

Abstract
Loss-of-function mutations of progranulin (PGRN) have been linked to frontotemporal dementia, but little is known about the effects of PGRN deficiency on the brain in health and disease. PGRN has been implicated in neurovascular development, inflammation, and Wnt signaling, a pathway involved in the formation of the blood–brain barrier (BBB). Because BBB alterations and inflammation contribute to ischemic brain injury, we examined the role of PGRN in the brain damage produced by ischemia-reperfusion. PGRN+/− and PGRN−/− mice underwent middle cerebral artery occlusion (MCAO) with monitoring of cerebral blood flow. Infarct volume and motor deficits were assessed 72 h later. Post-ischemic inflammation was examined by expression of inflammatory genes and flow cytometry. BBB structure and permeability were examined by electron microscopy (EM) and Evans blue (EB) extravasation, respectively. MCAO resulted in ∼60% larger infarcts in PGRN+/− and PGRN−/− mice, an effect independent of hemodynamic factors or post-ischemic inflammation. Rather, massive hemorrhages and post-ischemic BBB disruption were observed, unrelated to degradation of tight junction (TJ) proteins or matrix metalloproteinases (MMPs). By EM, TJ were 30–52% shorter, fewer, and less interlocking, suggesting a weaker seal between endothelial cells. Intracerebral injection of platelet-derived growth factor-CC (PDGF-CC), which increases BBB permeability, resulted in a more severe BBB breakdown in PGRN+/− and PGRN−/− than wild-type mice. We describe a previously unrecognized involvement of PGRN in the expression of key ultrastructural features of the BBB. Such a novel vasoprotective role of PGRN may contribute to brain dysfunction and damage in conditions associated with reduced PGRN function.
Keywords: blood–brain barrier, frontotemporal dementia, neurovascular unit, progranulin, stroke
Introduction
Progranulin (PGRN) is a 65 kDa secreted protein expressed ubiquitously throughout the body, including the brain, in which it is localized to neurons, microglia, and the developing vasculature (Daniel et al., 2003; Petkau et al., 2010; Matsuwaki et al., 2011). PGRN functions as a growth factor (Bateman and Bennett, 1998) and is thought to participate in embryogenesis, wound healing, cancer, and inflammation (Bateman and Bennett, 1998; De Muynck and Van Damme, 2011; Gass et al., 2012b). Mutations in the PGRN gene, located on chromosome 17q21.32 (Bhandari and Bateman, 1992), have been linked to frontotemporal dementia (FTD), a frequent cause of middle-age cognitive impairment characterized by selective neurodegeneration of the prefrontal and anterotemporal cortices (Rademakers et al., 2012). Microbleeds are also observed in areas of neurodegeneration, raising the possibility that the blood–brain barrier (BBB) is altered in FTD (De Reuck et al., 2012), but a link to PGRN mutations has not been established. Although it is well known that PGRN mutations induce haploinsufficiency of this protein (Baker et al., 2006; Cruts et al., 2006), the pathogenic mechanisms by which PGRN deficiency leads to brain dysfunction and damage remain unclear (Ward and Miller, 2011).
PGRN has strong neurotrophic (Gass et al., 2012a; De Muynck et al., 2013) and anti-inflammatory (Zhu et al., 2002; Kessenbrock et al., 2008; Tang et al., 2011) properties and is neuroprotective in several injury modalities, including oxygen glucose deprivation (Yin et al., 2010) and oxidative injury (Xu et al., 2011; Martens et al., 2012). Furthermore, PGRN is expressed in the developing cerebral microvasculature (Daniel et al., 2003) and may play a role in Wnt signaling (Rosen et al., 2011), a pathway involved in BBB development (Zlokovic, 2011; Reis and Liebner, 2013). These observations, in concert with the microvascular pathology observed in some cases of FTD (De Reuck et al., 2012), raise the possibility that loss of endogenous PGRN could alter the structure and function of cerebral microvessels and increase the susceptibility of the brain to BBB breakdown.
In the present study, we examined the role of PGRN in the mechanisms of brain injury produced by ischemia–reperfusion. We found that PGRN-deficient (PGRN-KO) mice have a profound disruption of the BBB in the early phase of ischemia–reperfusion, leading to brain hemorrhage and increased tissue damage. These alterations were linked to a constitutive defect in the ultrastructure of endothelial junctional complexes, resulting in an increased susceptibility to BBB breakdown. Our findings suggest a novel role of PGRN in the regulation of the BBB structure and function and raise the possibility that alterations in BBB contribute to brain pathologies associated with PGRN loss-of-function mutations.
Materials and Methods
Mice.
All procedures were approved by the institutional animal care and use committee of Weill Cornell Medical College. Experiments were performed in male PGRN heterozygous (PGRN+/−) (age, 6.9 ± 0.2 weeks; weight, 26.2 ± 0.4 g) and PGRN-KO (PGRN−/−) mice (age, 7.4 ± 0.2 weeks; weight, 24.4 ± 0.2 g) obtained from an in-house colony (B6-albino/CD1 mixed background) originally developed by Yin et al. (2010). Age-matched wild-type (PGRN+/+) littermates (WT: age, 7.2 ± 0.2 weeks; weight, 24.4 ± 0.3 g) served as controls. Group sizes were calculated based on power analysis. We used a power of 0.8 and significance levels of 0.05. Mice were randomly assigned to the various experimental groups, and analyses were performed by an investigator blinded to genotype and treatment.
Middle cerebral artery occlusion.
Transient focal cerebral ischemia was induced using an intraluminal filament model of middle cerebral artery occlusion (MCAO), as described previously (Jackman et al., 2011; Hochrainer et al., 2012). Briefly, mice were anesthetized with isoflurane (1.5–2%), and rectal temperature was maintained at ∼37°C. A heat-blunted suture was inserted via the right external carotid artery until it obstructed the MCA, and the common carotid artery simultaneously ligated for the duration of the ischemic period (33 min). Cerebral blood flow (CBF) was measured using transcranial laser Doppler flowmetry (Periflux System 5010; Perimed) in the center (bregma coordinates: 2 mm posterior, 5 mm lateral) and periphery (0 mm posterior, 2 mm lateral) of the ischemic territory, as determined in preliminary studies (Kunz et al., 2007). Only mice with >85% flow reduction during the ischemic period were included in this study. The number of excluded mice did not differ between groups. For sham MCAO, vessels were visualized and cleared of connective tissue. Functional impairment was assessed using the hanging-wire test (Cho et al., 2005; Abe et al., 2010). Infarct volume (swelling corrected) was quantified in cresyl violet-stained sections obtained throughout the infarcted territory, as described previously (Hochrainer et al., 2012). Post-ischemic swelling was assessed by measuring the difference in volume between the ischemic hemisphere and the contralateral one. Water content (edema) was assessed by the difference between the wet and dry weight of the ischemic hemisphere.
Measurement of resting CBF.
CBF was assessed quantitatively using arterial spin labeling magnetic resonance imaging (ASL-MRI), performed on a 7.0 Tesla 70/30 Bruker Biospec small-animal MRI system with 450 mT/m gradient amplitude and a 4500 T · m−1 · s−1 slew rate. A volume coil was used for transmission and a surface coil for reception. Anatomical localizer images were acquired to find the transversal slice approximately corresponding to bregma 0.5 mm. This position was used for subsequent ASL-MRI, which was based on a flow-sensitive alternating inversion recovery rapid aquisition with relaxation enhancement (FAIR-RARE) pulse sequence labeling the inflowing blood by global inversion of the equilibrium magnetization (Kim, 1995). One axial slice was acquired with a field of view of 15 × 15 mm, spatial resolution of 0.117 × 0.117 × 1 mm, TE of 5.368 ms, effective TE of 48.32 ms, recovery time of 10 s, and a RARE factor of 72. Twenty-two turbo inversion recovery values ranging from 30 to 2300 ms were used, and the inversion slab thickness was 4 mm. For computation of CBF, the Bruker ASL perfusion processing macro was used. The masked CBF images were exported and further processed using customized software. Data were spatially despiked by setting an upper cutoff value corresponding to the upper 0.1 percentile of data in each scan, and then the average value over the slice is reported as CBF (milliliters per 100 g per minute).
Cerebrovascular reactivity.
Changes in CBF were monitored in anesthetized, artificially ventilated mice using a laser-Doppler probe (Periflux System 5010; Perimed) positioned in a cranial window overlying the somatosensory cortex (Capone et al., 2012). After mean arterial pressure (70–80 mmHg) and blood gases (PCO2, 33–38 mmHg; PO2, 110–120 mmHg; pH 7.3–7.4) were stable, the cranial window was superfused with Ringer's solution, and CBF responses were recorded. The whisker-barrel cortex was activated by stroking the contralateral vibrissae, and the resulting changes in CBF were recorded (functional hyperemia). In addition, the endothelium-dependent vasodilators acetylcholine (10 μmol/L) and bradykinin (50 μmol/L) and the calcium ionophore A23187 (3 μmol/L) or the smooth muscle relaxant adenosine (400 μmol/L; Sigma-Aldrich) were superfused onto the exposed neocortex, and corresponding increases in CBF were recorded. CBF is expressed relative to baseline.
Real-time PCR.
A 1–2 mm strip of forebrain tissue was isolated from within the MCA territory (+1.2 to −0.8 mm bregma) and separated into hemispheres as described previously (Kunz et al., 2008). Total RNA was isolated using Trizol reagent (Invitrogen), and quantitative determination of gene expression was performed on a Chromo 4 detector (Peltier thermal cycler; MJ Research) using a two-step cycling protocol. Primers are listed in Table 1. Two microliters of diluted cDNA (1:10) were amplified by Platinum SYBR green qPCR supermix UDG (Invitrogen). The reactions were incubated at 50°C for 2 min and then 95°C for 10 min. A PCR cycling protocol consisting of 15 s at 95°C and 1 min at 60°C for 45 cycles was required for quantification. Relative expression levels were calculated according to Livak and Schmittgen (2001). Quantities of all targets in test samples were normalized to the mouse housekeeping hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene, and values were correlated to WT sham-treated samples (Kunz et al., 2008).
Table 1.
PCR primers used in the study
| Gene | MGI Symbol | Forward primer | Reverse primer |
|---|---|---|---|
| TNF | Tnf | TTGGAGTCATTGCTCTGTGAA | GGGTCAGAGTAAAGGGGTCAG |
| MCP-1 | Ccl2 | AGGTGTCCCAAAGAAGCTGTA | ATGTCTGGACCCATTCCTTCT |
| IL-6 | Il6 | ATGGATGCTACCAAACTGGAT | TGAAGGACTCTGGCTTTGTCT |
| IL-1β | Il1b | CTCTCCACCTCAATGGACAGA | TTTTGTCGTTGCTTGGTTCTC |
| ICAM-1 | Icam1 | GCCTTGGTAGAGGTGACTGAG | GACCGGAGCTGAAAAGTTGTA |
| ELAM-1 | Sele | CTCACTCCTGACATCGTCCTC | ACGTTGTAAGAAGGCACATGG |
| TGF-β1 | Tgfb1 | GAGAGCCCTGGATACCAACTA | GACAGAAGTTGGCATGGTAGC |
| TGF-β2 | Tgfb2 | TGGCCGAGCAGCGGATTGAAC | TGCACAGCGTCTGTCACGTCG |
| TGF-β3 | Tgfb3 | TGCTTCCGCAACCTGGAGGAGA | CTGCGCTGCGGAGGTATGGG |
| RANTES | Ccl5 | ATATGGCTCGGACACCACTC | GTGACAAACACGACTGCAAGA |
| SLPI | Slpi | GTGACGGCAAATACAAGTGCT | CTGGGAGCAGGGAAGTAGTTT |
| INOS | Nos2 | CAGCTGGGCTGTACAAACCTT | CATTGGAAGTGAAGCGTTTCG |
| NOX2 | Cybb | CCAACTGGGATAACGAGTTCA | GAGAGTTTCAGCCAAGGCTTC |
| COX-2 | COX2 | TGGTGCCTGGTCTGATGATG | GTGGTAACCGCTCAGGTGTTG |
| MMP-2 | Mmp2 | GAATGCCATCCCTGATAACCT | CAGCCAGTCTGATTTGATGCT |
| MMP-9 | Mmp9 | ATTCGCGTGGATAAGGAGTTC | CCTTGTTCACCTCATTTTGGA |
| CLDN5 | Cldn5 | TGGTGGCACTCTTTGTTACCT | AGTGCTACCCGTGCCTTAACT |
| VE-CADHERIN | Cdh5 | CTGTGGAGACCATAGGTGACAG | CTGGGATCTTTCTGGTGAGTG |
| PDGFB | Pdgfb | CGGCCGGTCCAGGTGAGAAAG | CACTTGCAGGCGAGGTGGTCC |
| PDGFRα | Pdgfra | CTCCTCACGCTGTGTGCGACC | GGCGGGATCCATCCAAGCCAT |
| PDGFRβ | Pdgfrb | ACTGCCGTGCAGCCCAATGAG | GGTGGAACTGGCAAGGCTGGG |
| VEGFA | Vegfa | CCAGGAGGACCTTGTGTGAT | GAGGAAGGGTAAGCCACTCAC |
| VEGFR1 | Ft2 | AGGAAGACGGTTCCTTCTGAG | CCCAGTGCACCTAGGTATGTC |
| VEGFR2 | Kdr | GGGGAACTGAAGACAGGCTAC | CGGCCAAGAGGTTTTCCTAGT |
| ANG1 | Angpt1 | GCTTCCCACCACATCTTTCTT | CCTGTAATGTCGGCACATACC |
| ANG2 | Angpt2 | ACTGGGAAGGCAACGAGGCG | GCGGTCCCCGTGAGTCCTGTAA |
| TIE2 | Tek | GGAAAATTTAACCCCATCTGC | CAGTGAATATGGCCACTGAGAA |
| tPA | Plat | AATAAAACCGTCACGAACAACA | TTTATTGATCATGCACACCAGAG |
| AQP4 | Aqp4 | TACATGGAGGTGGAGGACAAC | TCTTCTCTTCTCCACGGTCAA |
| HPRT | Hprt | CCGAGGATTTGGAAAAAGTGT | GGAAAATACAGCCAACACTGC |
SLPI, secretory leukocyte protease inhibitor; COX-2, cyclooxygenase-2; CLDN5, claudin-5; MGI, mouse gene identifiers.
Brain cell isolation and flow cytometry analyses.
Mice were transcardially perfused, and the right forebrain was dissected and gently triturated in 10 mm HEPES–HBSS buffer (in mm: 138 NaCl, 5 KCl, 0.4 Na2HPO4, 0.4 KH2PO4, and 5 d-glucose) using an MACS homogenizer (Miltenyi Biotec). The suspension was digested with 62.5 μg/ml Liberase DH (Roche Diagnostics) and 50 U/ml DNase I at 37°C for 45 min. Cells were washed by centrifugation and subjected to a 70–25% Percoll (GE Healthcare) density gradient centrifugation (25 min at 800 × g and 4°C). Enriched-mononuclear cells were collected from the interphase, washed with 10 mm HEPES–HBSS buffer, and resuspended in 2% fetal bovine serum in PBS (FACS buffer) for FACS analysis. Data were acquired on BD Accuri C6 flow cytometer (BD Biosciences) and analyzed using Accuri C6 software. Dead cells and debris were gated out by forward scatter and side scatter properties. Cell populations were separated based on CD45 and Ly6C expression and further sorted by CD11b and Gr1 expression on additional fluorescence channels. Infiltrating leukocytes were identified by CD45 high expression. Neutrophils were identified as CD45high followed by CD11b+/Gr+ gating. CD45high/CD11b+/Gr1− identified macrophages. CD45high/CD11b−/Gr1− gating was considered as non-myeloid cells.
Assessment of post-ischemic hemorrhage.
High-resolution images of coronal sections of perfused brain were obtained, and the area of hemorrhage was quantified using NIH Image J. In addition, 30 μm sections throughout the MCA territory were collected and processed for hematoxylin and eosin (H&E) staining, and hemorrhage was examined by light microscopy (Nikon eclipse 80i).
Post-ischemic EB quantification.
EB (2%) was injected intravenously. After 2 h circulation, mice were transcardially perfused with saline. The total ipsilateral forebrain was homogenized and diluted with trichloroacetic acid and centrifuged, and the supernatant was collected and diluted with ethanol. Samples were read in triplicate on a plate reader (620 nm excitation, 680 nm emission), and a standard curve was established using 0–10,000 ng/ml EB. No differences in EB concentration were observed between sham-operated mice and the non-ischemic hemisphere of mice subjected to MCAO. To reduce inter-animal variability, data were expressed as a ratio of nanograms of EB in ipsilateral hemisphere/contralateral hemisphere.
Western blotting.
A 1–2 mm strip of forebrain tissue was isolated from within the MCA territory (+1.2 to −0.8 mm bregma), separated into hemispheres, and homogenized in lysis buffer. Protein content was quantified (DC protein assay II; Bio-Rad), 50 μg of total protein was loaded in Laemmli's sample buffer and separated on 7% SDS-polyacrylamide gel, and proteins were transferred to a PVDF membrane. Membranes were incubated with primary antibodies [rabbit anti-zona occludens-1 (ZO-1; Mid; Invitrogen), rabbit anti-occludin (Invitrogen), and anti-β-actin (Sigma)] at 4°C overnight, followed by secondary antibodies (IgG HRP; Santa Cruz Biotechnology). Immunoreactive bands were visualized using luminol reagent and imaged (Kodak Image Station 2000R; Eastman Kodak). Each band was corrected relative to its β-actin loading control.
Gel zymography.
A 3 mm section of saline-perfused brain tissue within the MCA territory was isolated, separated into ipsilateral and contralateral hemispheres, frozen, and homogenized in lysis buffer (50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.05% Brij-35, 0.02% NaN3, and 1% Triton X-100). For matrix metalloproteinase (MMP) purification, 1.126 mg of protein was incubated with gelatin Sepharose 4B (GE Healthcare). After washing, beads were suspended in sample buffer for elution, and total eluent was separated on a 8% polyacrylamide gel containing 0.1% gelatin. Gels were then transferred to renaturing buffer (2.5% Triton X-100), followed by developing buffer (6.057 g of Tris base, 11.7 g of NaCl, 0.74 g of CaCl2, and 0.02% Brij 35) for 72 h at 37°C and stained in 0.25% Coomassie R-250 solution. Samples were expressed relative to mouse pro-MMP-9 standard (Anapsec). Linearity of gel degradation (MMP activity) to MMP concentration was confirmed over the range of 100–800 pg.
Electron microscopy.
Mice were deeply anesthetized and transcardially perfused with heparin (1000 U/ml) in saline, followed by acrolein (3.75%) in 2% paraformaldehyde. Brains were cut into coronal blocks, postfixed in paraformaldehyde (2%), and then sliced using a vibratome (Leica). Coronal sections were coprocessed for electron microscopic immunogold labeling for ZO-1 using a modified protocol (Chan et al., 1990). Tissue was processed and incubated for 48 h in primary ZO-1 antibody, followed by secondary donkey anti-rabbit colloidal gold (1 nm) IgG (GE Healthcare). Gold particles were further adhered to the tissue by glutaraldehyde and enhanced with a silver solution (IntenS-EM kit; GE Healthcare). Tissue was then postfixed with osmium tetraoxide and then ethanol dehydrated, followed by propylene oxide and incubation overnight in propylene oxide and Embed 812 Epon substitute (Electron Microscopy Sciences). Sections were then incubated in Epon substitute, embedded between two sheets of Aclar plastic, and placed in an oven. Ultrathin sections (70 nm) of cortex and striatum were cut using an ultratome (Leica) and diamond knife (Diatome). The sections were collected on copper grids, counterstained with uranyl acetate and lead citrate, and examined with a Phillips CM-10 electron microscope (FEI). The microscopic images were captured with an AMT Advantage HR/HR-B CCD Camera System (Advanced Microscopy Techniques). Micrographs of blood vessels (n = 73 in 9039.91 μm2 neocortical tissue and n = 108 in 9921.29 μm2 striatal tissue; n = 3 mice per group) were collected, and the length, number, and complexity of each tight junction (TJ) were analyzed using MCID (Imaging Research; Fig. 1). The ratio of pericyte coverage was quantified by tracing the pericyte length, including processes, in contact with the internal basal lamina and dividing the sum length by the circumference of the internal basal lamina (Frank et al., 1987).
Figure 1.

TJ tortuosity. Diagram depicting methods used to calculate the tortuosity index presented in Figure 8C. To calculate complexity of the TJ, TJ length (solid black line) is divided by the diagonal (dotted line) of the rectangle (dashed line) that contains the length and height of the complete TJ. EC, Endothelial cell; BL, basal lamina.
Brain microinjections of platelet-derived growth factor-CC.
Platelet-derived growth factor (PDGF-CC) disrupts the BBB when microinjected into the brain (Su et al., 2008). PDGF-CC is activated by tissue plasminogen activator (tPA; Fredriksson et al., 2004) and has been shown to open the BBB via activation of astrocytic PDGF receptor-α (Su et al., 2008). Using a glass micropipette, mouse recombinant PDGF-CC (R&D Systems; 72.6 ng) or 2 μl of vehicle (0.6% BSA) was injected into the striatum (bregma coordinates: 0.5 mm anterior, 2.5 mm lateral, 2.7 mm depth). The dose of PDGF-CC was determined in preliminary experiments to produce minimal or no BBB disruption in WT mice. Immediately after removal of the micropipette, EB (4%) was injected intravenously and allowed to circulate for 1 h. Serial coronal sections were obtained and EB-dependent fluorescence imaged using a fluorescence microscope equipped with appropriate filters (633 nm excitation, 660–700 nm emission). Total EB extravasation volume (cubic millimeters) was quantified using NIH Image J.
Statistical analyses.
Data are expressed as mean ± SE. Intergroup differences were analyzed using a Student's unpaired t test or one-way ANOVA with Bonferroni's post hoc analysis, as appropriate. Differences were considered statistically significant for p < 0.05.
Results
PGRN-deficient mice are more susceptible to ischemic brain injury
To assess the susceptibility of PGRN-deficient mice to cerebral ischemia, we examined the brain damage produced by transient MCAO. Both PGRN+/− and KO (−/−) mice had an increase in infarct volume compared with WT (+/+) mice (Fig. 2A). The infarct volume tended to be larger 24 h after MCAO (WT, 28.0 ± 6.8 mm3; PGRN-KO, 37.8 ± 6.7 mm3; p = 0.07, n = 14 per group) and reached statistical significance at 72 h, when the infarct volume was 50–60% larger in PGRN+/− and KO compared with WT (Fig. 2A). The increase in infarct volume was associated with a reduced latency to fall at the hanging wire test (Fig. 2B), reflecting impaired grip strength. In addition, PGRN deficiency increased post-ischemic hemispheric swelling at 72 h (WT, 3.2 ± 2.0 mm3; PGRN-KO, 12.8 ± 3.6 mm3; n = 11–13 per group; p < 0.05) and edema (hemispheric water content; WT, 80.2 ± 0.7% by weight; PGRN-KO, 83.5 ± 0.7%; n = 5–6 per group, p < 0.05). Thus, PGRN deficiency results in greater brain injury, edema, and motor deficits after MCAO.
Figure 2.

Ischemic injury is exacerbated in PGRN-deficient mice. A, B, Both PGRN+/− and PGRN-KO (−/−) have larger infarcts and have greater functional impairment than WT (+/+) mice 72 h after MCAO (n = 7–10 per group, *p < 0.05 from WT, ANOVA). C, D, The degree of cerebral ischemia and reperfusion are similar in the center and periphery of the ischemic territory in WT and PGRN-KO (p > 005). E, Resting CBF, measured using ASL-MRI, is similar in naive WT and PGRN-KO (n = 5 per group; p > 0.05, t test). F, Vascular responses to whisker stimulation (WS), the Ca2+ ionophore A23187 (CI), acetylcholine (ACh), bradykinin (BK), and adenosine (Ado) are similar in naive WT and PGRN-KO (−/−) (n = 5 per group; p > 0.05, t test).
Resting and intra-ischemic CBF and cerebrovascular reactivity are not different in PGRN-KO and WT mice
Next, we sought to determine if differences in the intensity of the ischemic insult could explain the larger infarct in PGRN-KO mice. To this end, we assessed the changes in CBF induced by MCAO in the center and periphery of the ischemic territory. However, the CBF reduction in these regions did not differ between WT (+/+) and PGRN-KO mice (Fig. 2C,D). To rule out the possibility that reductions in resting CBF contributed to the increased susceptibility of PGRN-KO mice to ischemic injury, we measured CBF quantitatively using ASL-MRI in non-ischemic mice. Resting CBF was similar in WT and PGRN-KO mice (Fig. 2E). Furthermore, in mice equipped with a cranial window, we found that the increases in CBF evoked by neural activity (whisker stimulation) and neocortical application of endothelium-dependent vasodilators or the smooth muscle relaxant adenosine were comparable in WT and PGRN-KO (Fig. 2F). Similarly, microvascular density assessed using staining for the endothelial marker Glut-1 did not differ between WT and PGRN-KO mice (WT, 15.0 ± 1.9 × 10 μm2; PGRN-KO, 19.3 ± 0.9 × 10 μm2; n = 5 per group, p > 0.05). Together, these observations indicate that hemodynamic factors resulting in more severe ischemia are unlikely to play a role in the increased ischemic damage in PGRN-KO mice.
Exaggerated post-ischemic inflammation does not contribute to increased injury in PGRN-KO mice
Because of the previously described anti-inflammatory effects of PGRN (Yin et al., 2010), we examined post-ischemic mRNA expression of selected inflammatory mediators involved in ischemic brain injury and/or PGRN signaling (Iadecola and Anrather, 2011; Cenik et al., 2012). These include cytokines [tumor necrosis factor (TNF), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), IL-1β, transforming growth factor-β1 (TGF-β1), and regulated on activation normal T-cell expressed and secreted/CCL5 (RANTES)], adhesion molecules [intracellular adhesion molecule-1 (ICAM-1) and endothelial leukocyte adhesion molecule-1 (ELAM-1)], and proinflammatory enzymes [inducible nitric oxide synthase (iNOS) and NADPH oxidase 2/gp91phox (Nox2)]. Overall, WT and PGRN-KO mice had comparable increases in mRNA for these mediators during the first 24 h after injury, the critical period for injury development (Fig. 3). There was a tendency for greater increases in the late post-ischemic period for ICAM-1 and MCP-1 (Fig. 3), which is consistent with secondary effects of the larger injury in these mice. To provide additional evidence that inflammation does not have a role in the increased injury in PGRN-KO mice, we used flow cytometry to quantify blood-borne inflammatory cells infiltrating the post-ischemic brain. The number of infiltrating leukocytes did not differ between WT and PGRN-KO mice 6 h after ischemia (Fig. 4A,B). Consistent with the mRNA data, leukocyte infiltration was higher in PGRN-KO mice at 72 h (Fig. 4A,C). Therefore, in PGRN-KO mice, there is no evidence of excessive inflammation in the early post-ischemic period when the damage develops.
Figure 3.
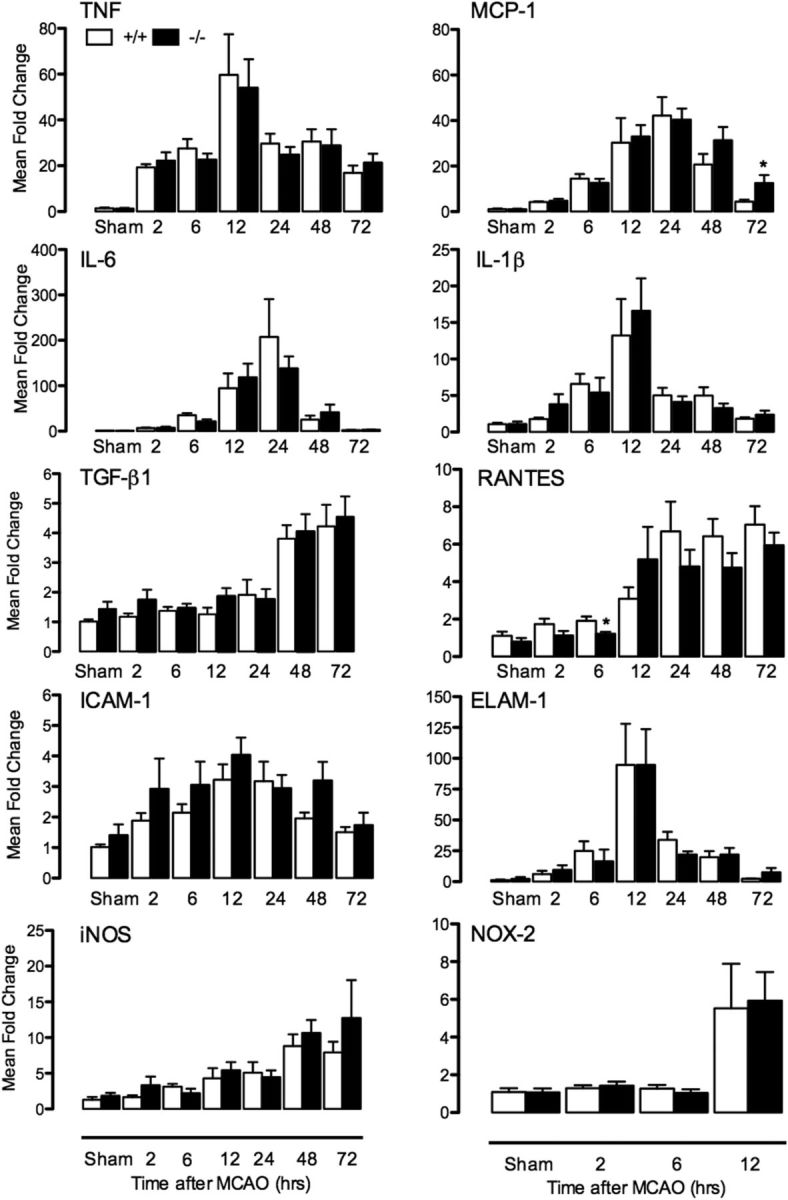
Post-ischemic inflammatory gene expression. Time course of mRNA expression of genes involved in post-ischemic inflammation in WT (+/+) and PGRN-KO (−/−) (n = 5–8 per group; *p < 0.05 from respective WT, t test).
Figure 4.

Post-ischemic inflammation. A, Representative density plots of brain mononuclear cells, expressed as percentage of total number of cells, in WT (+/+) and PGRN-KO (−/−) mice 6 and 72 h after MCAO measured with flow cytometry. B, C, Absolute numbers of infiltrating CD45high leukocytes are comparable in the post-ischemic brain of WT and PGRN-KO mice 6 h after MCAO (n = 5 per group; p > 0.05, t test). Leukocyte number is higher in PGRN-KO mice at 72 h, consistent with greater injury at this time (n = 6–8 per group; *p < 0.05, t test).
PGRN-KO mice exhibit increased hemorrhage and BBB breakdown after cerebral ischemia
Histological examination of the post-ischemic brain revealed that PGRN-KO mice have increased areas of hemorrhage in the ischemic territory. In WT mice, small petechial hemorrhages were occasionally observed in the striatum. However, in PGRN-KO mice, the large areas of hemorrhage were observed, starting 2–6 h after MCAO (Fig. 5A,B). Such pronounced hemorrhage soon after MCAO raised the possibility that the BBB breakdown produced by ischemia–reperfusion is exacerbated in PGRN-KO mice. To test this hypothesis, we examined BBB permeability using EB extravasation as a marker. Because hemorrhages were observed at 2–6 h, analyses were focused between 2 and 12 h after ischemia. In sham-operated mice, no differences in EB permeability were noticed between WT and PGRN-KO mice (Fig. 5D). In WT mice, post-ischemic EB extravasation was minimal and restricted to the striatum (Fig. 5C). In contrast, PGRN-KO mice exhibited a massive EB extravasation, which involved extensively both neocortex and striatum of the ischemic hemisphere (Fig. 5C). The EB extravasation was observed as early as 2 h after MCAO and was greatest at 6 h, when it was more than fivefold higher than in WT mice (Fig. 5D). TJ proteins are major components of the BBB, the disruption of which leads to BBB breakdown (Wolburg and Lippoldt, 2002). However, no differences in the expression of ZO-1, occludin, claudin-5 (CLDN5), and vascular endothelial (VE)-cadherin, critical proteins for TJ stability and endothelial permeability (Dejana et al., 2009; Daneman, 2012), were observed between WT and PGRN-KO mice, before as well as after MCAO (Fig. 6A–D). Furthermore, using immunogold electron microscopy, we did not observe differences in ZO-1 localization or number in junctional complexes (Fig. 6E), suggesting that mislocalization of this critical TJ protein was not causing the BBB disruption. Post-ischemic expression of MMP-2 and MMP-9, key mediators of the early and delayed opening of the BBB (Gasche et al., 1999; Yang et al., 2007; Fujimoto et al., 2008), did not differ between WT and PGRN-KO mice (Fig. 7A,B). There was a trend for MMP-9 expression to be higher in PGRN-KO mice at 48 and 72 h (Fig. 7A), a finding reflecting the more severe injury observed in PGRN-KO mice at this time. To confirm that MMP proteolytic activity was not enhanced in PGRN-KO mice early after ischemia, we measured MMP activity 6 h after MCAO, when hemorrhage was observed and BBB permeability was greatest. Pro-MMP-9 activity was observed at 6 h in both WT and PGRN-KO mice, but the levels were similar in the two groups (Fig. 7C). No MMP-2 activity was observed at 6 h. Therefore, differences in TJ proteins and post-ischemic MMP expression or activity cannot explain the increased BBB permeability in PGRN-KO mice.
Figure 5.

Post-ischemic hemorrhage and BBB disruption are enhanced in PGRN-KO. A, Representative H&E-stained brain section illustrating hemorrhage 6 h after MCAO in PGRN-KO mice. Early tissue injury is also seen as pallor in the dorsal striatum. Scale bar, 100 μm. B, The area of post-ischemic hemorrhage is greater in PGRN-KO (−/−) mice 6 h after MCAO compared with WT (+/+) mice (n = 8 per group; *p < 0.05, t test) C, Representative images of EB extravasation in WT and PGRN-KO mice 6 h after MCAO. Notice the increased BBB permeability in PGRN-KO mice. Scale bar, 5 mm. D, Temporal profile of EB extravasation after MCAO, expressed as ratio between ischemic and non-ischemic hemisphere, showing greater BBB permeability in PGRN-KO (n = 7–10 per group; *p < 0.05 from respective WT, t test).
Figure 6.

TJ proteins do not differ between WT and PGRN-KO mice after MCAO. A, Protein expression of ZO-1 is similar in WT (+/+) and PGRN-KO (−/−) mice at all time points after MCAO (S, sham). At 24 h, ZO-1 expression is reduced in both WT and PGRN-KO mice (n = 5–8 per group; *p < 0.05 from respective sham, ANOVA). B, Protein expression of occludin is similar in WT and PGRN-KO mice after MCAO (n = 5–8 per group; p > 0.05). C, D, No differences were detected in the mRNA expression of CLDN5 and VE-cadherin. E, Both the spatial distribution at the TJ cleft (representative images) and the ZO-1 immunogold density were similar in WT and PGRN-KO mice (n = 43–65 endothelial cells per group). Scale bar, 200 nm.
Figure 7.

MMP expression and activity do not differ between WT and PGRN-KO mice after MCAO. A, MMP-9 mRNA expression is similar in WT (+/+) and PGRN-KO (−/−) mice at early time points (2–24 h) but tends to be higher in PGRN-KO mice at 48 and 72 h (n = 5–8 per group; p > 0.05). B, MMP-2 mRNA is induced at 48 and 72 h after MCAO, but expression is similar in WT and PGRN-KO mice (n = 5–8 per group; *p < 0.05 from respective sham, ANOVA). C, Activity of pro-MMP-9 was assessed by gel zymography. Intensity of pro-MMP-9 in samples, expressed relative to pro-MMP-9 standard, is similar in WT and PGRN-KO mice 6 h after MCAO (n = 8 per group; p > 0.05).
Key ultrastructural features of the BBB are altered in naive PGRN-KO mice
To gain additional insight into the BBB characteristics of PGRN-KO mice, we examined the ultrastructure of endothelial junctional complexes in cerebral microvessels of the neocortex and striatum of naive mice. In WT mice, junctional complexes were characterized by a tortuous overlap of the membranes of adjacent endothelial cells (Fig. 8A). TJs appeared as areas of close apposition of neighboring membranes, with fusion of outer membrane leaflets (Fig. 8A). However, junctional complexes were 30–52% shorter and less tortuous in PGRN-KO than in WT mice, indicative of a less robust linkage between neighboring endothelial cells (Fig. 8B–D). Furthermore, TJ number was reduced in the neocortex of PGRN-KO mice (Fig. 8E). Pericytes play an important role in the development and maintenance of the BBB (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010). However, pericyte coverage, assessed by electron microscopy (Fig. 8F)—the gold standard for pericyte identification and quantification (Armulik et al., 2011)—and by expression of the pericyte marker PDGF-receptor-β (PDGFRβ; Fig. 9A), were similar in WT and PGRN-KO mice. These observations suggest profound alterations in the ultrastructure of the BBB in PGRN-KO mice.
Figure 8.

BBB ultrastructure is altered in PGRN-KO. A, B, Representative micrographs illustrating TJ morphology in WT (+/+) and PGRN-KO (−/−) mice. TJ complexity and length are reduced in PGRN-KO mice (arrows). Scale bar, 500 nm. C, TJ tortuosity (see Fig. 1) is reduced in PGRN-KO mice in neocortex (n = 108 to 129 TJ per group) and striatum (n = 65–109 TJ per group; *p < 0.05, t test). D, TJ are shorter in PGRN-KO in neocortex (n = 108–29 TJ per group) and striatum (n = 59–86 TJ per group; *p < 0.05, t test). E, TJ are fewer in PGRN-KO cortex (n = 42–47 vessels per group), but not striatum (n = 37–48 vessels per group; *p < 0.05, t test). F, Pericyte coverage is similar in WT and PGRN-KO cortex (n = 33–51 vessels per group) and striatum (n = 49–59 vessels per group). For all graphs, data was acquired in n = 3 WT and n = 3 PGRN-KO mice.
Figure 9.
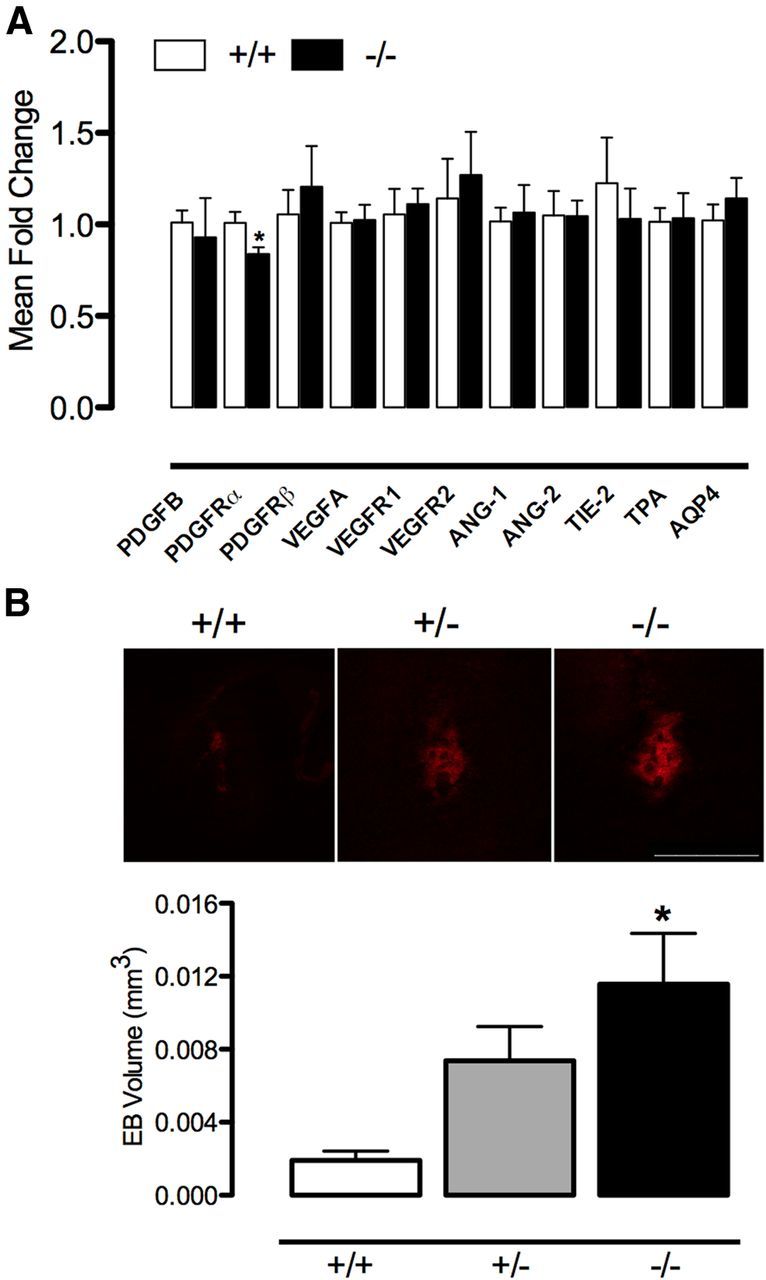
PGRN deficiency increases BBB permeability to PDGF-CC. A, No major differences were observed in the mRNA expression of factors involved in vascular development and stability, except for PDGFRα (n = 6 per group; *p < 0.05, t test). B, After injection of PDGF-CC, EB leakage/BBB permeability is enhanced in PGRN+/− and KO (−/−) mice compared with WT (+/+) (n = 6–7 per group; *p < 0.05 from WT, ANOVA). Scale bar, 500 μm. AQP4, Aquaporin-4.
The susceptibility to BBB breakdown is enhanced in naive PGRN-KO mice
No differences were observed in the mRNA expression of factors involved in vascular development and stability [vascular endothelial growth factor (VEGF), VEGF receptors (VEGFRs), angiopoietin-1 (ANG-1), ANG-2, angiopoietin receptor TIE-2, tissue plasminogen activator (tPA), etc.] between WT and PGRN-KO mice (Fig. 9A). To determine whether the observed reductions in TJ length and tortuosity in naive PGRN-KO mice are associated with an increased susceptibility to BBB breakdown, we assessed the effect of intrastriatal injection of PDGF-CC, an agent that increases BBB permeability (Su et al., 2008). Microinjection of PDGF-CC, at a dose that produced minimal or no BBB opening in WT mice, significantly increased EB extravasation in naive PGRN-KO mice (Fig. 9B). PGRN+/− mice had an intermediate phenotype (Fig. 9B). These findings demonstrate that PGRN-KO mice have an increased susceptibility to BBB disruption.
Discussion
We demonstrated that PGRN+/− and PGRN-KO mice have more extensive brain injury after transient focal cerebral ischemia. The increased damage in PGRN-KO mice is associated with a profound breakdown of the BBB, which occurs early after reperfusion and leads to hemorrhage in the ischemic territory. Underlying the BBB breakdown is an alteration of endothelial TJ number and morphology. Indeed, naive PGRN+/− and PGRN-KO mice exhibited increased susceptibility to BBB damage after intracerebral injection of PDGF-CC, providing evidence that the ultrastructural alterations were sufficient to disrupt the BBB when challenged. These observations demonstrate for the first time that PGRN deficiency results in structural alteration of endothelial junctional complexes that render the BBB more susceptible to damage, leading to hemorrhage and increased brain injury after ischemia–reperfusion.
Hemodynamic factors and post-ischemic inflammation do not contribute to increased injury in PGRN-deficient mice
PGRN-KO and PGRN+/− mice have larger cerebral infarcts and functional impairment after cerebral ischemia. Intra-ischemic CBF measurements confirmed that the ischemic insult was comparable in WT and PGRN-KO mice. Furthermore, resting CBF and CBF reactivity to key vasomotor stimuli did not differ between naive PGRN-KO and WT mice. Collectively, these findings suggest that hemodynamic factors cannot explain the increased susceptibility of PGRN-KO mice to ischemic brain injury, as observed in other conditions (Zhang et al., 1997). In addition, PGRN has been implicated in inflammatory signaling (Cenik et al., 2012). However, no differences in post-ischemic inflammation were observed between WT and PGRN-KO mice at early time points after ischemia when the tissue damage develops. Therefore, exaggerated inflammation is unlikely to play a role in the exacerbation of the damage afforded by PGRN deficiency.
BBB breakdown and hemorrhage occur early after ischemia in PGRN-deficient mice
We observed a substantial increase in brain hemorrhage in PGRN-KO mice 2–6 h after ischemia–reperfusion. Because loss of BBB integrity is considered a prerequisite for post-ischemic brain hemorrhage (Latour et al., 2004), we evaluated the temporal profile of BBB opening in these mice. Opening of the BBB was greatest at 6 h, consistent with the hemorrhages observed at this time point. Interestingly, the area of EB extravasation observed at 6 h after ischemia is larger than the infarcted area assessed at 72 h and incorporates the ischemic core as well as “penumbral” areas that were spared from infarction. Vascular inflammation is unlikely to be responsible for the BBB disruption, because the upregulation of vascular inflammatory genes, such as ELAM-1, was not enhanced in PGRN-KO mice. Several studies have reported a key role for TJ proteins and MMP-2 and MMP-9 and in the development of hemorrhage and BBB breakdown in the early stages of cerebral ischemia (Liu et al., 2012; Suofu et al., 2012). However, despite the profound BBB disruption 6 h after MCAO, post-ischemic expression of ZO-1, CLDN5, and occludin, key TJ proteins (Daneman, 2012), was preserved in PGRN-KO mice. In addition, there were no differences in levels of TJ proteins in the absence of ischemia (sham surgery). MMP-9 expression and activity were also similar in WT and PGRN-KO mice at early time points after ischemia. These data suggest that BBB permeability observed in PGRN-KO mice is not attributable to a generalized degradation of the BBB by MMPs or to enhanced post-ischemic suppression of TJ proteins.
PGRN is required for normal endothelial TJ ultrastructure
To explore the possibility that PGRN could be involved in the formation of endothelial junctional complexes, we assessed cerebral microvascular ultrastructure in PGRN-KO mice. Because naive PGRN-KO mice do not suffer from spontaneous hemorrhages or BBB permeability defects, as shown by lack of increased permeability to EB, we were not surprised by the absence of major structural alterations in microvascular structure, such as pericyte loss or detachment of endothelial TJ (Lindahl et al., 1997; Nico et al., 2003; Zlokovic, 2011). However, we did observe distinctive alterations in the morphology of endothelial junctional complexes. Deficiency of PGRN resulted in a transformation of the TJ from an intricate, elongated interaction of adjacent membrane leaflets in WT mice to a simplified abutment of adjacent cells in PGRN-KO mice. Furthermore, we found the TJ cleft to be substantially shorter in PGRN-KO mice, resulting in a corresponding reduction in the size of the physical barrier formed by the TJ in these mice. Reduced TJ complexity and length are consistent with a structural weakening of the BBB, which could contribute to the increased BBB permeability and hemorrhage observed 6 h after ischemia. Accordingly, striatal injection of PDGF-CC induced larger increases in BBB permeability in PGRN-KO than in WT mice, supporting the hypothesis that the BBB is intrinsically more susceptible to disruption and damage in these mice. Although the increase in PDGF-CC-induced BBB permeability was less pronounced in PGRN+/− mice, such alteration was sufficient to increase infarct volume, attesting to the critical importance of BBB integrity to the outcome of cerebral ischemia. We did not examine TJ ultrastructure in PGRN+/− mice, but based on our findings of comparable increases in infarct volume and in BBB permeability after PDGF-CC injection, it is reasonable to assume that PGRN+/− mice also have TJ alterations.
To our knowledge, the present study provides the first evidence implicating PGRN in the normal development of TJ morphology and in the susceptibility of the BBB to injury. The molecular mechanisms of the BBB alterations remain to be established. PGRN was implicated recently in the regulation of the Wnt/β-catenin pathway (Rosen et al., 2011), a major mediator of BBB development and maintenance (Dejana et al., 2009). The Wnt signaling pathway is upregulated by PGRN deficiency in PGRN-KO mice, as well as FTD attributable to PGRN mutations (Rosen et al., 2011; Wexler et al., 2011). However, it remains unclear whether upregulation of Wnt alters the ultrastructure of the BBB, resulting in increased permeability. In addition, the Wnt signaling pathway may contribute to neurogenesis and post-stroke recovery (Shruster et al., 2012) and regulate post-ischemic vascular remodeling and angiogenesis (Liebner and Plate, 2010). Therefore, it is conceivable that PGRN deficiency may also influence recovery at later time points after stroke.
Vascular expression of PGRN was not assessed in this study. Although PGRN has been reported in the developing brain microvasculature (Daniel et al., 2003), the regional distribution throughout the vessel wall and cellular localization have yet to be characterized. Endothelial localization is likely, however PGRN is a secreted protein and could gain access to the endothelium via the plasma or from other cells of the neurovascular unit. Thus, additional studies are required to define the sources and targets of PGRN within the vascular wall and associated cells.
Implications for neurological diseases associated with PGRN deficiency
Mutations in the PGRN gene are a common cause of familial FTD and are associated with accumulation of the transactive response DNA binding protein 43 (Rademakers et al., 2012). These pathogenic mutations, of which at least 69 have now been described (Cruts et al., 2012), are all loss of function, resulting from PGRN haploinsufficiency and reduced levels of active PGRN (Sieben et al., 2012). The mechanisms by which loss of PGRN function leads to pathology remain to be established. Protein mishandling with accumulation of ubiquitinated aggregates, excessive inflammation, or loss of neurotrophic support have been suggested as potential pathogenic mechanisms in several disease models (Ward and Miller, 2011). However, we found that exaggerated inflammation does not contribute to the enhanced damage in ischemia–reperfusion. Rather, our studies suggest the novel possibility that PGRN deficiency leads to constitutive microvascular alterations that increase the susceptibility of the brain to BBB disruption. Although other PGRN prosurvival effects cannot be ruled out (Almeida et al., 2012; Gass et al., 2012a; Tao et al., 2012; De Muynck et al., 2013), since PGRN-KO are also protected from the hippocampal damage produced by oxygen-glucose deprivation in vitro, in which BBB permeability has no role (Yin et al., 2010). However the present findings link for the first time PGRN deficiency to alterations in BBB ultrastructure and function and raise the possibility that BBB breakdown plays a role in neurological conditions associated with PGRN deficiency. Now that a role of PGRN in BBB structure and function has been established in young mice, in the absence of confounding effects of aging on the BBB (Zlokovic, 2008), CBF regulation (Park et al., 2007), and ischemic injury (Nagayama et al, 1999). However, now that a role of PGRN in BBB structure and function has been established in young mice, it would be informative to examine the BBB in middle-aged PGRN-KO mice, because FTD is observed in this age group. Despite differences in PGRN expression, similar FTD-associated abnormalities in behavior have been reported in both PGRN-KO and PGRN+/− mice in the absence of excessive inflammation (Filiano et al., 2013). This observation, in concert with our finding of a similarly increased susceptibility to cerebral ischemia in the absence of major early inflammatory changes, suggests that our observations in PGRN-KO mice may be relevant to human diseases with PGRN loss of function.
Conclusions
In summary, we demonstrated that ischemic injury is enhanced in PGRN+/− and PGRN-KO mice. The effect is not related to hemodynamic dysfunction or, as suggested by previous studies, enhancement of post-ischemic inflammation. Rather, the BBB integrity is weakened in PGRN-KO mice, resulting in massive BBB breakdown and increased hemorrhage after ischemia–reperfusion. The mechanisms of the BBB disruption involve a constitutive alteration in endothelial junctional complexes resulting in reduced TJ tortuosity, number, and length. These findings provide novel evidence that PGRN deficiency leads to major alterations in BBB structure and function that predispose PGRN-KO mice to BBB breakdown, hemorrhage, and increased ischemic brain injury. Furthermore, the data raise the intriguing possibility that loss of the vasoprotective effects of PGRN may play a pathogenic role in brain diseases associated with PGRN loss-of-function mutations.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institutes of Health Grants NS34179 (C.I.) and NS73666 (C.I.), a National Health and Medical Research Council C. J. Martin Overseas training fellowship (K.J.) and USA to Australia Fellowship by the American Australian Association (K.J.), and German Research Foundation Grant KA2279/4-1 (T.K.).
References
- Abe T, Shimamura M, Jackman K, Kurinami H, Anrather J, Zhou P, Iadecola C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke. 2010;41:898–904. doi: 10.1161/STROKEAHA.109.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, Geschwind MD, Tartaglia MC, Gao F, Gianni D, Sena-Esteves M, Geschwind DH, Miller BL, Farese RV, Jr, Gao F-B. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012;2:789–798. doi: 10.1016/j.celrep.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. Granulins: the structure and function of an emerging family of growth factors. J Endocrinol. 1998;158:145–151. doi: 10.1677/joe.0.1580145. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V, Bateman A. Structure and chromosomal location of the human granulin gene. Biochem Biophys Res Commun. 1992;188:57–63. doi: 10.1016/0006-291X(92)92349-3. [DOI] [PubMed] [Google Scholar]
- Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, Davisson RL, Iadecola C. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106–113. doi: 10.1161/HYPERTENSIONAHA.112.193672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem. 2012;287:32298–32306. doi: 10.1074/jbc.R112.399170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Aoki C, Pickel VM. Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods. 1990;33:113–127. doi: 10.1016/0165-0270(90)90015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat. 2012;33:1340–1344. doi: 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012;72:648–672. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–599. doi: 10.1002/dvdy.10341. [DOI] [PubMed] [Google Scholar]
- Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- De Muynck L, Van Damme P. Cellular effects of progranulin in health and disease. J Mol Neurosci. 2011;45:549–560. doi: 10.1007/s12031-011-9553-z. [DOI] [PubMed] [Google Scholar]
- De Muynck L, Herdewyn S, Beel S, Scheveneels W, Van Den Bosch L, Robberecht W, Van Damme P. The neurotrophic properties of progranulin depend on the granulin E domain but do not require sortilin binding. Neurobiol Aging. 2013;34:2541–2547. doi: 10.1016/j.neurobiolaging.2013.04.022. [DOI] [PubMed] [Google Scholar]
- De Reuck J, Deramecourt V, Cordonnier C, Auger F, Durieux N, Bordet R, Maurage CA, Leys D, Pasquier F. Detection of microbleeds in post-mortem brains of patients with frontotemporal lobar degeneration: a 7.0-Tesla magnetic resonance imaging study with neuropathological correlates. Eur J Neurol. 2012;19:1355–1360. doi: 10.1111/j.1468-1331.2012.03776.x. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, Jiao J, Zhang Z, Huang EJ, Gao FB, Farese RV, Jr, Roberson ED. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013;33:5352–5361. doi: 10.1523/JNEUROSCI.6103-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RN, Dutta S, Mancini MA. Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Invest Ophthalmol Vis Sci. 1987;28:1086–1091. [PubMed] [Google Scholar]
- Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Takagi Y, Aoki T, Hayase M, Marumo T, Gomi M, Nishimura M, Kataoka H, Hashimoto N, Nozaki K. Tissue inhibitor of metalloproteinases protect blood–brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28:1674–1685. doi: 10.1038/jcbfm.2008.59. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen-West K, Lewis J, Link CD, Rademakers R, Nykjaer A, Petrucelli L. Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener. 2012a;7:33. doi: 10.1186/1750-1326-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, Prudencio M, Stetler C, Petrucelli L. Progranulin: an emerging target for FTLD therapies. Brain Res. 2012b;1462:118–128. doi: 10.1016/j.brainres.2012.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrainer K, Jackman K, Anrather J, Iadecola C. Reperfusion rather than ischemia drives the formation of ubiquitin aggregates after middle cerebral artery occlusion. Stroke. 2012;43:2229–2235. doi: 10.1161/STROKEAHA.112.650416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman K, Kunz A, Iadecola C. Modeling focal cerebral ischemia in vivo. Methods Mol Biol. 2011;793:195–209. doi: 10.1007/978-1-61779-328-8_13. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Fröhlich L, Sixt M, Lämmermann T, Pfister H, Bateman A, Belaaouaj A, Ring J, Ollert M, Fässler R, Jenne DE. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (FAIR) technique: application to functional mapping. Magn Reson Med. 1995;34:293–301. doi: 10.1002/mrm.1910340303. [DOI] [PubMed] [Google Scholar]
- Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz A, Abe T, Hochrainer K, Shimamura M, Anrather J, Racchumi G, Zhou P, Iadecola C. Nuclear factor-κB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J Neurosci. 2008;28:1649–1658. doi: 10.1523/JNEUROSCI.5205-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour LL, Kang DW, Ezzeddine MA, Chalela JA, Warach S. Early blood-brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56:468–477. doi: 10.1002/ana.20199. [DOI] [PubMed] [Google Scholar]
- Liebner S, Plate KH. Differentiation of the brain vasculature: the answer came blowing by the Wnt. J Angiogenes Res. 2010;2:1. doi: 10.1186/2040-2384-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood–brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martens LH, Zhang J, Barmada SJ, Zhou P, Kamiya S, Sun B, Min SW, Gan L, Finkbeiner S, Huang EJ, Farese RV., Jr Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. J Clin Invest. 2012;122:3955–3959. doi: 10.1172/JCI63113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuwaki T, Asakura R, Suzuki M, Yamanouchi K, Nishihara M. Age-dependent changes in progranulin expression in the mouse brain. J Reprod Dev. 2011;57:113–119. doi: 10.1262/jrd.10-116S. [DOI] [PubMed] [Google Scholar]
- Nagayama M, Aber T, Nagayama T, Ross ME, Iadecola C. Age-dependent increase in ischemic brain injury in wild-type mice and in mice lacking the inducible nitric oxide synthase gene. J Cereb Blood Flow Metab. 1999;19:661–666. doi: 10.1097/00004647-199906000-00009. [DOI] [PubMed] [Google Scholar]
- Nico B, Frigeri A, Nicchia GP, Corsi P, Ribatti D, Quondamatteo F, Herken R, Girolamo F, Marzullo A, Svelto M, Roncali L. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia. 2003;42:235–251. doi: 10.1002/glia.10216. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- Petkau TL, Neal SJ, Orban PC, MacDonald JL, Hill AM, Lu G, Feldman HH, Mackenzie IRA, Leavitt BR. Progranulin expression in the developing and adult murine brain. J Comp Neurol. 2010;518:3931–3947. doi: 10.1002/cne.22430. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423–434. doi: 10.1038/nrneph.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis M, Liebner S. Wnt signaling in the vasculature. Exp Cell Res. 2013;319:1317–1323. doi: 10.1016/j.yexcr.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Rosen EY, Wexler EM, Versano R, Coppola G, Gao F, Winden KD, Oldham MC, Martens LH, Zhou P, Farese RV, Jr, Geschwind DH. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shruster A, Ben-Zur T, Melamed E, Offen D. Wnt signaling enhances neurogenesis and improves neurological function after focal ischemic injury. PLoS One. 2012;7:e40843. doi: 10.1371/journal.pone.0040843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P, De Deyn PP, Santens P, Van Broeckhoven C, Cruts M. The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol. 2012;124:353–372. doi: 10.1007/s00401-012-1029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su EJ, Fredriksson L, Geyer M, Folestad E, Cale J, Andrae J, Gao Y, Pietras K, Mann K, Yepes M, Strickland DK, Betsholtz C, Eriksson U, Lawrence DA. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suofu Y, Clark JF, Broderick JP, Kurosawa Y, Wagner KR, Lu A. Matrix metalloproteinase-2 or -9 deletions protect against hemorrhagic transformation during early stage of cerebral ischemia and reperfusion. Neuroscience. 2012;212:180–189. doi: 10.1016/j.neuroscience.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Lu Y, Tian QY, Zhang Y, Guo FJ, Liu GY, Syed NM, Lai Y, Lin EA, Kong L, Su J, Yin F, Ding AH, Zanin-Zhorov A, Dustin ML, Tao J, Craft J, Yin Z, Feng JQ, Abramson SB, Yu XP, Liu CJ. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332:478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Ji F, Wang F, Liu B, Zhu Y. Neuroprotective effects of progranulin in ischemic mice. Brain Res. 2012;1436:130–136. doi: 10.1016/j.brainres.2011.11.063. [DOI] [PubMed] [Google Scholar]
- Ward ME, Miller BL. Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci. 2011;45:574–582. doi: 10.1007/s12031-011-9622-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler EM, Rosen E, Lu D, Osborn GE, Martin E, Raybould H, Geschwind DH. Genome-wide analysis of a Wnt1-regulated transcriptional network implicates neurodegenerative pathways. Sci Signal. 2011;4:ra65–ra65. doi: 10.1126/scisignal.2002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323–337. doi: 10.1016/S1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- Xu J, Xilouri M, Bruban J, Shioi J, Shao Z, Papazoglou I, Vekrellis K, Robakis NK. Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol Aging. 2011;32:2326e5–e16. doi: 10.1016/j.neurobiolaging.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, Nathan C, Ding A. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/S0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]