurons. A, Schematic representation of the mouse PVN highlighting the anatomical organization of magno- and parvocellular neurons and representative Iclamp recordings made from putative preautonomic (dp) and neuroendocrine (mpd) neurons based upon their anatomical location within the PVN. Note that, in agreement with studies in rats (Luther et al., 2002), putative preautonomic dp neurons expressed a prominent low-threshold Ca2+ spike, whereas putative neuroendocrine mpd cells did not. Scale bars: y = 20 mV, x = 250 ms. The highlighted region (red) in the mpd region corresponds to the area where electrophysiological recordings were conducted. B, Confirmation of the location and neurochemical profile of the recorded neurons. Top, Representative image providing an overview of labeling for CRF (red) in a coronal mouse brain slice containing the PVN showing the location of two recorded neurons (in green), which were filled with biocytin during the recordings. Bottom, High-resolution image of one of the recorded neurons with immunoreactivity for biocytin and CRF pseudocolored blue and red, respectively. Scale bars: Top, 100 μm; bottom, 5 μm. C, Illustrated are sections (5 s) of whole-cell voltage-clamp recordings of sIPSCs (top) and sEPSCs (bottom) recorded from the same representative mpd neuron derived from a CTRL (left) and ELS (right) WT mouse. A section (shaded area, 0.5 s) of the recordings is displayed on an expanded time scale below. Scale bars: IPSCs, y = 20 pA, EPSCs y = 10 pA, x = 500 ms and 50 ms for top and bottom traces, respectively. Bar graphs illustrate the mean frequency of sIPSCs and mIPSCs (n = 9–25; D) and sEPSCs and mEPSCs (n = 14–32; E) recorded from mpd neurons derived from CTRL (black) and ELS (red) WT mice. *p < 0.05; **p < 0.01 CTRL vs ELS, unpaired Student's t test. F, Representative images demonstrating the upregulation of vGLUT2 expression in the PVN of ELS compared with CTRL WT neonatal (P22–P28) mice. F1, F5, HuC, a pan-neuronal marker used to delineate the PVN. F2, F6, Overviews demonstrating the increase in vGLUT2 immunoreactivity throughout the PVN and lateral hypothalamus in tissue from ELS mice. Magnified views of the PVN demonstrating the relative increase in vGLUT2 (F3,F7) and vGLUT2-CRF (F4,F8) expression in ELS tissue. Quantification of vGLUT2 clusters within the PVN revealed a significant increase in ELS tissue (p = 0.001 WT vs ELS, unpaired Student's t test). CRF immunoreactivity in ELS tissue was significantly greater than that detected in CTRL tissue (p < 0.001, unpaired Student's t test). Scale bars, 100 μm. Note that tissue from CTRL and ELS WT mice was reacted and imaged under identical conditions. Scale bars: F1, F2, F5, F6, 100 μm; F3, F7, 10 μm; F4, F8, 10 μm.
Action currents.
Cell-attached recordings of spontaneous truncated action currents were made at 30–32°C in ECS as per Zaki and Barrett-Jolley (2002). Patch pipettes with a resistance of 4–6 MΩ were filled with an intracellular solution containing the following (in mm); 130 K-gluconate, 10 HEPES, 10 EGTA, 1 CaCl2 and 1 MgCl2, pH 7.2 with KOH. A seal was obtained (>50 MΩ) and the action current frequency allowed to stabilize for at least 10 min before recording, although a qualitative assessment of the tonic versus bursting nature of the firing (see Data analysis; Action currents) could be made after 5 min of recording. Action currents were recorded in the voltage-clamp configuration at a holding potential that maintained a 0 pA leak current (Perkins, 2006) and filtered at 5 kHz using an 8-pole low-pass Bessel filter.
Recordings of mIPSCs, sIPSCs, mEPSCs, and sEPSCs.
Whole-cell voltage-clamp recordings of mIPSCs were obtained from mpd neurons at 35°C in ECS containing 2 mm kynurenic acid (Sigma Chemicals) and 0.5 μm TTX (Tocris Bioscience) to block ionotropic glutamatergic transmission and action potentials, respectively. Patch pipettes with a resistance of 4–6 MΩ were filled with an intracellular solution containing the following (in mm); 135 CsCl, 10 HEPES, 10 EGTA, 1 MgCl2, 1 CaCl2, 2 Mg-ATP, and 5 QX-314 (Alamone Labs), 290–300 mOsm, pH 7.2–7.3 with CsOH. mIPSCs were recorded at a holding potential of −60 mV. In a subset of experiments, sEPSCs/sIPSCs and mEPSCs/mIPSCs were recorded from the same mpd neuron at 35°C in ECS in the absence and presence of TTX, respectively, using a low Cl− (12 mm) intracellular solution. In these recordings, patch pipettes were filled with an intracellular solution containing the following (in mm): 135 CH3O3SCs, 8 CsCl, 10 HEPES, 10 EGTA, 1 MgCl2, 1 CaCl2, 300–310 mOsm, pH 7.2–7.3 with CsOH. Under such recording conditions, the calculated (pClamp Version 8.2) and experimentally verified EGABA and EGlutamate were −63 and 0 mV, respectively. Therefore, EPSCs and IPSCs (miniature and spontaneous) were recorded at a holding potential of −63 and 0 mV, respectively. In both sets of experiments, currents were filtered at 2 kHz using an 8-pole low-pass Bessel filter. The series resistance was between 8 and 20 MΩ with up to 80% compensation. Only cells with a stable access resistance were used and experiments were aborted for series resistance changes > 20%.
Recording of tonic GABA and glutamate currents.
Whole-cell voltage-clamp recordings to determine the presence of tonic GABA (IGABA) and glutamate (IGlutamate) currents in mpd neurons were made at 35°C in normal or low Mg2+ (0.5 mm) ECS, respectively, also containing the nonselective ionotropic glutamate antagonist kynurenic acid (2 mm) or the GABAAR antagonist bicuculline (30 μm), respectively. For IGABA measurements, recordings were made at a holding potential of −60 mV with an intracellular solution containing the following (in mm): 135 CsCl, 10 HEPES, 10 EGTA, 1 MgCl2, 1 CaCl2, 2 Mg-ATP and 5 QX-314, 290–300 mOsm, pH 7.2–7.3 with CsOH. For IGlutamate measurements, recordings were made at a holding potential of +40 mV with an intracellular solution containing the following (in mm): 135 CH3O3SCs, 1 EGTA, 10 HEPES, 5 TEA-Cl, 1 MgCl, 0.5 Na-GTP, 2 Mg-ATP, and 5 Tris phosphocreatine, 280–290 mOsm, pH 7.2–7.3 with CsOH. Bicuculline (30 μm) and kynurenic acid (2 mm) were used to define IGABA and IGlutamate, respectively.
Perforated patch recording.
Perforated patch recordings were made from mpd neurons at 28–30°C (to minimize the likelihood of membrane rupture and whole-cell configuration) in ECS additionally containing 1 μm CGP53432 to block metabotropic GABAB receptors. Patch pipettes with a resistance of 4–6 MΩ were tip filled with a prefiltered intracellular solution containing the following (in mm): 130 KCl, 10 HEPES, 0.1 EGTA, 1 MgCl2, 0.1 CaCl2, pH 7.2 with KOH, and then back-filled with the same solution also containing 75 μg ml−1 gramicidin (diluted from a fresh 75 mg ml−1 stock solution in DMSO). The gramicidin-containing pipette solution was sonicated for >10 min before experimentation and continually vortexed gently throughout the experimentation period. Because of precipitation of the antibiotic at this high concentration, the intracellular solution was prepared fresh every 2–3 h. Upon acquisition of a high-resistance seal (>1 GΩ), the progress of perforation was monitored in voltage-clamp mode (holding potential −60 mV) by observing the slow development (typically <45 min) of capacitance transients in response to −5 mV hyperpolarizing steps. The high Cl− concentration of the pipette solution ensured that undesired membrane rupture could be monitored as a sudden increase in the amplitude of the capacitance transients and the subsequent appearance of inwardly directed GABAergic synaptic currents. Such recordings were immediately discarded. Once fully perforated (<60 MΩ), a current (I) − voltage (V) plot was generated to estimate the EGABA of the recorded cell. Briefly, the recorded cell was held at holding potentials ranging from −50 to −90 mV and GABA (1 mm in pipette) was locally applied from a second patch pipette (situated ∼30–40 μm from cell) to the cell soma by pressure injection (10 ms duration, 2–5 psi) using a Picospritzer II system (Intracel). At each holding potential, the response to GABA was measured from a minimum of three consecutive applications presented at ∼10 s intervals to prevent GABAAR desensitization. Throughout the recording, the GABA response at a holding potential of −60 mV was assessed continually (i.e., after each change of holding potential) to ensure that the perforated patch had not ruptured or the perforation had not improved. The liquid junction potential, estimated to be 3.4 mV (using pClamp Version 8.2), was left uncorrected. The recording configuration was switched to the current-clamp mode to confirm the EGABA measured under voltage-clamp conditions and to measure the resting membrane potential (VMembrane). Note that TTX (0.5 μm) was bath applied to spontaneously firing neurons and the VMembrane measured immediately after cessation of action potential firing. The voltage drop arising from the series resistance was corrected using the simulated bridge balance circuitry of the amplifier. All GABA-induced currents were filtered at 5 kHz using an 8-pole low-pass Bessel filter.
Data analysis
All recordings were analyzed offline using the Strathclyde Electrophysiology Software (Electrophysiology Data Recorder [WinEDR] and Whole Cell analysis Program [WinWCP]; courtesy of Dr J. Dempster, University of Strathclyde).
Whole-cell current-clamp recordings
Using WinEDR, the presence or absence of an LTS after the application of a hyperpolarizing current injection was confirmed by the inspection of individual recordings. The input resistance of mpd neurons was calculated offline using Ohm's law (RInput = VMembrane/IInject). The mean input resistance was calculated from three to five current injections/cell. Current pulses were rejected if a steady plateau phase was not reached or if the charging of the cell membrane was contaminated by truncated action potentials.
Action currents
Using WinEDR, events were detected (offline) using a method based on a rate of rise threshold (60–120 pA/ms) that was specific for individual cells. Any electrical noise detected as an event was removed and any undetected action currents were included after a manual inspection. Firing frequency and interevent interval (IEI) were calculated using WinEDR. In common with the rat, the firing of mouse mpd neurons was heterogeneous, displaying “burst-like” or “tonic” patterns of firing. To determine and quantify the steroid effect reliably, the action of 5α3α-THPROG was restricted to “tonically” firing cells. Cells with a coefficient of variation (calculated as the ratio of the SD of the IEI to the mean IEI) of ≤ 0.5 were deemed to display tonic firing (21% of all cells; Yang et al., 2007). A mean frequency and IEI before and after drug application was calculated for a minimum of 6 min of recording time using 90 s bins (i.e., 4 bins) for each experimental condition. In experiments investigating the actions of 5α3α-THPROG and 4,5,6,7-tetrahydroisoxazolo[4,5-c]pyridin-3-ol (THIP), a minimum perfusion period of 10 and 4 min, respectively, were allowed before the drug effect upon the action current frequency was assessed. Note that THIP required a reduced perfusion time due to the more rapid onset of action compared with 5α3α-THPROG as assessed in voltage-clamp experiments of THIP-sensitive neurons (e.g., thalamic ventrobasalis neurons; Belelli et al., 2005). To allow a meaningful quantitative comparison across experiments, data were normalized to the control frequency. The effects of 5α3α-THPROG and THIP upon the spontaneous firing were quantified after 10 and 4 min of bath application, respectively. Specifically, for every cell, the percentage change in action current firing relative to a 6 min control section was calculated for each 90 s bin throughout the recording. For each cell, sensitivity to drug action was determined by comparing the percentage change of 4 90 s bins (i.e., 6 min) under control conditions and after drug application. For each drug treatment, a mean percentage change in action current firing was then calculated and statistically analyzed.
Voltage-clamp recordings: analysis of synaptic currents
Individual events were detected in WinEDR using an amplitude threshold detection algorithm (−4 pA threshold, 3 ms duration) and visually inspected for validity (e.g., any noise that reached the threshold or traces that contained multiple events were rejected). Accepted events (a minimum of 40 for each experimental condition) were digitally averaged. mIPSCs were analyzed with regard to peak amplitude, rise time, and decay kinetics (T70% values, the time taken for the amplitude of each event to decay to 70% of peak). Events with rise times >0.7 ms (representing <1% of total) were discarded to eliminate from the analysis events subject to dendritic filtering. The decay phase of the digitally averaged event was then fitted with single exponential, [Y(t) = A*exp(−t/τ)], and biexponential functions, [Y(t) = A1*exp(−t/τ1)+ A2*exp(−t/τ2)]. An F test was then used to establish whether the decay was best described by a mono- or biexponential fit, as indicated by a decrease in the SD of the residuals. Because all resultant averaged mIPSCs were best described by a biexponential equation, a mean weighted decay constant (τw) was then calculated to describe the relative contribution of each decay component according to the following equation: τw = [A1/(A1 + A2)]*τ1 + [A2/(A1 + A2)]*τ2, where A1 and A2 describe the relative contribution that τ1 and τ2 make to the overall decay time course, respectively.
The frequency of mIPSCs, sIPSCs, and sEPSCs was determined for each cell in the absence and presence of drug (e.g., 5α3α-THPROG and THIP) by counting the number of events in 20 s bins over a minimum of 2 separate 1 min periods (first and last minute) of a 5 min control and drug section. Events were detected (offline) in WinEDR using a method based on a rate of rise time threshold that was specific for individual cells and that allows detection of the slowest events (∼40 pA/ms). Recordings were then visually inspected to ensure that all events were included and any detected spurious noise removed. A mean frequency and IEI was then calculated. The sensitivity of each IPSC/EPSC parameter to a drug action was determined for each neuron by comparing their cumulative distribution before and after drug treatment with the Kolmogorov–Smirnov (KS) test (see Statistical analysis).
Analysis of the tonic and drug-evoked current
The GABAergic and glutamatergic tonic current and the drug-induced current were calculated as the difference in the mean baseline current before and after application of bicuculline (30 μm IGABA), kynurenic acid (2 mm IGlutamate) or the drug, respectively. The holding current and root mean square (RMS; i.e., SD) were sampled every 102.4 ms (bicuculline and THIP) or 51.2 ms (AP-5 and kynurenic acid) over a 1 min period for each recording condition. At the sampling rate of 10 kHz, 1024 baseline points for each 102.4 ms and 512 baseline points for each 51.2 ms provided one data point, respectively. Epochs containing synaptic events or an unstable baseline were excluded from the analysis. A minimum of 100 data points were measured for each recording condition (i.e., in the absence and presence of drug). To ensure that a drug effect was genuine and not simply due to a temporal “drift” in the holding current, two separate 1 min sections of the holding current were analyzed during the control period (C1 and C2). Similarly, a 1 min section was analyzed after drug application (D) once the drug effect had reached a plateau. Similar temporal intervals were selected between C1 and C2 and C2 and D to control for temporally comparable drift during the control period and after drug application. The mean DC values for each epoch of C1 and C2 were pooled and the SD calculated. A drug effect was considered genuine if the absolute value of change in the holding current (i.e., D − C2) was greater than twice the SD associated with the DC measurements of the control period.
Perforated patch recordings; determination of EGABA and VMembrane
Current injections were manually detected offline using WinEDR and the input resistance of cells calculated in a manner similar to that described above using WinWCP. At each holding potential tested (cell specific), the GABA-induced currents after local pressure application were manually detected in WinEDR and events exported to WinWCP, where the peak amplitude was measured. For each holding potential, the average amplitude of a minimum of three GABA-evoked currents was calculated. For each cell, the GABA-induced currents were normalized to the holding potential at which the maximum current amplitude was measured to permit a direct comparison of the I–V plots for CTRL WT, ELS WT, and δ0/0 neurons. The reversal potential of the GABA response was calculated from the x-axis intercept of the current (x-axis)–voltage (y-axis) relationship for each neuron. Epochs containing action potentials or an unstable baseline were excluded from the analysis. A minimum of 100 data points were measured for each cell. To calculate the resting membrane potential (VMembrane), it was sampled every 102.4 ms over a stable 1 min recording period. At the sampling rate of 10 kHz, 1024 baseline points for each 102.4 ms provided one data point. The GABA driving force was calculated as the difference between the resting VMembrane and the EGABA for each cell.
Reagents and drugs
All reagents were obtained from either Sigma or Tocris Bioscience unless stated otherwise. Stock solutions of the neurosteroid 5α3α-THPROG (a generous gift from Dr K. Gee, University of California–Irvine) and CGP54432 and TFB-TBOA (both Tocris Bioscience) were prepared as a concentrated stock (1000×) in DMSO and then diluted to the required concentration in extracellular solution. The maximum vehicle concentration (0.1%) had no effect upon receptor function. Stock solutions of bicuculline (Alexis Biochemicals); D-AP-5, RO256981, and kynurenic acid (all Ascent Scientific); tetrodotoxin (Tocris Bioscience); and the δ-preferring agent THIP (a generous gift from Bjarke Ebert, K. Lundbeck, Copenhagen Valby, Denmark) were prepared similarly as concentrated stock solutions (1000×) in distilled water and then diluted to the necessary concentration in the perfusion system. 5α3α-THPROG was applied to the brain slice via the perfusion system (3–6 ml/min) and allowed to infiltrate the slice for a minimum of 10 min before recordings were acquired; THIP and TFB-TBOA were perfused for a shorter time of at least 4 min.
Statistical analysis
All data are presented as the arithmetic mean ± SEM unless stated otherwise. Statistical comparisons were made using Student's t test (paired and unpaired) and regular and repeated-measures ANOVA (one- and two-way), followed by the post hoc Newman–Keuls test, as appropriate; the KS test was used to compare populations of individual events before and after drug application to a given cell using a stringent criterion for significance (p < 0.01). When normalized data are presented, mean values have been calculated averaging the normalized changes calculated for each individual cell. The effects of drug (e.g., 5α3α-THPROG and THIP) upon the pooled normalized frequency of action currents relative to the control were analyzed statistically using a one-way repeated measure ANOVA with post hoc testing. Specifically, the mean percentage change in action current firing was calculated for each 90 s bin throughout the recording for every cell tested and then averaged. The effects of 5α3α-THPROG and THIP upon the spontaneous firing were then quantified after 10 and 4 min, respectively.
Preparation of lysates and Western blotting
Hypothalamic and cerebellum samples were dissected from whole brains and PVN punches were obtained from hypothalamic slices (900 μm thick) prepared as described above. In each instance, samples were immediately placed in ice-cold lysis buffer containing the following (in mm): 500 NaCl, 250 NaF, 125 Tris HCl, 50 NaPPi, 25 EGTA, 5 EDTA, and 1 Na3VO4, along with 0.1% (v/v) mercaptoethanol, 1% Triton X-100 (v/v), and 86 mg ml−1 sucrose. The tissue was homogenized on ice and centrifuged for 10 min at 12,000 rpm at 4°C. The resulting supernatant was collected and the pellet discarded. The protein content of the lysate was determined by the method of Bradford (1976). Proteins of interest (10 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were subsequently incubated for 1 h at room temperature in blocking buffer (5% nonfat dried milk in TBST 20 mm Tris HCl, 150 mm NaCl, 0.5% Tween, pH 7.4). After this, membranes were incubated in a phosphospecific antibody targeted to the N-terminal domain of the GABAAR δ-subunit (1:500; Millipore) overnight at 4°C with gentle shaking. The membranes were washed 5 times with 1× TBST and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5000) for 1 h at room temperature. Subsequently, the membranes were washed with 1× TBST and incubated with a monoclonal anti β-actin antibody (1:5000; Sigma) for 2 h at room temperature with gentle shaking, which acted as an internal loading control. After further washing with 1× TBST, the membranes were again incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5000) for 1 h at room temperature and then washed once more. Total protein was visualized by enhanced chemiluminescence detection (GE Healthcare).
Immunohistochemistry
Visualization of biocytin-filled cells
To confirm that the recorded cells were CRF+ve, biocytin (0.1%) was added to the intracellular electrolyte. Immunohistochemistry for biocytin was performed according to previous protocols (Swinny et al., 2010).
Tissue preparation
Anaesthesia was induced with isoflurane and maintained with urethane (1.25 mg/kg bodyweight, i.p.). The animals were perfused transcardially with 0.9% saline solution for 3 min, followed by 15 min fixation with a fixative consisting of 1% paraformaldehyde and 15% v/v saturated picric acid in 0.1 m phosphate buffer (PB), pH 7.4. This weak fixation protocol allows for the visualization of somatic CRF without the need for pretreating animals with colchicine. The weak fixation also allows for the visualization of integral membrane proteins such as NMDA subunits. The brains were kept in the same fixative solution overnight at 4°C. Coronal sections of the PVN, 50 μm thick, were prepared on a Vibratome and stored in 0.1 m PB containing 0.05% sodium azide.
Immunohistochemical reaction
Nonspecific binding of secondary antibodies was blocked by incubating sections with 20% normal horse serum dissolved in Tris-buffered saline containing 0.3% Triton X-100 (TBS-Tx) for 2 h at room temperature. The following primary antibodies were used: guinea pig anti CRF (1:2000; Bachem); rabbit anti-excitatory amino acid transporter 1 (EAAT1, 1:250; Abcam); rabbit anti GABAAR δ-subunit (1:500, a gift from Werner Sieghart); mouse anti-glial fibrillary acid protein (GFAP, 1:2000; Neuromab); mouse anti-HuC (1:500; Invitrogen); rabbit anti-vGLUT2 (1:1000; Synaptic Systems); mouse anti-vGLUT2 (1:4000; Neuromab; mouse anti-GluN1 (1:1000; Neuromab); and mouse anti-GluN2B (Neuromab). The primary antibodies were diluted in TBS-Tx overnight at 4°C. After washing with TBS-Tx, sections were incubated in a mixture of appropriate secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen), indocarbocyanine (Cy3; Jackson ImmunoResearch), and indodicarbocyanine (Cy5; Jackson ImmunoResearch) for 2 h at room temperature. Sections were washed in TBS-Tx and mounted in Vectashield (Vector Laboratories). Method specificity was also tested by omitting the primary antibodies in the incubation sequence. To confirm the absence of crossreactivity between IgGs in double- and triple-immunolabeling experiments, some sections were processed through the same immunocytochemical sequence, except that only an individual primary antibody was applied with the full complement of secondary antibodies.
Image acquisition
Sections were examined with a confocal laser-scanning microscope (LSM710; Zeiss) using either a Plan Apochromatic 63× DIC oil objective (numerical aperture [NA] 1.4) or a Plan Apochromatic 100× DIC oil objective (NA 1.46). Z-stacks were used for routine evaluation of the labeling. All images presented represent a single optical section. These images were acquired using sequential acquisition of the different channels to avoid cross talk between fluorophores, with the pinholes adjusted to one airy unit for all channels. Images were processed with Zen 2008 Light Edition software (Zeiss) and exported into Adobe Photoshop. Only brightness and contrast were adjusted for the whole frame and no part of a frame was enhanced or modified in any way.
Quantification of vGLUT2, CRF, GFAP, EAAT1, GLUN1, and GLUN2B immunoreactivity in the PVN of CTRL and ELS WT mice and δ0/0 and WT mice
All sections were processed and imaged under identical conditions and analyses were performed blind. vGLUT2, GLUN1, and GLUN2B immunoreactivity presented as distinct, individual puncta. We therefore were able to quantify the density of expression, expressed as the number of individual puncta per 10 μm2 for vGLUT2 and puncta per 1000 μm2 for GLUN. In contrast to the individual or discrete punctate immunoreactivity pattern of vGLUT2, CRF immunoreactivity pattern within the PVN presented as a combination of signal located within somata and axonal varicosities. To estimate changes in the levels of CRF within both somata and axonal varicosities, which would infer total CRF content of the PVN, we quantified the fluorescence intensity in control and ELS tissue. Similarly, because GFAP signal is not located in astrocytic cell bodies, fluorescence intensity was also used to estimate the relative levels of GFAP and EAAT1 expression. The imaging and quantification was performed as follows: within a tissue section, 3 fields of view (FOVs) were selected within the PVN. Z-stacks consisting of 3 optical sections spaced 5 μm apart in the Z-plane were acquired for each FOV. The dimensions of each optical section were 85 μm × 85 μm × 1 μm in the X-Y-Z planes. Within an optical section, the number of vGLUT2 puncta was manually counted or the fluorescence intensity for CRF, GFAP, and EAAT1 was measured using ImageJ software. A value for each FOV was obtained by computing the average from the optical sections contained within a FOV. The degree of background labeling for individual experiments was determined in tissue sections reacted with only the secondary antibodies. Background fluorescence was then subtracted from the values obtained for individual markers. The means ± SD (vGLUT2 in puncta/10 μm2; GLUN1 and GLUN2B in puncta per 1000 μm2; or CRF, GFAP, or EAAT1 fluorescence intensity in arbitrary units) for all FOV between sections and between either control or ELS animals was compared for statistical differences using Kruskal–Wallis one-way ANOVA. These values were then pooled because there were no statistical differences (p > 0.05) between the values for FOV between sections and between animals. All quantitative immunohistochemical data were thus derived from three FOVs per tissue section, three tissue sections per animal (except for GLUN1 and GLUN2B immunoreactivity analysis in WT control, when two tissue sections per animal were used), three control and three ELS animals (all from six distinct litters). The average value from all FOVs and sections per animal were computed for individual animals. This average value for an individual animal was then considered an n of 1.
Results
Validation of a mouse ELS model
To investigate the impact of ELS on mouse hypothalamic neurotransmitter function and neurosteroid modulation, we adopted a model of fragmented maternal care, as described previously (Rice et al., 2008 and see Materials and Methods). Confirming the validity of the model and in agreement with Rice et al. (2008), dams from the ELS group more frequently left the nest (i.e., exhibited an increased number of sorties) compared with controls (F(1,52) = 10.9, p < 0.01, two-way ANOVA, n = 7 litters for each group; Fig. 1A), whereas the total duration of the dam–pup interactions did not differ between the ELS and control groups (effect of day: F(5,52) = 0.26, p = 0.93; effect of treatment: F(1,52) = 1.04, p = 0.31, two-way ANOVA; Fig. 1B). In agreement with abnormal maternal care, the ELS pup weight was significantly reduced at P9, at weaning, and at adult age compared with CTRL (p < 0.01, unpaired Student's t test; Fig. 1C). Moreover, CRF somatic immunoreactivity in the PVN of ELS-exposed neonatal mice (P22–P28) was significantly greater than that detected in control tissue (fluorescence intensity in arbitrary units for control: 16.13 ± 0.07; for ELS: 24.67 ± 0.05; n = 3 for both; p < 0.001, unpaired Student's t test; Fig. 1D) and the upregulation was maintained through to adulthood (>2 months; control: 21.06 ± 0.04; ELS: 26.74 ± 0.09; n = 3 for both; p = 0.001, unpaired Student's t test). Collectively, these data show that the ELS paradigm used in this study results in enduring metabolic and neurochemical changes that appear to manifest as a consequence of impoverished maternal care in early life.
Figure 1.

Maternal and CRF expression profile in WT and ELS mice. A, Graph summarizing the mean number of sorties from the nest for control (CTRL: black, n = 7) and ELS (red, n = 7) WT dams from P3–P8. A significant difference in the number of maternal sorties was observed between the two treatment groups (F(1,52) = 10.9, p < 0.01), whereas the treatment day had no significant effect (F(5,52) = 1.01, p = 0.42). B, Graph summarizing the mean dam–pup interaction time over P3–P8 for the same litters as above. There was no effect of day (F(5,52) = 0.26, p = 0.93) or treatment (F(1,52) = 1.04, p = 0.31) upon the time spent in the nest (two-way ANOVA). C, Bar graphs summarizing the mean weights of CTRL (black) and ELS (red) WT at P9, P18–P24, and 8 weeks of age (CTRL, n = 36–42; ELS, n = 35–63. D, Representative images of CRF expression in the neonatal (P22–P28) PVN of CTRL and ELS WT mice. Note upregulation of somatic CRF expression in the PVN of neonatal ELS WT mice. Tissue sections were reacted and imaged under identical conditions. D1, D3, HuC (a pan-neuronal marker) was used at all times to confirm that the regions investigated are comparable. D2, CRF immunoreactivity within CTRL WT tissue is expressed within isolated somata and varicose processes. D4, In contrast, somatic CRF immunoreactivity is evident to a greater extent throughout the PVN of ELS WT tissue. CRF immunoreactivity in ELS tissue was significantly greater than that detected in CTRL tissue (p < 0.001, unpaired Student's t test). Scale bars, 100 μm. Furthermore, this observed increase in CRF expression was maintained through to adulthood (> 2 months; see Results).
ELS enhances glutamatergic drive onto neonatal mpd neurons and impairs astrocytic glutamate reuptake
As a prelude to investigating the actions of ELS on neurotransmitter function of the hypothalamic stress neurocircuitry, identification of distinct parvocellular neuronal populations was achieved initially using a combination of anatomical, electrophysiological, and immunohistochemical criteria, as described in detail in the Materials and Methods. Our analysis suggests that similar anatomical and electrophysiological criteria to those previously applied to rat hypothalamic slices (Luther et al., 2002; Melnick et al., 2007) can be used to characterize mouse parvocellular neurons. Specifically, mouse parvocellular neurons located in the mpd and dp/mpv regions were confirmed to correspond to the neuroendocrine CRF-releasing (LTS−ve) and preautonomic (LTS-expressing, LTS+ve) neurons respectively (Luther et al., 2002; Fig. 2A). Additional confirmation that the recorded cells were CRF+ve was obtained by post hoc immunohistochemical analysis of biocytin-filled cells (Fig. 2B).
An altered glutamatergic drive in the PVN has previously been inferred from molecular studies in rodent models of chronic stress (Flak et al., 2009; Miklós and Kovács, 2012). Therefore, we investigated whether ELS altered the balance of excitatory (Boudaba et al., 1997; Herman et al., 2002; Ulrich-Lai et al., 2011) to inhibitory drive (Zaki and Barrett-Jolley, 2002; Biancardi et al., 2010) onto mpd neurons. Miniature and spontaneous EPSCs (mediated by glutamate) and IPSCs (mediated by GABA) were recorded from the same neuron by adjusting the holding potential to EGABA (−63 mV) and EGlutamate (0 mV), respectively (see Materials and Methods). The frequency of the mIPSCs was similar for the control and ELS groups (p > 0.05, unpaired Student's t test) whereas the amplitude exhibited an ∼30% decrease (p < 0.05, unpaired Student's t test; Fig. 2D, Table 1). The ELS sIPSC frequency appeared increased compared with control, but only approached significance (p = 0.06, unpaired Student's t test) with no changes in the sIPSC amplitude (p > 0.05, unpaired Student's t test; Fig. 2C,D, Table 1). In contrast, the frequency of sEPSCs was significantly increased for ELS compared with control mpd neurons (p < 0.05, unpaired Student's t test; Fig. 2C,E, Table 1). Note that no difference in the frequency of sEPSCs was observed between male or female mice bred under control conditions (male: 2.7 ± 0.5 Hz, n = 16; female: 2.4 ± 0.3 Hz, n = 16, p = 0.47, unpaired Student's t test) or for those exposed to ELS (male: 4.2 ± 0.6 Hz, n = 13; female: 3.9 ± 0.8 Hz, n = 13, p = 0.75, unpaired Student's t test), so the data were pooled. In the presence of TTX, the mEPSC frequency for ELS mpd neurons was similarly increased compared with the WT (p < 0.05, unpaired Student's t test; Fig. 2E, Table 1). In addition, the amplitude of the sEPSCs, but not mEPSCs, appeared modestly increased (p < 0.05, unpaired Student's t test; Table 1). In agreement with the altered function, immunohistochemistry of the PVN revealed a significant increase in the expression of the vesicular glutamate transporter vGLUT2, a selective marker of presynaptic glutamatergic input, in ELS compared with control (vGLUT2 clusters per 10 μm2; control: 1.53 ± 0.08; ELS: 2.44 ± 0.09; n = 3 animals for both; p < 0.05, unpaired Student's t test; Fig. 2F). Next, we explored possible additional alterations in glutamatergic transmission mediated by extrasynaptic receptors (Groc et al., 2009; Paoletti et al., 2013). The tonic glutamatergic conductance, revealed by the magnitude of the current induced by kynurenic acid (2 mm), for ELS mpd neurons was significantly greater than control (p < 0.05, unpaired Student's t test; Fig. 3A, Table 1). Note that no difference in the tonic conductance was observed between male or female mice bred under control conditions (male: −20 ± 7 pA, n = 6; female: −17 ± 9 pA, n = 6; p = 0.84, unpaired Student's t test) or exposed to ELS (male: −35 ± 11 pA, n = 3; female: −34 ± 7 pA, n = 4; p = 0.71, unpaired Student's t test) conditions, so the data were pooled. Regardless of magnitude, for both control and ELS neurons, this current was primarily mediated by tonic activation of NMDA receptors (NMDARs); that is, the percentage of the current induced by 2 mm kyneurenic acid that was AP-5 sensitive (control: 73 ± 39, n = 6; ELS: 80 ± 10, n = 4; p > 0.05 ELS vs CTRL, one-way repeated-measure ANOVA [RMA]). In contrast, no resident GABAAR-mediated tonic current was detected in control or ELS neurons (bicuculline 30 μm-evoked current: control: 4.4 ± 2.8 pA, n = 7; ELS: 3.6 ± 2.4 pA, n = 4). Impaired glial glutamate transporter function has been implicated in glutamatergic dysfunction (Oliet et al., 2001; Fleming et al., 2011; Tasker et al., 2012; Gómez-Galán et al., 2013). The transporter inhibitor TFB-TBOA (200 nm; Shimamoto et al., 2004) produced an inward, kynurenic acid-sensitive current that was significantly greater for control neurons compared with the ELS group (p < 0.05, unpaired Student's t test; Fig. 3B,C, Table 1). In accordance with the functional data above, the level of immunoreactivity (mean ± SD, arbitrary units) for the EAAT1 was significantly lower in the PVN of tissue obtained from ELS mice compared with control (control: 69 ± 2 vs ELS: 59 ± 2, n = 3 animals for both; p = 0.004, unpaired Student's t test; Fig. 3D). Low-power overview examination of the immunoreactivity pattern for GFAP, the cytoskeleton protein found in astrocytes, revealed no apparent differences in the distribution of the signal throughout the PVN of control and ELS mice (Fig. 3D1,D4). This suggests that the number of astrocytes may not be significantly different in control and ELS mice. This could not be confirmed quantitatively because the GFAP signal is not located in astrocytic cell bodies. However, high-resolution quantification of the intensity of GFAP immunoreactivity revealed it to be significantly lower in the PVN of tissue obtained from ELS mice compared with control (control: 5.5 ± 0.4 vs ELS, 2.3 ± 0.2, n = 3 animals for both; p = 0.001, unpaired Student's t test; Fig. 3D3,D6). Furthermore, at the high-resolution visualization, GFAP-immunoreactive arbors were noticeably thinner in ELS compared with control sections, an ELS-induced alteration in astrocyte morphology that likely results in the altered levels of EAAT1 expression. The changes in the EAAT1 and GFAP expression patterns suggest a combined structural and neurochemical remodeling of astrocytes as a result of ELS. These findings are consistent with an impaired glial glutamate transporter function in the PVN of ELS mice (Fleming et al., 2011). Together, our observations suggest that ELS alters the ratio of excitatory to inhibitory inputs onto neonatal (P18–P26) mpd neurons in favor of the former, primarily as a consequence of a considerably greater glutamate-mediated excitatory drive.
Table 1.
Summary of the properties of excitatory and inhibitory phasic currents and extrasynaptic currents recorded from mpd neurons of WT CTRL, WT ELS, and δ0/0 mice
| Excitatory and inhibitory phasic currents | ||||||
|---|---|---|---|---|---|---|
| WT CTRL |
WT ELS |
δ0/0 |
||||
| mEPSC (n = 15) | sEPSC (n = 32) | mEPSC (n = 14) | sEPSC (n = 26) | mEPSC (n = 16) | sEPSC (n = 32) | |
| Peak amplitude (pA) | −32 ± 3 | −26 ± 1 | −32 ± 2 | −30 ± 2† | 31 ± 2 | 28 ± 1 |
| Rise time (ms) | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.1 |
| τW (ms) | 1.6 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.7 ± 0.1 | 1.7 ± 0.2 | 1.6 ± 0.1 |
| Frequency (Hz) | 1.9 ± 0.3 | 2.5 ± 0.3 | 5 ± 1.5† | 4 ± 0.5† | 2.3 ± 0.5 | 6.1 ± 1*†† |
| mIPSC (n = 9) | sIPSC (n = 25) | mIPSC (n = 11) | sIPSC (n = 18) | mIPSC (n = 11) | sIPSC (n = 18) | |
|---|---|---|---|---|---|---|
| Peak amplitude (pA) | 60 ± 2 | 49 ± 3 | 41 ± 3† | 45 ± 5 | 54 ± 5 | 49 ± 3 |
| Rise time (ms) | 0.4 ± 0.1 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 |
| τW (ms) | 6.8 ± 0.6 | 7.7 ± 0.4 | 7.1 ± 0.5 | 6.7 ± 0.5 | 6.8 ± 0.4 | 7.4 ± 0.5 |
| Frequency (Hz) | 1.7 ± 0.7 | 2.1 ± 0.4 | 2.4 ± 0.9 | 3.8 ± 0.8 | 1.9 ± 0.7 | 2.7 ± 0.5 |
| Extrasynaptic currents | |||
|---|---|---|---|
| WT CTRL | WT ELS | δ0/0 | |
| IKynurenic acid (pA) | 18 ± 5 | 37 ± 6‡ | 40 ± 5‡‡ |
| IAP-5 (pA) | 11 ± 7 | 28 ± 5 | 30 ± 10 |
| ITFB-TBOA (pA) | 55 ± 6 | 24 ± 8‡‡ | 32 ± 5‡ |
| ITHIP (pA) | −18 ± 3 | - | −15 ± 5 |
*p < 0.05, **p < 0.01 unpaired Student's t test mEPSC versus sEPSC for δ0/0 neurons.
†p < 0.05,
††p < 0.01 unpaired Student's t test WT ELS or δ0/0 versus WT CTRL. Note that recordings were made using low intracellular Cl− (12 mm; see SI) so that EPSCs and IPSCs could be recorded from the same cell at a holding potential of −63 and 0 mV, respectively.
‡p < 0.05,
‡‡p < 0.01 WT ELS or δ0/0 versus WT CTRL (WT CTRL, n = 6–12; WT ELS, n = 4–7; δ0/0, n = 4–17). Drug concentrations used: kynurenic acid 2 mm, AP-5 50 μm, TFB-TBOA 200 nm, and THIP 1 μm.
Figure 3.
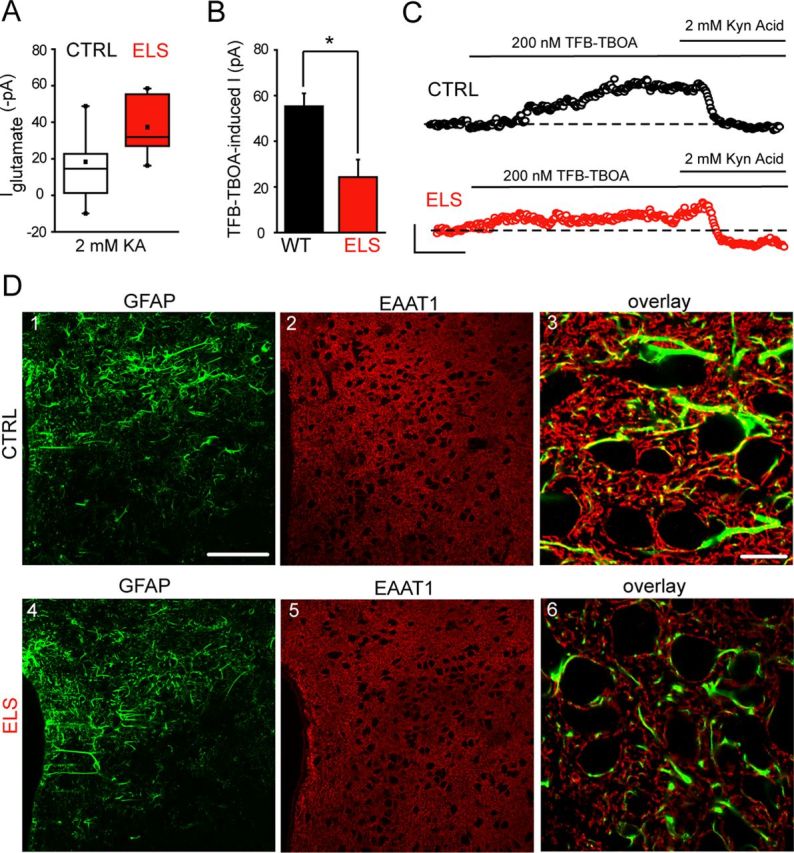
Exposure to ELS greatly increases mpd neuron glutamatergic conductance and impairs astrocytic glutamate reuptake. A, Box plot showing the mean IGlutamate recorded from mpd neurons derived from control (CTRL; white) and ELS (red) WT mice upon the bath application of kynurenic acid (2 mm). Note that the mean IGlutamate is greater for ELS mpd neurons compared with CTRL WT. p < 0.05, unpaired Student's t test (CTRL, n = 12; ELS, n = 7). B, Bar graph illustrating the mean TFB-TBOA (200 nm)-induced current recorded from CTRL (black) and ELS (red) WT mpd neurons. *p < 0.05 CTRL vs ELS, unpaired Student's t test (CTRL, n = 6; ELS, n = 7). C, Representative whole-cell voltage-clamp recordings of mpd neurons derived from CTRL (black) and ELS (red) WT mice under control conditions after the bath application of TFB-TBOA (200 nm) and in the combined presence of TFB-TBOA (200 nm) and kynurenic acid (2 mm). The TFB-TBOA-induced current was reversed in both cases by the subsequent bath application of kynurenic acid (2 mm). Note that the tonic IGlutamate after kynurenic application was significantly greater for the ELS compared with CTRL WT neuron. The representative traces were generated by averaging the mean DC of 100 ms epochs every 2 s with points falling on to the decay of sEPSCs or those containing spurious noise being discarded from the analysis. The dotted line represents the holding current before drug application. Scale bars: y = 50 pA, x = 100 s. D, Immunolocalization of the astrocytic cytoskeleton protein GFAP and the glial-specific glutamate transporter EAAT1 demonstrating their relative levels of expression in the PVN of tissue from control and ELS mice reacted and imaged under identical conditions. Low-power overview examination indicated that the distribution of GFAP-immunoreactive profiles throughout the PVN was not noticeably different in tissue from control (D1) and ELS (D4) mice. However, high resolution inspection of GFAP immunoreactive arbours revealed those in control tissue (D3) to be noticeably thicker compared with those in ELS tissue (D6), suggesting ELS-induced structural plasticity of astrocytes. The levels of EAAT1 immunoreactivity are consistently higher in control (D2,D3) compared with ELS (D5,D6) tissue in accordance with the functional data above. Scale bars: D1, D2, D4, D5, 100 μm; D3, D6, 10 μm.
ELS indirectly compromises inhibitory transmission: a role for GABAAR-active neurosteroids?
Although the ELS experience appeared to affect modestly GABAAR-mediated inhibitory transmission compared with glutamate-mediated excitatory transmission, additional inhibitory mechanisms may fail. It is well established that certain endogenously occurring neurosteroids (e.g., 5α3α-THPROG) potently and selectively enhance GABAAR function at low nanomolar concentrations (Belelli and Lambert, 2005). Intriguingly, neurosteroid levels change dynamically in response to stress challenges (Purdy et al., 1991; Barbaccia et al., 1996; Biggio et al., 2007) and therefore may “fine-tune” GABAAR function of PVN neurons to influence HPA axis activity. Therefore, we explored how ELS may influence the actions of 5α3α-THPROG upon the frequency of truncated action currents recorded from spontaneously, tonically firing mpd neurons of the neonatal hypothalamus (P18–P26), as described previously (Perkins, 2006; Yang et al., 2007). For WT control, ∼2/3 of all mouse parvocellular neurons tested exhibited spontaneous action current firing (Table 2). Such activity was completely abolished by the ionotropic glutamate receptor antagonist (2 mm) kynurenic acid (n = 3; data not shown) and their frequency was increased by the GABAAR antagonist bicuculline (30 μm; 254 ± 56% of control, n = 10; p < 0.05, one-way RMA). Similar to spinally projecting parvocellular neurons (Womack et al., 2006), the action current frequency of control mpd neurons was decreased by physiological concentrations of 5α3α-THPROG (10 and 100 nm) to 60 ± 7% (5 of 8 cells) and 24 ± 3% of control (8 of 8 cells), respectively (p < 0.01 for both concentrations, one-way RMA; Fig. 4A,B, left), an effect that was reversed by bicuculline (data not shown). No differences in the effect of 100 nm 5α3α-THPROG were observed between neurons derived from either male (21 ± 6% of control, n = 4) or female (34 ± 7% of control, n = 4) mice, so the data were pooled (p = 0.24 two-way RMA). Moreover, dp/mpv neurons were similarly sensitive to 5α3α-THPROG 100 nm (p > 0.05, two-way RMA; Fig. 4A, right). We subsequently focused on the CRF+ve mpd neurons. For these neurons, the experience of fragmented dam–pup interactions (i.e., ELS behavioral paradigm) severely compromised the potent inhibitory actions of the neurosteroid. Therefore, although the frequency of action currents was similar for control and ELS WT neurons (Table 2), in ELS mpd neurons, 5α3α-THPROG (100 nm) was ineffective (p > 0.05 vs ELS control, but p < 0.01 for ELS vs WT control, two-way RMA; Fig. 4A,B). Note that, similar to the control, no difference in the effect of 100 nm 5α3α-THPROG was observed upon neurons derived from male (89 ± 5% of control, n = 3) or female (98 ± 13% of control, n = 3) ELS mice, so the data were pooled (p = 0.55, two-way RMA).
Table 2.
Summary of the spontaneous action current firing properties recorded from mpd neurons of WT CTRL, WT ELS, and δ0/0 mice
| Cells firing spontaneously | Cells tonically firing | Mean frequency tonically firing cells (Hz) | |
|---|---|---|---|
| WT CTRL | 69% (327/476 cells) | 21% | 6.1 ± 0.5 |
| WT ELS | 65% (59/91 cells) | 25% | 5.1 ± 0.7 |
| δ0/0 | 69% (141/204 cells) | 22% | 5.7 ± 0.7 |
| CTRL | +TBOA (200 nm) | +LiAc (1–1.5 mm) | +RO-256981 (100 nm) | |
|---|---|---|---|---|
| WT | 6.5 ± 0.8 | 7.4 ± 1.9 | — | — |
| ELS | 5.1 ± 0.7 | — | 5 ± 0.7 | 5.9 ± 0.8 |
| δ0/0 | 5.7 ± 0.7 | — | 6.6 ± 0.9 | 5.5 ± 0.9 |
p > 0.05 unpaired Student's t test WT ELS or δ0/0 versus WT CTRL mean frequency (WT CTRL, n = 92; WT ELS, n = 20; δ0/0, n = 37).
p > 0.05 unpaired Student's t test for each treatment group versus respective control. (WT CTRL, n = 10–13; WT ELS, n = 12–20; δ0/0, n = 9–37).
Figure 4.

Exposure to ELS blunts 5α3α-THPROG suppression of mpd neuron firing and alters Cl− homeostasis in these neurons. A, Left, Summary graph depicting the time course of the effect (expressed as percentage of the control frequency) of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from control (CTRL; black, n = 8) and ELS (red, n = 6) WT mpd neurons. Right: Bar graph summarizing the effect of 100 nm 5α3α-THPROG upon the spontaneous firing of WT CTRL (black, n = 8), ELS (red, n = 6) mpd, and CTRL dp/mpv neurons (gray, n = 3). *p < 0.05 one-way RMA vs control; †p < 0.001 ELS vs CTRL WT mpd neurons, two-way RMA. B, Sections (10 s) of cell-attached recordings obtained from representative CTRL and ELS WT mpd neurons before and 10 min after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 2 s. C, Cumulative probability plots of the decay of all mIPSCs (described by T70 values) collected from steroid-sensitive CTRL (left, n = 8) and ELS (right, n = 5) WT mpd neurons before (CTRL: black, ELS: red) and after (gray) the bath application of 100 nm 5α3α-THPROG. The insets illustrate the superimposed normalized ensemble averages of mIPSCs recorded from representative CTRL (left) and ELS (right) WT mpd neurons before and after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 10 ms. D, Paired plots of the τW value before and after treatment with 100 nm 5α3α-THPROG for each CTRL (left; black) and ELS (right; red) WT neuron tested. ELS neurons deemed neurosteroid insensitive (T70, p > 0.01, KS test) are shown in gray. C, control; NS, in the presence of neurosteroid. E, Bar graph illustrating the impact of 5α3α-THPROG (100 nm) upon the sEPSC (neurosteroid-sensitive) and sIPSC frequency for CTRL (black, n = 6–7) and ELS (red, n = 4–5) WT mpd neurons. *p < 0.05 vs control; p > 0.05 CTRL vs ELS WT, two-way RMA. F, I–V curves of the GABA-evoked response (normalized to the maximum outward current for each individual cell) derived from perforated-patch recordings for CTRL (black, n = 7) and ELS (red, n = 7) WT mpd neurons. Note the rightward shift in the EGABA for mpd neurons derived from ELS mice. The insets illustrate representative current traces recorded at different holding potentials from CTRL (black) and ELS (red) mpd neurons. Scale bars: y = 20 pA, x = 250 ms. G, Box plot showing the mean EGABA obtained in mpd neurons derived from CTRL (white, n = 7) and ELS (red, n = 7) WT mice during perforated patch recordings. The box extends from the 25th percentile to the 75th percentile with the line designating the median value and the point the mean EGABA value. The upward and downward bars illustrate the upper and lower limits of the range of values. p < 0.05 CTRL vs ELS, unpaired Student's t test. H, Bar graph illustrating the mean GABA-driving force (calculated as Vmemb − EGABA) obtained from perforated patch recording of CTRL (black, n = 7) and ELS (red, n = 7) WT mpd neurons. *p < 0.05, unpaired Student's t test.
The neurosteroid insensitivity observed may result from ELS-induced changes in excitatory or inhibitory transmission. Given that GABAARs are the specific target of neurosteroid actions (Belelli and Lambert, 2005), we initially investigated whether ELS altered the interaction of neurosteroids with synaptic GABAARs (e.g., their prolongation of the mIPSC decay; Cooper et al., 1999; Belelli and Lambert, 2005). The sensitivity of mpd neuron GABAARs to neurosteroid action was assessed individually for each cell in paired recordings using equal intra- and extracellular Cl− concentrations (see Materials and Methods). In agreement with the recordings with low intracellular Cl− described above (ELS enhances glutamatergic drive onto mpd neurons and impairs astrocytic glutamte reuptake; Fig. 2, Table 1), ELS caused a significant decrease (∼ 30%) of the mIPSC amplitude compared with control neurons, but had no significant impact on their rise time or decay kinetics (Table 3). 5α3α-THPROG (100 nm) did not affect RMS noise, mIPSC peak amplitude, or rise time in either group (Table 3), but prolonged the mIPSC decay of both control and ELS mpd neurons (% τw increase, control: 43 ± 9, n = 8; ELS: 34 ± 10, n = 7; p < 0.001, one-way RMA vs respective control for both groups, but p > 0.05 for WT control vs ELS; two-way RMA). Analysis of the individual cell sensitivity (p < 0.01 KS test for T70 values, see Materials and Methods) revealed that all control cells (n = 8) and 70% (5 of 7 tested) from the ELS group were sensitive to the neurosteroid (Fig. 4C,D). However, given that the action current discharge of all ELS neurons was insensitive to 5α3α-THPROG (100 nm), changes to the neurosteroid sensitivity of synaptic GABAARs of the ELS group do not account for the loss of steroid efficacy. We investigated next whether a putative GABAAR-mediated presynaptic steroid action (Poisbeau et al., 1997; Haage and Johansson, 1999; Uchida et al., 2002; Ruiz et al., 2010) influenced network-driven transmission (i.e., sIPSC and sEPSC frequency) and if it was affected by the ELS experience. For control neurons, 5α3α-THPROG (100 nm) decreased the sEPSC frequency to 70 ± 3% of control in 7 of 10 cells tested (p < 0.001 vs control, one-way RMA; Fig. 4E), an action likely to contribute to the neurosteroid-evoked suppression of mpd neuronal firing (Fig. 5A and Discussion). In contrast, 5α3α-THPROG (100 nm) had no effect upon the sIPSC frequency (p > 0.05 vs control, one-way RMA; Fig. 4E). However, importantly, the action of 5α3α-THPROG upon sEPSC frequency (mediated by presynaptic GABAARs) and the lack of effect of the steroid upon the sIPSC frequency were not influenced by the ELS experience (p > 0.05 control vs ELS WT, two-way RMA for both sIPSC and sEPSC neurosteroid effects; Fig. 4E), thus suggesting that the potent GABAAR-mediated inhibitory presynaptic actions upon the frequency of glutamatergic events were unaltered. An alteration of Cl− homeostasis to convert the actions of GABA from inhibitory to excitatory could contribute to the ELS-induced impairment of 5α3α-THPROG actions on neuronal firing (Hewitt et al., 2009). Gramicidin-perforated patch recordings revealed that EGABA was significantly more depolarized for ELS compared with control (p < 0.05, unpaired Student's t test; Fig. 4F,G), but VMembrane was unaffected (p > 0.05, unpaired Student's t test; Table 4). Therefore, the driving force was significantly reduced compared with control, but remained hyperpolarizing for the ELS group (p < 0.05, unpaired Student's t test; Fig. 4H, Table 4).
Table 3.
Summary of the properties of mIPSCs and the effect of 5α3α-THPROG upon mIPSC properties & RMS recorded from mpd neurons of WT CTRL, WT ELS, and δ0/0 mice
| WT CTRL | WT ELS | δ0/0 | |
|---|---|---|---|
| Peak amplitude (pA) | −116 ± 17 | −80 ± 10* | −136 ± 11 |
| Rise time (ms) | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 |
| τW (ms) | 6.4 ± 0.2 | 6.0 ± 0.4 | 6.8 ± 0.3 |
| T70 (ms) | 8.4 ± 0.5 | 7.4 ± 0.6 | 8.8 ± 0.5 |
| Control | 100 nm | Control | 100 nm | Control | 100 nm | |
|---|---|---|---|---|---|---|
| Peak amplitude (pA) | −150 ± 17 | −139 ± 15 | −83 ± 6 | −89 ± 9 | −116 ± 19 | −115 ± 21 |
| Rise time (ms) | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.4 ± 0.1 |
| τW (ms) | 7.0 ± 0.3 | 10.1 ± 0.9†† | 5.9 ± 0.8 | 8.0 ± 1.6† | 7.9 ± 1.2 | 11.1 ± 1.3† |
| T70 (ms) | 8.9 ± 0.4 | 13.3 ± 1.1†† | 7.7 ± 1.2 | 9.7 ± 1.3† | 10.0 ± 1.2 | 15.8 ± 1.9†† |
| ΔRMS (pA) | 0.56 ± 0.2 | 0.13 ± 0.1 | 0.63 ± 0.2 |
*p < 0.05 unpaired Student's t test WT ELS or δ0/0 versus WT CTRL (WT CTRL, n = 29; WT ELS, n = 17; δ0/0, n = 19).
†p < 0.05 paired Student's t test,
††p < 0.01 paired Student's t test (WT CTRL, n = 8; WT ELS, n = 7; δ0/0, n = 5).
Figure 5.
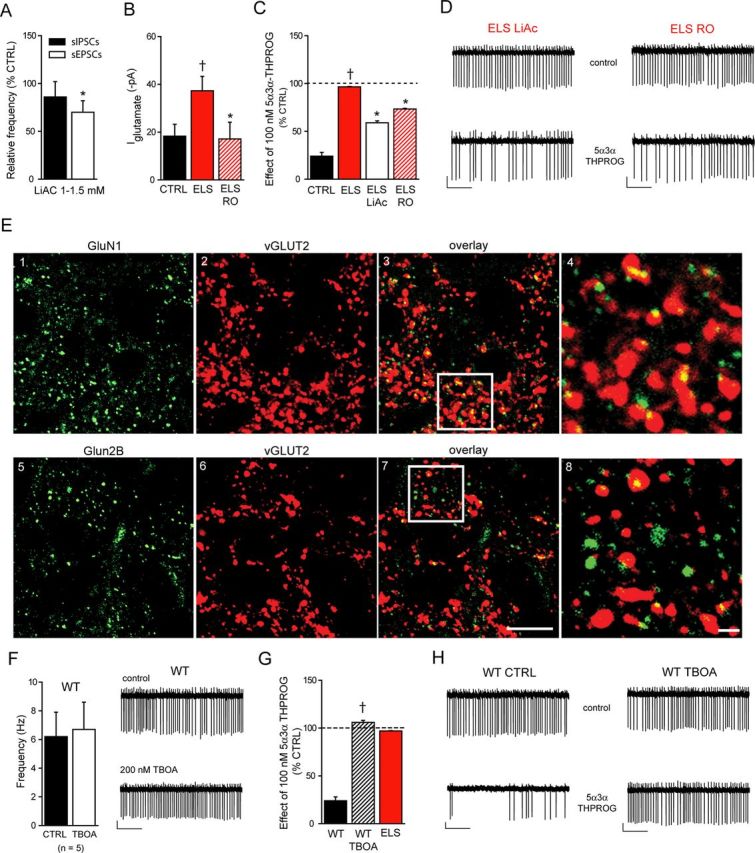
Inhibition of mpd neuronal firing by 5α3α-THPROG can be partially restored in ELS neurons or attenuated in WT neurons by decreasing or increasing the glutamatergic tone, respectively. A, Bar graph illustrating the relative frequency (expressed as percentage control) of sIPSCs and sEPSCs recorded from ELS (n = 16 and 20, respectively) mpd neurons preincubated in 1–1.5 mm LiAc for 1–2 h before recording. *p < 0.05 control vs LiAc, one-way RMA for ELS. B, Bar graph illustrating the mean kynurenic acid (2 mm)-induced IGlutamate recorded from control (CTRL; black, n = 12), ELS (red, n = 7), and RO-256981(100 nm)-treated ELS (red hatched, n = 6) mpd neurons. *p < 0.05 RO-256981-treated ELS vs untreated control; †p < 0.05 vs CTRL, one-way ANOVA; p > 0.05 CTRL vs RO-256981-treated ELS, one-way ANOVA. C, Bar graph summarizing the effect of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from CTRL (black, n = 8), ELS (red, n = 6), LiAc-treated (white, n = 4), and RO-256981-treated (red hatched, n = 5) ELS mpd neurons. Note that 5α3α-THPROG caused a significant reduction in the action current frequency of ELS neurons preincubated with LiAc (1–1.5 mm) or RO-256981 (100 nm) compared with their untreated controls. *p < 0.05 LiAc-treated or RO256981-treated ELS vs untreated control; †p < 0.01 vs WT, two-way RMA. D, Sections (10 s) of cell-attached recordings obtained from representative LiAc-treated (left) and RO-256981-treated ELS (right) mpd neurons before and 10 min after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 2 s. E, NMDA receptor subunit expression in the neonatal PVN. E1, GluN1 subunit signal appears as distinct clusters throughout the PVN and are closely apposed to vGLUT2 clusters. Quantification of the density of GluN1-immunoreactive clusters revealed that of the 23 ± 1 clusters per 1000 μm2 (mean ± SD), 20 ± 1 clusters per 1000 μm2 (∼90%) were apposed to vGLUT2 immunoreactive clusters (n = 3). E4, Magnified view of the boxed area in E3. E5, Intense GluN2B signal throughout the PVN that appears to be located to a lesser extent in the proximity of vGLUT2. Quantification of the density of GluN2B-immunoreactive clusters revealed that, of the 21 ± 1 clusters per 1000 μm2 (mean ± SD), 15 ± 1 clusters per 1000 μm2 (∼70%) were apposed to vGLUT2-immunoreactive clusters (n = 3). E8, Magnified view of the boxed area in E7. Scale bars: E1–E3, E5–E7, 10 μm; E4, E8, 1 μm. F, Left, Bar graph illustrating the mean firing frequency of WT mpd neurons before (black) and after the bath application of TFB-TBOA (200 nm, white, n = 5), p > 0.05, paired Student's t test. Right, Sections (10 s) of cell-attached recordings obtained from a representative WT mpd neuron before (top) and after (bottom) the bath application of TFB-TBOA. Scale bars: y = 10 pA, x = 2 s. G, Bar graph summarizing the effect of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from control WT (black, n = 8), TBOA-treated WT (black hatched, n = 6), and ELS (red, n = 6) mpd neurons. Note that 5α3α-THPROG induced a significant reduction in the action current frequency only in WT neurons. †p < 0.001 vs WT, two-way RMA. H, Sections (10 s) of cell-attached recordings obtained from representative control (left) and TBOA-treated WT (right) mpd neurons before and 10 min after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 10 pA, x = 2 s.
Table 4.
Summary of the perforated patch recording measurements for mpd neurons of WT CTRL, WT ELS, and δ0/0 mice
| WT CTRL | WT ELS | δ0/0 | |
|---|---|---|---|
| Resting Vmembrane (mV) | −63 ± 3 | −61 ± 2 | −57 ± 4 |
| EGABA (mV) | −79 ± 4 | −67 ± 3* | −78 ± 3 |
| GABA driving force (Vmemb − EGABA) (mV) | 16 ± 4 | 6 ± 3* | 21 ± 5 |
| RInput (MΩ) | 1643 ± 222 | 1442 ± 349 | 1452 ± 210 |
*p < 0.05 unpaired Student's t test versus WT CTRL (WT CTRL, n = 7; WT ELS, n = 7; δ0/0, n = 5)
Does the increased glutamatergic drive contribute to the impaired neurosteroid actions upon mpd firing?
The findings described above indicate for mpd neurons a modest decrease in GABAAR-mediated inhibition (i.e., lower mIPSC amplitude and a reduced hyperpolarizing driving force), but also suggest that altered GABAAR properties and/or their neurosteroid sensitivity cannot account for the blunted neurosteroid action upon action potential discharge. Therefore, we next considered whether the loss of neurosteroid efficacy may be an indirect consequence of the increased excitatory drive, which would neutralize neurosteroid inhibition (Prescott et al., 2006). We first investigated whether reducing either the synaptic or tonic excitatory drive to ELS mpd neurons to levels comparable to WT could rescue the inhibitory action of 5α3α-THPROG upon neuronal firing. The vGLUT2 inhibitor lithium acetoacetate (LiAc) reduced the synaptic drive (Juge et al., 2010), with a 30% decrease in the sEPSC frequency for ELS mpd neurons (p < 0.05, control vs LiAc, one-way RMA; Fig. 5A). This treatment partially restored 5α3α-THPROG inhibition of ELS (Fig. 5C,D, left) mpd neuronal firing (p < 0.05 for LiAc-treated ELS vs respective untreated control and p < 0.01 vs WT, two-way RMA). Similarly, RO-256981 (100 nm), a selective antagonist of GLUN2B-containing receptors (which are abundantly expressed extrasynaptically in mpd neurons; Fig. 5E5–E8), reduced the tonic current of ELS to a level comparable to WT mpd neurons (p > 0.05 WT vs RO-256981-treated ELS, one-way ANOVA; Fig. 5B). Similar to LiAc, this treatment partially rescued the inhibition of mpd discharge by 100 nm 5α3α-THPROG for ELS (p < 0.05 both for RO-256981-treated ELS vs respective controls and vs WT, two-way RMA; Fig. 5C,D, right). Note that the firing frequency of ELS mpd neurons was unaffected by treatment with either LiAc or RO-256981 (Table 2). To further corroborate the proposal that increased glutamatergic drive suppresses the inhibitory action of 5α3α-THPROG on mpd neuronal firing, the neurosteroid action on mpd firing was tested in WT slices treated with TFB-TBOA (200 nm). In common with ELS, in paired recordings, the bath application of TFB-TBOA did not affect the firing frequency of WT mpd neurons (p > 0.05, WT vs WT + TFB-TBOA, paired Student's t test; Fig. 5F), presumably because of a ceiling for the frequency of tonically firing mpd neurons. However, and consistent with the above proposal, the actions of 5α3α-THPROG (100 nm) were similarly blunted for TFB-TBOA-treated mpd neurons (Fig. 5G,H). These findings with treatments to reduce or enhance glutamatergic transmission are consistent with the proposal that the increased excitatory drive contributes significantly to the loss of the neurosteroid action in the ELS PVN.
δ0/0 mice exhibit some phenotypic signatures common to ELS mice
Interestingly, δ0/0 mice suffer from impaired maternal care (Maguire and Mody, 2008) and, in common with ELS, exhibit blunted actions by another GABAAR-active neurosteroid, 5α,3α tetrahydrodeoxycorticosterone (i.e., 5α3α-THDOC), upon neuronal firing of CRF+ve neurons (Sarkar et al., 2011). In agreement, we found that 100 nm 5α3α-THPROG was ineffective upon cell firing of δ0/0 mpd neurons (p > 0.05 vs own control but p < 0.05 WT vs δ0/0, two-way RMA; Fig. 6A–C). No differences in the effect of 100 nm 5α3α-THPROG were observed between neurons derived from male (99 ± 10% of control, n = 4) or female (96 ± 4% of control, n = 3) δ0/0 mice, so the data were pooled (p = 0.79, two-way RMA).
Figure 6.
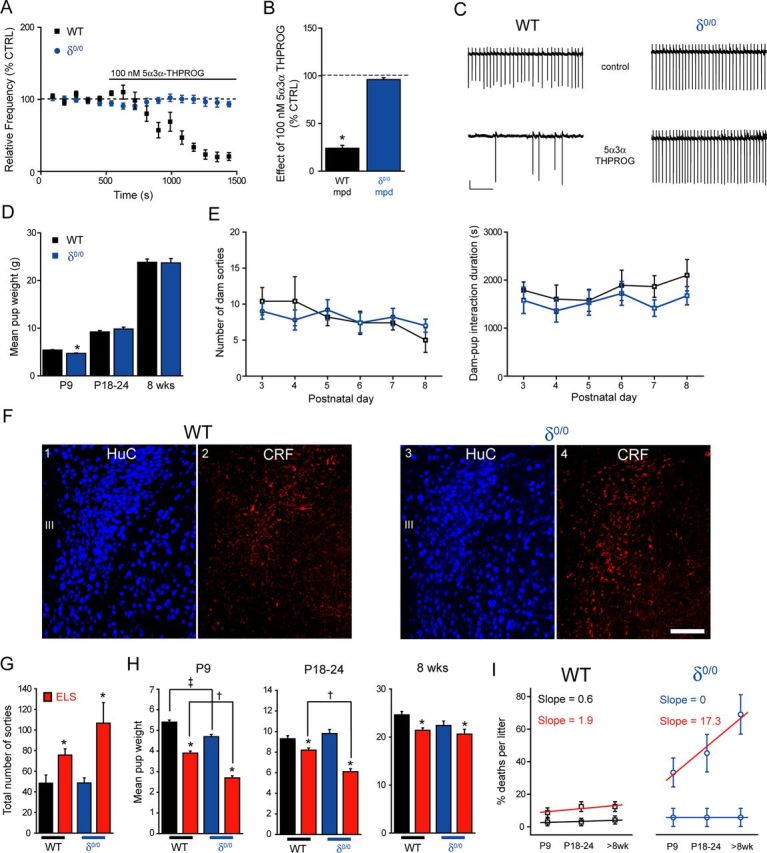
δ0/0 mice exhibit blunted neurosteroid actions upon mpd firing, reduced body weight neonatally, elevated CRF expression, and increased vulnerability to ELS exposure. A, Summary graph depicting the time course of the effect of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from mpd neurons of WT (black, n = 8) and δ0/0 (blue, n = 7) mice. B, Bar graph summarizing the effect of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from mpd neurons derived from WT (black) and δ0/0 (blue) mice. *p < 0.01 vs control, one-way RMA. C, Sections (10 s) of cell-attached recordings obtained from representative WT and δ0/0 mpd neurons before and 10 min after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 2 s. D, Bar graphs summarizing the mean weights of CTRL WT (black) and δ0/0 (blue) at P9, P18–P24, and 8 weeks of age (WT, n = 26–31; δ0/0, n = 24–36). Note that weight was significantly lower at P9 for δ0/0 vs WT *p < 0.05, unpaired Student's t test. E, Left: Graph summarizing the mean number of sorties from the nest for control (black, n = 5) and δ0/0 (blue, n = 5) WT dams from P3–P8. There was no significant effect of genotype (F(1,47) = 0.001, p = 0.97) or day (F(5,47) = 1.27, p = 0.29) on the number of maternal sorties. Right: Graph summarizing the mean dam–pup interaction time over P3–P8 for the same litters as above. There was no effect of day (F(5,47) = 0.73, p = 0.61) or genotype (F(1,47) = 3.2, p = 0.08) upon the time spent in the nest (two-way ANOVA). F, Representative images of CRF expression in the PVN of WT and δ0/0 neonatal (P22–P28) mice using immunohistochemistry. F1, F3, HuC (a pan-neuronal marker) was used to confirm that the regions investigated are comparable. F2, CRF immunoreactivity within WT tissue is expressed within somata and varicose processes. F4, Compared with WT tissue, CRF expression within the PVN of δ0/0 mice is increased significantly and more widely distributed throughout the PVN (p = 0.001, unpaired Student's t test). Tissue sections were reacted and imaged under identical conditions. Scale bar, 100 μm. G, Bar graph illustrating the total number of maternal sorties for control WT (black) and δ0/0 (blue) mice and those housed under ELS (red) conditions. There was only a main effect of treatment upon the number of sorties. *p < 0.001 ELS vs respective CTRL, either WT or δ0/0, two-way ANOVA (for details, see Table 5). H, Bar graphs summarizing the mean weights of control WT (black) and δ0/0 (blue) mice and those housed under ELS (red) conditions at P9, P18–P24, and 8 weeks of age (WT: CTRL, n = 36–42; ELS, n = 35–63; δ0/0: CTRL, n = 24–36; ELS, n = 12–24). At P9 and P18–P24, there was a significant effect of both genotype and treatment, whereas at >8 weeks, only a main effect of treatment was observed (two-way ANOVA; for details, see Table 5). However, note mortality rate below. At P9, the mean weight of control δ0/0 pups is significantly reduced compared with WT. ‡p < 0.05. In addition, at all ages, the mean weight of both WT and δ0/0 pups exposed to ELS is significantly reduced compared with their respective controls. *p < 0.001. Importantly, the mean weight of δ0/0 mice exposed to ELS is significantly lower than that of WT ELS mice both at P9 and P18–P24 (†p < 0.05), but was not different at 8 weeks of age (p > 0.05). For details, see Table 5. I, Plot summarizing the mean percentage deaths per litter for WT and δ0/0 animals housed under control (WT, black; δ0/0, blue) and ELS (red both for WT and δ0/0) conditions at P9, P24, and 8 weeks of age. The mortality rate of control WT and δ0/0 pups did not significantly change with age (p > 0.05, one-way RMA). In contrast, the mortality rate of WT and δ0/0 pups exposed to ELS increased with age (p < 0.05, one-way RMA) and was dramatically more pronounced for δ0/0 compared with WT ELS (slope of 17.3 vs 1.9, respectively). III, Third ventricle.
In view of the apparent similarities shared by the ELS and δ0/0 models, we first investigated the maternal behavior of δ0/0 dams and the ensuing effect on the offspring. In common with WT ELS mice, the δ0/0 pup weight was significantly reduced at P9 compared with WT (p < 0.05 unpaired Student's t test), but reached levels comparable to control mice at weaning and adult age (p > 0.05, unpaired Student's t test; Fig. 6D). However, the number of maternal sorties of δ0/0 dams was not significantly different compared with WT dams (F(1,47) = 0.001, p = 0.97, two-way ANOVA; n = 5 litters for both genotypes; Fig. 6E, left), suggesting the maternal behavior of δ0/0 to be distinct from WT ELS dams (Kaffmann and Meaney, 2007). Further, as for WT ELS, the duration of the dam–pup interactions was not different from the WT control (F(1,47) = 3.2, p = 0.08, two-way ANOVA; n = 5 litters for both genotypes; Fig. 6E, right). Importantly, and in common with WT ELS mice, δ0/0 mice exhibited a significant increase of CRF immunoreactivity in the soma of mpd neurons (fluorescence intensity in arbitrary units: WT tissue: 10.7 ± 0.3; δ0/0 tissue: 13.53 ± 0.2; n = 3 animals for both, p = 0.001, unpaired Student's t test; Fig. 6F). The increased CRF expression in δ0/0 mpd neurons suggested a potential susceptibility of the δ0/0mouse strain to stressful challenges. To corroborate this proposal, we explored the impact of ELS on δ0/0 mouse breeding. Consistent with our hypothesis, the ELS paradigm resulted in a significantly more severe phenotype cf. WT ELS. In common with the influence of ELS on WT mice, there was a significant effect of ELS exposure upon the number of δ0/0 maternal sorties (F(1,20) = 20.3, p = < 0.001, two-way ANOVA; Fig. 6G), although the effect of ELS exposure on this parameter was not influenced by genotype (i.e., there was no significant interaction between genotype and treatment: F(1,20) = 2.6, p = 0.12; two-way ANOVA; for detailed statistics, see Table 5). However, the weight of the δ0/0 ELS pups was significantly lower at both at P9 and P21–P24 compared with the control δ0/0, WT control, or WT ELS experimental groups, indicating a significant interaction between genotype and ELS exposure (P9: F(1,161) = 5.3, p < 0.05; P18–P24: F(1,128) = 14.1, p < 0.001; two-way ANOVA; for detailed statistics, see Table 5; Fig. 6H). Most significantly, although the δ0/0 ELS pup weight was comparable to WT ELS by their adult stage, ELS breeding resulted in an increased mortality rate for δ0/0 mice from P9 to adulthood compared with δ0/0 control and WT ELS mice (Fig. 6I).
Table 5.
Summary of statistical analysis of ELS treatment upon the sorties and weight of WT and δ0/0 mice
| Sorties (n) F-value | p-value | Weight F-value | p-value | |
|---|---|---|---|---|
| Effect of genotype (G) | ||||
| P9 | F(1,20) = 2.7 | 0.116 | F(1,161) = 49.9 | <0.001 |
| P18–P24 | — | — | F(1,128) = 6.2 | 0.014 |
| >8 wk | — | — | F(1,103) = 1.1 | 0.293 |
| Effect of treatment (T) | ||||
| P9 | F(1,20) = 20.3 | <0.001 | F(1,161) = 161.9 | <0.001 |
| P18–P24 | — | — | F(1,128) = 49.3 | <0.001 |
| >8 wk | — | — | F(1,103) = 14.7 | <0.001 |
| G × T interaction | ||||
| P9 | F(1,20) = 2.6 | 0.12 | F(1,161) = 5.3 | 0.02 |
| P18–P24 | — | — | F(1,128) = 14.1 | <0.001 |
| >8 wk | — | — | F(1,103) = 0.007 | 0.935 |
Given the apparent similarity of the molecular phenotypes of the stress axis (i.e., increased CRF expression) and the shared neurosteroid insensitivity of mpd neuron firing exhibited by WT ELS and δ0/0 mice, we investigated whether they also shared the same functional plasticity (i.e., increased glutamatergic drive). In common with WT ELS, neonatal δ0/0 mpd neurons also exhibited a significantly greater frequency of sEPSCs (p < 0.05, unpaired Student's t test; Fig. 7A,B, left), but an unaltered sIPSC frequency (p > 0.05, unpaired Student's t test; Fig. 7A,B, right, Table 1). Note that no differences were observed in the frequency of sEPSCs between the sexes (male: 5 ± 1.5 Hz, n = 13; female: 6.8 ± 1.3 Hz, n = 19, p = 0.4, unpaired Student's t test). However, in contrast to WT ELS mpd neurons, the δ0/0 mEPSC frequency was unaltered compared with control WT (p > 0.05, unpaired Student's t test; Fig. 7B, left). In agreement with this, there were no significant differences between vGLUT2 labeling for δ0/0 compared with WT mpd neurons (vGLUT2 clusters per 10 μm2 WT: 1.48 ± 0.03; δ0/0: 1.60 ± 0.05; n = 3 animals for both; p = 0.05, unpaired Student's t test; Fig. 7C). As described for mpd neurons from WT ELS mice, the frequency of mIPSCs was similar for WT and δ0/0 mice (p > 0.05, unpaired Student's t test; Fig. 7B, right). Moreover, in common with WT ELS, δ0/0 mpd neurons exhibited an enhanced glutamatergic tonic conductance (p < 0.01, unpaired Student's t test. Fig. 7D, Table 1), which was largely mediated by NMDARs (AP-5-sensitive component: 66 ± 10%, n = 4; p > 0.05 vs WT control and WT ELS, one-way RMA) and the magnitude of this current did not exhibit any differences between the sexes (male: −42 ± 9 pA, n = 8; female: −38 ± 5 pA, n = 9, p = 0.69 unpaired Student's t test). Furthermore, the transporter inhibitor TFB-TBOA (200 nm) produced an inward, kynurenic acid-sensitive current that was significantly reduced compared with WT (p < 0.05, unpaired Student's t test; Fig. 7E, Table 1). Importantly, and in common with ELS, treatment with either LiAc to produce a 25% reduction in sEPSCs frequency (Fig. 7F) or RO-256981 (100 nm) to reduce the NMDAR-mediated tonic current of δ0/0 to values comparable to WT mpd neurons (p > 0.05 WT vs RO-256981-treated δ0/0, one-way ANOVA; Fig. 7G) partially restored 5α3α-THPROG inhibition of δ0/0 mpd neuronal firing (p < 0.05 for LiAc-treated δ0/0 vs respective untreated control and p < 0.01 vs WT, two-way RMA; p < 0.05 for RO-256981-treated δ0/0 vs respective controls and vs WT, two-way RMA; Fig. 7H,I). Note that, in common with ELS, the firing frequency of δ0/0 mpd neurons was also unaffected by treatment with either LiAc or RO-256981 (Table 2).
Figure 7.

δ0/0 mice display alterations in glutamatergic transmission similar to WT ELS mice, which blunt neurosteroid suppression of mpd neuron firing. A, Illustrated are sections (5 s) of whole-cell voltage-clamp recordings of sIPSCs (top) and sEPSCs (bottom) obtained from the same representative mpd neurons for WT (left) and δ0/0 (right) neonatal (P18–P26) mice. A section (shaded area, 0.5 s) of the recordings is displayed on an expanded time scale below. Scale bars: IPSCs, y = 20 pA; EPSCs y = 10 pA; x = 500 ms and 50 ms for top and bottom traces, respectively. B, Bar graphs illustrating the mean frequency of sEPSCs and mEPSCs (left, n = 15–32) and sIPSCs and mIPSCs (right, n = 9–25) for mpd neurones derived from WT (black) and δ0/0 (blue) mice. Note that the frequency of sEPSCs is significantly greater for δ0/0 compared with WT CTRL (*p < 0.01, unpaired Student's t test), whereas the mEPSC, sIPSC and mIPSC frequency are unchanged (p > 0.05, unpaired Student's t test). C, Representative images of vGLUT2 expression in the PVN of WT and δ0/0 mice. The density of vGLUT2 puncta was similar for WT and δ0/0 mice (p = 0.05, unpaired Student's t test). Scale bar, 10 μm. D, Box plot showing the mean IGlutamate for WT (white, n = 12) and δ0/0 (blue, n = 17) mpd neurons revealed by the bath application of kynurenic acid (2 mm). Note that the mean IGlutamate is greater for δ0/0 mpd neurons compared with WT (p < 0.01 unpaired Student's t test). E, Bar graph illustrating the mean TFB-TBOA (200 nm)-induced current recorded from mpd neurons derived from CTRL WT (black) and δ0/0 (blue) mice upon the bath application of kynurenic acid (2 mm). *p < 0.05 vs WT, unpaired Student's t test (WT, n = 6; δ0/0, n = 4). F, Bar graph illustrating the relative frequency (expressed as a percentage of control) of sIPSCs and sEPSCs recorded from δ0/0 (n = 13 and 17 respectively) mpd neurons preincubated in 1–1.5 mm LiAc for 1–2 h before recording. *p < 0.05 control vs LiAc, one-way RMA. G, Bar graph illustrating the mean kynurenic acid (2 mm)-induced IGlutamate recorded from WT (black, n = 12), δ0/0 (blue, n = 17), and RO-256981(100 nm)-treated δ0/0 (blue hatched, n = 4) mpd neurons. *p < 0.05 RO-256981-treated δ0/0 vs untreated control; †p < 0.05 vs WT, one-way ANOVA; p > 0.05 WT vs RO-256981-treated δ0/0, one-way ANOVA. H, Bar graph summarizing the effect of 5α3α-THPROG (100 nm) upon the frequency of action currents recorded from WT (black, n = 8), δ0/0 (blue, n = 7), LiAc-treated (white, n = 6), and RO-256981-treated (blue hatched, n = 6) δ0/0 mpd neurons. Note that 5α3α-THPROG caused a significant reduction in the action current frequency of δ0/0 neurons preincubated with LiAc (1–1.5 mm) or RO-256981 (100 nm) compared with their untreated controls. *p < 0.05 LiAc or RO-256981-treated δ0/0 vs untreated control; †p < 0.01 vs WT, two-way RMA. I, Sections (10 s) of cell-attached recordings obtained from representative LiAc-treated (left) and RO-256981-treated (right) δ0/0 mpd neurons before and 10 min after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 2 s.
Because neurosteroid actions are reported to be greatly influenced by the presence of the δ-subunit (Mihalek et al., 1999; Belelli et al., 2002; Brown et al., 2002; Sarkar et al., 2011), the lack of δ-GABAARs could conceivably contribute to the impaired steroid effect on mpd neuron firing. However, in agreement with previous studies (Wisden et al., 1992; Fritschy and Mohler, 1995; Hörtnagl et al., 2013 but see Sarkar et al., 2011), we found no immunohistochemical or molecular evidence for δ-subunit expression or labeling in either neonatal (P22–P28; Fig. 8A–C) or adult (> 2 months; data not shown) mpd neurons. In support of this finding, whole-cell voltage-clamp recordings revealed that the δ-GABAAR-preferring agonist THIP (1 μm) only induced a modest bicuculline-sensitive inward current that was similar for both WT and δ0/0 neonatal (P18–P26) mpd neurons (p > 0.05 δ0/0 vs WT, two-way ANOVA; Fig. 8D,E), presumably reflecting the limited activation of other (e.g., α5βγ2 or ε-containing) receptor isoforms in both the WT and δ0/0 strains (Ranna et al., 2006; Stórustovu and Ebert, 2006). Furthermore, for WT neurons, THIP (1 μm), in clear contrast to the neurosteroid, had no effect upon the frequency of action currents, mIPSCs, or the frequency of sEPSCs (p > 0.05, one-way RMA; Fig. 8F, Table 6). Impairment of neurosteroid actions on δ0/0 mpd neuronal discharge may nevertheless implicate an alteration in the properties and/or sensitivity of inhibitory currents to neurosteroids (Vicini et al., 2002). However, neither the properties of mIPSCs (p > 0.05, unpaired Student's t test) nor the neurosteroid sensitivity of GABAAR-mediated mIPSCs (p > 0.05 WT vs δ0/0, two-way RMA) nor the presynaptic actions of the neurosteroid on the frequency of sEPSCs (reduction) and sIPSCs (no effect) for δ0/0 mpd neurons were significantly different from WT (p > 0.05 WT vs δ0/0, two-way RMA for both measurements; Fig. 9A–C, Table 3). Furthermore, in contrast to WT ELS neurons, perforated patch recordings did not reveal any difference in EGABA or VMembrane, and thus the driving force, between δ0/0 and WT mpd neurons (p > 0.05, unpaired Student's t test for all 3 measurements; Fig. 9D–F, Table 4).
Figure 8.
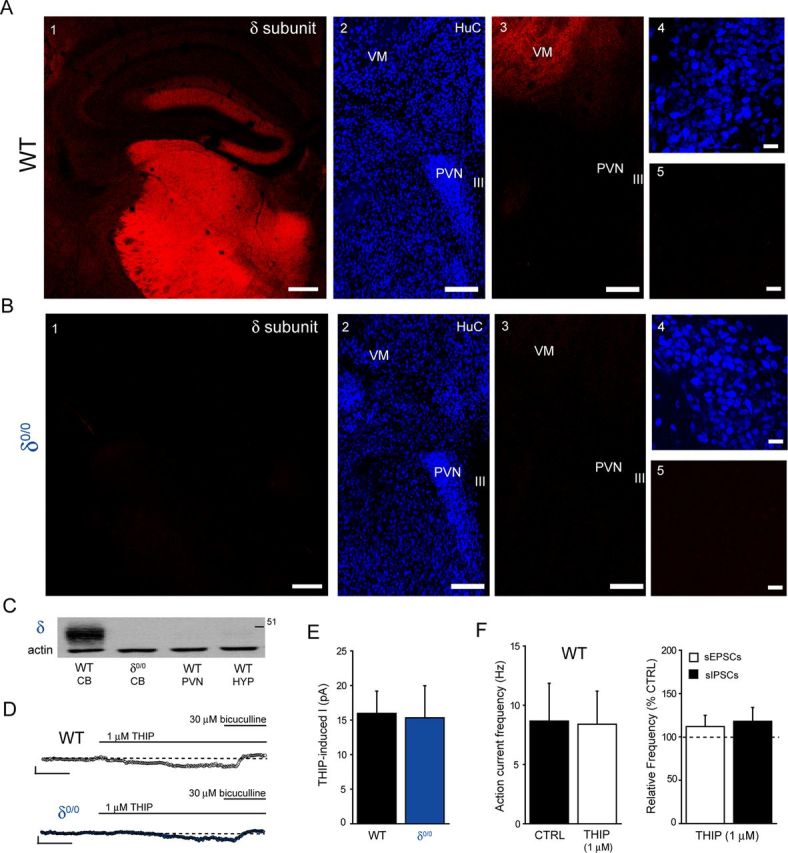
δ-GABAARs are not expressed in the PVN. A, B, Overviews (A1–A3) show the characteristic expression pattern of the δ subunit in tissue from neonatal (P22–P28) WT mouse, which is not detectable in tissue from δ0/0 mice (B1–B3). Magnified views of the PVN of WT (A4,A5) and δ0/0 (B4,B5) mice demonstrate that, even at high resolution, no δ-subunit signal is detectable in this brain region compared with thalamic ventromedial nucleus (VM). Scale bars: A1–A3, B1–B3, 100 μm; A4, A5, B4, B5, 10 μm. C, Representative Western blot detecting the expression of the δ-subunit in homogenates prepared from WT and δ0/0 “punches” of mouse brain tissue. A total of 10 μg of protein was loaded per lane and a clear immunoreactive signal was detected in WT samples for cerebellum (CB), but not for PVN and hypothalamus (HYP). In addition, note the lack of δ-signal detected in CB sample derived from a δ0/0 mouse. β-actin was used as a loading control. D, Representative whole-cell voltage-clamp recordings (Vh = −60 mV) from WT (top) and δ0/0 (bottom) mpd neurons under control, after the bath application of THIP (1 μm), and after the combined application of THIP (1 μm) and bicuculline (30 μm). Note that THIP (1 μm) induced only a similar, modest inward current in WT and δ0/0 mpd neurons, an effect reversed by the subsequent application of bicuculline (30 μm). Scale bars: y = 10 pA, x = 100 s. The representative traces were generated by averaging the mean DC of 100 ms epochs every 2 s with points falling on to the decay of mIPSCs or containing spurious noise being discarded from the analysis. The dotted line represents the holding current before drug application. E, Summary bar chart illustrating the effect of THIP (1 μm) upon the Iholding (expressed as THIP-induced current) of WT (black; n = 6) and δ0/0 (blue; n = 4) mpd neurons. p > 0.05, unpaired Student's t test. F, Left, Summary graph depicting the lack of an effect of THIP (1 μm) upon the action current frequency recorded from WT mpd neurons (n = 7). p > 0.05, paired Student's t test. Right, Summary bar chart illustrating the lack of effect of THIP (1 μm) upon the frequency of either sEPSCs or sIPSCs. p > 0.05, one-way RMA; n = 4 for both measurements. III, Third ventricle.
Table 6.
Summary of the effect of THIP (1 μm) upon the properties of mIPSCs recorded from WT mpd neurons
| WT |
||
|---|---|---|
| Control | THIP | |
| Peak amplitude (pA) | −157 ± 12 | −177 ± 17 |
| Rise time (ms) | 0.4 ± 0.1 | 0.4 ± 0.1 |
| τW (ms) | 6.8 ± 0.8 | 7.3 ± 0.8 |
| Frequency (Hz) | 1.1 ± 0.4 | 1.0 ± 0.4 |
p > 0.05 paired Student's t test (n = 5).
Figure 9.

Deletion of the δ-subunit does not alter the neurosteroid sensitivity of synaptic GABAARs or the polarity of the GABA-evoked response of mpd neurons. A, Cumulative probability plots of the decay of all mIPSCs (expressed as T70 values) collected from WT (right, n = 8) and δ0/0 (left, n = 5) mpd neurons before (WT: black, δ0/0: blue) and after (gray) the bath application of 100 nm 5α3α-THPROG. The insets illustrate the superimposed normalized ensemble averages of mIPSCs recorded from representative mpd neurons before and after the bath application of 100 nm 5α3α-THPROG. Scale bars: y = 20 pA, x = 10 ms, applicable to both traces. B, Paired plots of the τW value before and after treatment with 100 nm 5α3α-THPROG for WT (right, black) and δ0/0 (left, blue) mpd neurons tested. Note that all neurons were deemed neurosteroid sensitive (T70, p < 0.01 KS test). C, Control; NS, in the presence of neurosteroid. C, Bar graph illustrating the impact of 5α3α-THPROG (100 nm) upon the sEPSC (neurosteroid-sensitive) and sIPSC frequency for WT (black, n = 6–7) and δ0/0 (blue, n = 4–6) mpd neurons. *p < 0.05 vs CTRL, one-way RMA; p > 0.05 WT vs δ0/0, two-way RMA. D, I–V curves of the GABA-evoked response (normalized to the maximum current obtained for each cell) derived from perforated-patch recordings for WT (black, n = 7) and δ0/0 (blue, n = 5) mpd neurons. Each point represents the mean ± SEM (p > 0.05, unpaired Student's t test). The insets illustrate representative current traces obtained at different holding potentials for WT (black) and δ0/0 (blue) mpd neurons. Scale bars: WT, y = 20 pA, x = 250 ms; δ0/0, y = 10 pA, x = 250 ms. E, Box plot showing the mean EGABA obtained for WT (white, n = 7) and δ0/0 (blue, n = 5) mpd neurons subject to perforated patch recordings (p > 0.05 unpaired Student's t test WT vs δ0/0; for details, see Table 4). F, Bar graph illustrating the mean GABA-driving force (calculated as Vmemb − EGABA) for WT (black, n = 7) and δ0/0 (blue, n = 5) mpd neurons (p > 0.05 unpaired Student's t test; for details, see Table 3).
In summary, our findings offer compelling evidence that adverse early-life experiences are associated with a dramatic upregulation of excitatory transmission within stress-activated neuronal circuits, leading to a failure of inhibitory mechanisms, one of which we show to be dependent on GABAAR-active, endogenously occurring steroids.
Discussion
The impact of early-life experiences on neuronal development is well documented and adverse events are known to program an abnormal stress response by encoding long-lasting molecular changes in the brain (Heim et al., 2010). Such adaptations in turn have a negative impact on mental health and cognitive abilities in both humans and animals (Lupien et al., 2009). The specific neurobiological mechanisms underpinning the detrimental and persistent effects of early-life adversity are not fully elucidated, but they are recognized to operate on a background of genetic susceptibility (i.e., genetic diathesis stress theory; Heim et al., 2010). Here, we offer an insight into such mechanisms by providing functional and molecular evidence for a novel form of hypothalamic neuronal plasticity in an environmentally-induced (ELS) mouse and a genetic mouse model (δ0/0) of ELS. Although there are clear differences in the nature of the maternal care, both models exhibited a significantly increased synaptic and extrasynaptic glutamatergic drive impinging upon CRF+ve mpd neurons. The increased excitatory drive produces a dramatic loss of the potent inhibitory actions of the stress-protective, GABAAR-active neurosteroid 5α3α-THPROG on the firing of CRF+ve neurons.
Abnormal maternal care alters the hypothalamic excitatory/inhibitory balance
Environmental stress alters glutamatergic transmission in cortical/limbic areas via an increase in glutamate release and/or dysfunction of glial and synaptic transmission (de Vasconcellos-Bittencourt et al., 2011; Sanacora et al., 2012). In the hypothalamus, increased somatic excitatory drive for mpd neurons was indirectly inferred for rodent models of chronic stress (Flak et al., 2009; Miklós and Kovács, 2012), whereas a reverse adaptation (i.e., a decrease) was associated with a model of augmented maternal care (Korosi et al., 2010). The present study provides for the first time direct functional evidence that ELS causes an upregulation of the synaptic glutamatergic drive in the stress centers of the neonatal hypothalamus and reveals a novel role for a tonic, NMDAR-mediated extrasynaptic conductance. Furthermore, pertinent to the genetic diathesis stress theory, this molecular signature is shared by δ0/0 mice, which display some phenotypic features suggestive of abnormal maternal care (albeit clearly distinct from ELS mice). Furthermore, in common with ELS mice, the δ0/0 mice exhibited an upregulated somatic CRF expression in mpd neurons. These findings apparently contrast with those of Rice et al. (2008) regarding decreased CRF mRNA, but may be accounted for by the lower rate of mRNA transcription compared with protein translation in mammals (Vogel and Marcotte, 2012); that is, mRNA levels may not accurately reflect changes in peptide levels. Consistent with a stress-susceptible phenotype, when the ELS paradigm was imposed upon δ0/0 mouse breeding, the pups exhibited an exaggerated response with an increased perinatal and adult mortality rate compared with WT ELS or δ0/0 controls. This increased susceptibility of the δ0/0 mice to the breeding conditions may contribute to the more severe behavioral phenotype described for δ0/0 mice (Maguire and Mody, 2008). However, cross-fostering experiments are required to establish the relative contribution of genetic and maternal care abnormalities (Priebe et al., 2005) to the described δ0/0 phenotype (present study and Maguire and Mody, 2008). Nevertheless, our findings raise the prospect that polymorphisms of the GABRD gene may underlie an increased susceptibility to stressful experiences and, therefore, to the development of psychopathology in humans (Feng et al., 2010).
Although some changes in neural inhibition were induced by ELS (reduced mIPSC amplitude and hyperpolarizing GABA-ergic driving force), these were not evident for the δ0/0 mouse. Importantly, we demonstrated that the deficit in 5α3α-THPROG efficacy for both ELS and δ0/0 mice is not caused by the neurosteroid being less effective as a GABAAR modulator, but in both cases, appears to be a consequence of the increased glutamatergic drive. In support of this, drugs that enhanced (in WT) or inhibited (in both ELS and δ0/0 mice) glutamatergic transmission blunted or facilitated, respectively, the effect of the neurosteroid on AP discharge in the PVN. These findings indicate that the increased excitatory drive in δ0/0 mice is sufficient to negate the steroid inhibitory actions upon neuronal firing by converting it from probabilistic to deterministic (Prescott et al., 2006). Nevertheless, it cannot be excluded that additional changes in GABAA, glutamate receptor expression/function, or, indeed, additional proteins in other stress-responsive regions of the hypothalamus or telencephalon contribute to the ELS or δ0/0 phenotype. For example, permanent alterations in hippocampal GABAAR function and expression after early-life adversity have been described previously (Hsu et al., 2003).
Molecular targets of neurosteroid action and relevance to their stress-protective effects
Although the blunted neurosteroid action in the δ0/0 mouse appears to agree with a potent direct interaction of neurosteroids with recombinant δ-GABAARs (Belelli et al., 2002; Brown et al., 2002), our molecular, immunohistochemical, and functional (the δ-preferring agonist THIP) analysis is consistent with the lack of expression of δ-GABAAR in the hypothalamus reported previously (Wisden et al., 1992; Fritschy and Mohler, 1995; Hörtnagl et al., 2013), albeit apparently inconsistent with the report by Sarkar et al. (2011). The reasons for the discrepancy are not evident, although the use of different mouse lines may be a contributing factor. Here, we speculate that the potent inhibitory action of the neurosteroid on neuronal discharge is mediated by γ2-GABAARs expressed by mpd neurons and/or by those “upstream” in the PVN circuitry; for example, those located on glutamatergic terminals. Interestingly, the development of a hyperactive HPA axis during the neonatal period is associated with an anxious and depressive adult phenotype when a genetically engineered γ2-GABAAR deficit is introduced embryonically, but not in the fourth postnatal week (Shen et al., 2010). Whether neurosteroids contribute to this phenotype is unknown.
Inhibition of mpd firing by physiological concentrations of 5α3α-THPROG may be relevant to the documented anxiolytic and stress-protective actions of neurosteroids (Patchev et al., 1994, 1996; Biggio et al., 2007; Gunn et al., 2011). Specifically, with regard to the impact of early-life events, modulation of GABAARs by neurosteroids during neonatal development may be important for normal maturation of the HPA axis, thus influencing the excitability of stress-responsive PVN neurons (Korosi and Baram, 2009) in a manner similar to that described previously for the prefrontal cortex (Grobin et al., 2003). Providing indirect support for this, neonatal administration of the GABAAR-active neurosteroid 5α,3α-THDOC (at P2–P12) counteracts the neuroendocrine and behavioral dysregulation of the HPA axis evident in adult rats subjected as neonates to maternal separation (Patchev et al., 1997). Therefore, impaired neonatal neurosteroid signaling may contribute to HPA dysfunction and associated mood disorders (Pariante and Lightman, 2008).
Programming of the stress response and the incidence of mental disorders: relevance of the increased glutamatergic drive in the hypothalamus
This report reveals for the first time a novel role for increased glutamatergic NMDAR-mediated conductance in mpd neuron hyperexcitability of both neonatal (P18–P26) ELS and δ0/0 mice, specifically highlighting a dysfunction of the astrocytic glutamate transporters. Because an aberrant stress response associates with depression and in agreement, normalization of the HPA axis is one of the major targets of antidepressant treatment (Schüle et al., 2009), these findings are of specific significance in the quest for uncovering the biological basis of mental disorders for two reasons. First, they inform on a specific molecular mechanism whereby adverse early-life events program an abnormal development of the neuronal stress circuit. Second, and importantly, they add to a growing body of evidence that endorses important roles for both environmental stress and increased glutamatergic transmission as a consequence of astrocytic dysfunction in the pathophysiology of depression (Sanacora et al., 2012; Gómez-Galán et al., 2013). Furthermore, they provide support for the proposal that ionotropic glutamate receptors represent a novel therapeutic target for the treatment of stress-related psychiatric disturbances such as depression. Indeed, in agreement with animal studies (Li et al., 2011), recent clinical investigations with NMDAR antagonists, including GluN2B-receptor-selective ligands, have reported promising results in trials of treatment-resistant major depressive subjects (Preskorn et al., 2008; Li et., 2011; Ibrahim et al., 2012; Sanacora et al., 2012; Zarate et al., 2013). A detailed characterization of the behavioral profile of adult ELS compared with δ0/0 mice provides scope for future studies. Although our analysis indicates no sex differences in the hypothalamic glutamatergic alterations of ∼3- to 4-week-old mice experiencing early-life stress, it remains to be determined whether both sexes will be similarly affected in adulthood given the documented sex differences in the incidence of psychiatric disorders.
At present, we do not know whether a specific relationship exists between ELS-induced upregulation of glutamatergic transmission and neurosteroid production within the PVN or telencephalic regions. However, NMDARs are implicated in the endogenous synthesis of neurosteroids in CA1 hippocampal neurons (Tokuda et al., 2011; Zorumski et al., 2013). The present study raises the prospect that a similar scenario may apply to the neonatal PVN, where the neurosteroid enzymatic machinery is expressed abundantly (Eechaute et al., 1999). Moreover, a novel form of regulation of the excitatatory-inhibitory balance involving cross talk between NMDA and GABAA receptors in preautonomic parvocellular neurons has been reported (Potapenko et al., 2013). Future studies will explore the relationship and possible interdependence of early-life dysfunction of the stress neurocircuitry, NMDAR activity, neurosteroid synthesis, and behavior.
Footnotes
This work was supported by the Wellbeing of Women (Grant RG1265), the Biotechnology and Biological Sciences Research Council (Case Award Studentship 11426 to B.G.G.), and Tenovus Scotland. We thank Hilary Norburn for skillful assistance with the ELS experiments and Dr. Murray Herd for helpful discussion of the manuscript.
The authors declare no competing financial interests.
References
- Alon T, Zhou L, Pérez CA, Garfield AS, Friedman JM, Heisler LK. Transgenic mice expressing green fluorescent protein under the control of the corticotropin-releasing hormone promoter. Endocrinology. 2009;150:5626–5632. doi: 10.1210/en.2009-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Davis EP, Obenaus A, Sandman CA, Small SL, Solodkin A, Stern H. Fragmentation and unpredictability of early-life experience in mental disorders. Am J Psychiatry. 2012;169:907–915. doi: 10.1176/appi.ajp.2012.11091347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaccia ML, Roscetti G, Trabucchi M, Mostallino MC, Concas A, Purdy RH, Biggio G. Time-dependent changes in rat brain neuroactive steroid concentrations and GABAA receptor function after acute stress. Neuroendocrinology. 1996;63:166–172. doi: 10.1159/000126953. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ. Neurosteroids: endogenous regulators of the GABAA receptor. Nat Rev Neurosci. 2005;6:565–575. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- Belelli D, Casula A, Ling A, Lambert JJ. The influence of subunit composition on the interaction of neurosteroids with GABAA receptors. Neuropharmacology. 2002;43:651–661. doi: 10.1016/S0028-3908(02)00172-7. [DOI] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neurosci. 2005;25:11513–11520. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biag J, Huang Y, Gou L, Hintiryan H, Askarinam A, Hahn JD, Toga AW, Dong HW. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J Comp Neurol. 2012;520:6–33. doi: 10.1002/cne.22698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancardi VC, Campos RR, Stern JE. Altered balance of γ-aminobutyric acidergic and glutamatergic afferent inputs in rostral ventrolateral medulla-projecting neurons in the paraventricular nucleus of the hypothalamus of renovascular hypertensive rats. J Comp Neurol. 2010;518:567–585. doi: 10.1002/cne.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggio G, Concas A, Follesa P, Sanna E, Serra M. Stress, ethanol, and neuroactive steroids. Pharmacol Ther. 2007;116:140–171. doi: 10.1016/j.pharmthera.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaba C, Szabó K, Tasker JG. Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J Neurosci. 1996;16:7151–7160. doi: 10.1523/JNEUROSCI.16-22-07151.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaba C, Schrader LA, Tasker JG. Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. J Neurophysiol. 1997;77:3396–3400. doi: 10.1152/jn.1997.77.6.3396. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EJ, Johnston GA, Edwards FA. Effects of a naturally occurring neurosteroid on GABAA IPSCs during development in rat hippocampal or cerebellar slices. J Physiol. 1999;521:437–449. doi: 10.1111/j.1469-7793.1999.00437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan WE, Ziegler DR, Herman JP. Functional role of local GABAergic influences on the HPA axis. Brain Struct Funct. 2008;213:63–72. doi: 10.1007/s00429-008-0192-2. [DOI] [PubMed] [Google Scholar]
- Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- de Vasconcellos-Bittencourt AP, Vendite DA, Nassif M, Crema LM, Frozza R, Thomazi AP, Nieto FB, Wofchuk S, Salbego C, da Rocha ER, Dalmaz C. Chronic stress and lithium treatments alter hippocampal glutamate uptake and release in the rat and potentiate necrotic cellular death after oxygen and glucose deprivation. Neurochem Res. 2011;36:793–800. doi: 10.1007/s11064-011-0404-7. [DOI] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and γ-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- Eechaute WP, Dhooge WS, Gao CQ, Calders P, Rubens R, Weyne J, Kaufman JM. Progesterone-transforming enzyme activity in the hypothalamus of the male rat. J Steroid Biochem Mol Biol. 1999;70:159–167. doi: 10.1016/S0960-0760(99)00106-5. [DOI] [PubMed] [Google Scholar]
- Feng Y, Kapornai K, Kiss E, Tamás Z, Mayer L, Baji I, Daróczi G, Benák I, Kothencné VO, Dombovári E, Kaczvinszk E, Besnyo M, Gádoros J, Székely J, Kovács M, Vetró A, Kennedy JL, Barr CL. Association of the GABRD gene and childhood-onset mood disorders. Genes Brain Behav. 2010;9:668–672. doi: 10.1111/j.1601-183X.2010.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Ostrander MM, Tasker JG, Herman JP. Chronic stress-induced neurotransmitter plasticity in the PVN. J Comp Neurol. 2009;517:156–165. doi: 10.1002/cne.22142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming TM, Scott V, Naskar K, Joe N, Brown CH, Stern JE. State-dependent changes in astrocyte regulation of extrasynaptic NMDA receptor signalling in neurosecretory neurons. J Physiol. 2011;589:3929–3941. doi: 10.1113/jphysiol.2011.207340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin TB, Saab BJ, Mansuy IM. Neural mechanisms of stress resilience and vulnerability. Neuron. 2012;75:747–761. doi: 10.1016/j.neuron.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Gómez-Galán M, De Bundel D, Van Eeckhaut A, Smolders I, Lindskog M. Dysfunctional astrocytic regulation of glutamate transmission in a rat model of depression. Mol Psychiatry. 2013;18:582–594. doi: 10.1038/mp.2012.10. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Morrow AL. 3α-hydroxy-5α-pregnan-20-one levels and GABAA receptor-mediated 36Cl− flux across development in rat cerebral cortex. Brain Res Dev Brain Res. 2001;131:31–39. doi: 10.1016/S0165-3806(01)00242-5. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Heenan EJ, Lieberman JA, Morrow AL. Perinatal neurosteroid levels influence GABAergic interneuron localization in adult rat prefrontal cortex. J Neurosci. 2003;23:1832–1839. doi: 10.1523/JNEUROSCI.23-05-01832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Bard L, Choquet D. Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience. 2009;158:4–18. doi: 10.1016/j.neuroscience.2008.05.029. [DOI] [PubMed] [Google Scholar]
- Gunn BG, Brown AR, Lambert JJ, Belelli D. Neurosteroids and GABAA Receptor Interactions: A focus on stress. Front Neurosci. 2011;5:131. doi: 10.3389/fnins.2011.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haage D, Johansson S. Neurosteroid modulation of synaptic and GABA-evoked currents in neurons from the rat medial preoptic nucleus. J Neurophysiol. 1999;82:143–151. doi: 10.1152/jn.1999.82.1.143. [DOI] [PubMed] [Google Scholar]
- Heim C, Shugart M, Craighead WE, Nemeroff CB. Neurobiological and psychiatric consequences of child abuse and neglect. Dev Psychobiol. 2010;52:671–690. doi: 10.1002/dev.20494. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Ziegler DR, Tasker JG. Role of the paraventricular nucleus microenvironment in stress integration. Eur J Neurosci. 2002;16:381–385. doi: 10.1046/j.1460-9568.2002.02133.x. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Hörtnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013;236:345–372. doi: 10.1016/j.neuroscience.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu FC, Zhang GJ, Raol YS, Valentino RJ, Coulter DA, Brooks-Kayal AR. Repeated neonatal handling with maternal separation permanently alters hippocampal GABAA receptors and behavioral stress responses. Proc Natl Acad Sci U S A. 2003;100:12213–12218. doi: 10.1073/pnas.2131679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L, Diaz Granados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, Potter WZ, Zarate CA., Jr A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J Clin Psychopharmacol. 2012;32:551–557. doi: 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, Uneyama H, Edwards RH, Nicoll RA, Moriyama Y. Metabolic control of vesicular glutamate transport and release. Neuron. 2010;68:99–112. doi: 10.1016/j.neuron.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman A, Meaney MJ. Neurodevelopmental sequelae of postnatal maternal care in rodents: clinical and research implications of molecular insights. J Child Psychol Psychiatry. 2007;48:224–244. doi: 10.1111/j.1469-7610.2007.01730.x. [DOI] [PubMed] [Google Scholar]
- Korosi A, Baram TZ. The pathways from mother's love to baby's future. Front Behav Neurosci. 2009;3:27. doi: 10.3389/neuro.08.027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korosi A, Shanabrough M, McClelland S, Liu ZW, Borok E, Gao XB, Horvath TL, Baram TZ. Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J Neurosci. 2010;30:703–713. doi: 10.1523/JNEUROSCI.4214-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG. Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol. 2002;14:929–932. doi: 10.1046/j.1365-2826.2002.00867.x. [DOI] [PubMed] [Google Scholar]
- Maguire J, Mody I. GABAAR plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick I, Pronchuk N, Cowley MA, Grove KL, Colmers WF. Developmental switch in neuropeptide Y and melanocortin effects in the paraventricular nucleus of the hypothalamus. Neuron. 2007;56:1103–1115. doi: 10.1016/j.neuron.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE. Attenuated sensitivity to neuroactive steroids in γ-aminobutyrate type A receptor δ subunit knockout mice. Proc Natl Acad Sci U S A. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklós IH, Kovács KJ. GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience. 2002;113:581–592. doi: 10.1016/S0306-4522(02)00147-1. [DOI] [PubMed] [Google Scholar]
- Miklós IH, Kovács KJ. Reorganization of synaptic inputs to the hypothalamic paraventricular nucleus during chronic psychogenic stress in rats. Biol Psychiatry. 2012;71:301–308. doi: 10.1016/j.biopsych.2011.10.027. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-C. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31:464–468. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Shoaib M, Holsboer F, Almeida OF. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience. 1994;62:265–271. doi: 10.1016/0306-4522(94)90330-1. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Hassan AH, Holsboer DF, Almeida OF. The neurosteroid tetrahydroprogesterone attenuates the endocrine response to stress and exerts glucocorticoid-like effects on vasopressin gene transcription in the rat hypothalamus. Neuropsychopharmacology. 1996;15:533–540. doi: 10.1016/S0893-133X(96)00096-6. [DOI] [PubMed] [Google Scholar]
- Patchev VK, Montkowski A, Rouskova D, Koranyi L, Holsboer F, Almeida OF. Neonatal treatment of rats with the neuroactive steroid tetrahydrodeoxycorticosterone (THDOC) abolishes the behavioral and neuroendocrine consequences of adverse early life events. J Clin Invest. 1997;99:962–966. doi: 10.1172/JCI119261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisbeau P, Feltz P, Schlichter R. Modulation of GABAA receptor-mediated IPSCs by neuroactive steroids in a rat hypothalamo-hypophyseal coculture model. J Physiol. 1997;500:475–485. doi: 10.1113/jphysiol.1997.sp022034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu IR, Morton LA, Franco A, Di S, Ueta Y, Tasker JG. Synchronized bursts of miniature inhibitory postsynaptic currents. J Physiol. 2010;588:939–951. doi: 10.1113/jphysiol.2009.181461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapenko ES, Biancardi VC, Zhou Y, Stern JE. Astrocytes modulate a postsynaptic NMDA-GABAA-receptor crosstalk in hypothalamic neurosecretory neurons. J Neurosci. 2013;33:631–640. doi: 10.1523/JNEUROSCI.3936-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SA, Sejnowski TJ, De Koninck Y. Reduction of anion reversal potential subverts the inhibitory control of firing rate in spinal lamina I neurons: towards a biophysical basis for neuropathic pain. Mol Pain. 2006;2:32. doi: 10.1186/1744-8069-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Priebe K, Romeo RD, Francis DD, Sisti HM, Mueller A, McEwen BS, Brake WG. Maternal influences on adult stress and anxiety-like behavior in C57BL/6J and BALB/cJ mice: a cross-fostering study. Dev Psychobiol. 2005;47:398–407. doi: 10.1002/dev.20098. [DOI] [PubMed] [Google Scholar]
- Purdy RH, Morrow AL, Moore PH, Jr, Paul SM. Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci U S A. 1991;88:4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranna M, Sinkkonen ST, Möykkynen T, Uusi-Oukari M, Korpi ER. Impact of ε and θ subunits on pharmacological properties of α3β1 GABAA receptors expressed in Xenopus oocytes. BMC Pharmacol. 2006;6:1. doi: 10.1186/1471-2210-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology. 2008;149:4892–4900. doi: 10.1210/en.2008-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Campanac E, Scott RS, Rusakov DA, Kullmann DM. Presynaptic GABAA receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat Neurosci. 2010;13:431–438. doi: 10.1038/nn.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar J, Wakefield S, MacKenzie G, Moss SJ, Maguire J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J Neurosci. 2011;31:18198–18210. doi: 10.1523/JNEUROSCI.2560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüle C, Baghai TC, Eser D, Rupprecht R. Hypothalamic-pituitary-adrenocortical system dysregulation and new treatment strategies in depression. Expert Rev Neurother. 2009;9:1005–1019. doi: 10.1586/ern.09.52. [DOI] [PubMed] [Google Scholar]
- Shen Q, Lal R, Luellen BA, Earnheart JC, Andrews AM, Luscher B. γ-Aminobutyric acid-type A receptor deficits cause hypothalamic-pituitary-adrenal axis hyperactivity and antidepressant drug sensitivity reminiscent of melancholic forms of depression. Biol Psychiatry. 2010;68:512–520. doi: 10.1016/j.biopsych.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Stórustovu SI, Ebert B. Pharmacological characterization of agonists at δ-containing GABAA receptors: Functional selectivity for extrasynaptic receptors is dependent on the absence of γ2. J Pharmacol Exp Ther. 2006;316:1351–1359. doi: 10.1124/jpet.105.092403. [DOI] [PubMed] [Google Scholar]
- Swinny JD, O'Farrell E, Bingham BC, Piel DA, Valentino RJ, Beck SG. Neonatal rearing conditions distinctly shape locus coeruleus neuronal activity, dendritic arborization, and sensitivity to corticotrophin-releasing factor. Int J Neuropsychopharmacol. 2010;13:515–525. doi: 10.1017/S146114570999037X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker JG, Oliet SH, Bains JS, Brown CH, Stern JE. Glial regulation of neuronal function: from synapse to systems physiology. J Neuroendocrinol. 2012;24:566–576. doi: 10.1111/j.1365-2826.2011.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda K, Izumi Y, Zorumski CF. Ethanol enhances neurosteroidogenesis in hippocampal pyramidal neurons by paradoxical NMDA receptor activation. J Neurosci. 2011;31:9905–9909. doi: 10.1523/JNEUROSCI.1660-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Noda E, Kakazu Y, Mizoguchi Y, Akaike N, Nabekura J. Allopregnanolone enhancement of GABAergic transmission in rat medial preoptic area neurons. Am J Physiol Endocrinol Metab. 2002;283:E1257–1265. doi: 10.1152/ajpendo.00049.2002. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Jones KR, Ziegler DR, Cullinan WE, Herman JP. Forebrain origins of glutamatergic innervation to the rat paraventricular nucleus of the hypothalamus: differential inputs to the anterior versus posterior subregions. J Comp Neurol. 2011;519:1301–1319. doi: 10.1002/cne.22571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkuyl JM, Joëls M. Effect of adrenalectomy on miniature inhibitory postsynaptic currents in the paraventricular nucleus of the hypothalamus. J Neurophysiol. 2003;89:237–245. doi: 10.1152/jn.00401.2002. [DOI] [PubMed] [Google Scholar]
- Verkuyl JM, Karst H, Joëls M. GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur J Neurosci. 2005;21:113–121. doi: 10.1111/j.1460-9568.2004.03846.x. [DOI] [PubMed] [Google Scholar]
- Vicini S, Losi G, Homanics GE. GABAA receptor δ subunit deletion prevents neurosteroid modulation of inhibitory synaptic currents in cerebellar neurons. Neuropharmacology. 2002;43:646–650. doi: 10.1016/S0028-3908(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Pyner S, Barrett-Jolley R. Inhibition by α-tetrahydrodeoxycorticosterone (THDOC) of pre-sympathetic parvocellular neurones in the paraventricular nucleus of rat hypothalamus. Br J Pharmacol. 2006;149:600–607. doi: 10.1038/sj.bjp.0706911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, Li LH, Lee S, Jo IH, Lee SY, Ryu PD. Effects of adrenalectomy on the excitability of neurosecretory parvocellular neurones in the hypothalamic paraventricular nucleus. J Neuroendocrinol. 2007;19:293–301. doi: 10.1111/j.1365-2826.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- Zaki A, Barrett-Jolley R. Rapid neuromodulation by cortisol in the rat paraventricular nucleus: an in vitro study. Br J Pharmacol. 2002;137:87–97. doi: 10.1038/sj.bjp.0704832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Mathews D, Ibrahim L, Chaves JF, Marquardt C, Ukoh I, Jolkovsky L, Brutsche NE, Smith MA, Luckenbaugh DA. A randomized trial of a low-trapping nonselective N-methyl-D-aspartate channel blocker in major depression. Biol Psychiatry. 2013;74:257–264. doi: 10.1016/j.biopsych.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, Paul SM, Izumi Y, Covey DF, Mennerick S. Neurosteroids, stress and depression: potential therapeutic opportunities. Neurosci Biobehav Rev. 2013;37:109–122. doi: 10.1016/j.neubiorev.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]