

Abstract
Slow waves represent one of the prominent EEG signatures of non-rapid eye movement (non-REM) sleep and are thought to play an important role in the cellular and network plasticity that occurs during this behavioral state. These slow waves of natural sleep are currently considered to be exclusively generated by intrinsic and synaptic mechanisms within neocortical territories, although a role for the thalamus in this key physiological rhythm has been suggested but never demonstrated. Combining neuronal ensemble recordings, microdialysis, and optogenetics, here we show that the block of the thalamic output to the neocortex markedly (up to 50%) decreases the frequency of slow waves recorded during non-REM sleep in freely moving, naturally sleeping-waking rats. A smaller volume of thalamic inactivation than during sleep is required for observing similar effects on EEG slow waves recorded during anesthesia, a condition in which both bursts and single action potentials of thalamocortical neurons are almost exclusively dependent on T-type calcium channels. Thalamic inactivation more strongly reduces spindles than slow waves during both anesthesia and natural sleep. Moreover, selective excitation of thalamocortical neurons strongly entrains EEG slow waves in a narrow frequency band (0.75–1.5 Hz) only when thalamic T-type calcium channels are functionally active. These results demonstrate that the thalamus finely tunes the frequency of slow waves during non-REM sleep and anesthesia, and thus provide the first conclusive evidence that a dynamic interplay of the neocortical and thalamic oscillators of slow waves is required for the full expression of this key physiological EEG rhythm.
Introduction
Slow waves and their neuronal counterpart, the cortical and thalamic oscillations between depolarized UP states and hyperpolarized DOWN states (Steriade et al., 1993a; Contreras and Steriade, 1995; Petersen et al., 2003; Sirota and Buzsáki, 2005; Crunelli et al., 2012), are the main EEG hallmark of non-rapid eye movement (non-REM) sleep (Crunelli and Hughes, 2010; Brown et al., 2012) and are also observed during anesthesia (Chauvette et al., 2011). The physiological importance of these waves of natural sleep is emphasized by their ability to group together other EEG rhythms of non-REM sleep (Steriade, 1997) and by their putative role in the consolidation of recently acquired memories (Tononi and Cirelli, 2001; Marshall et al., 2006; Ji and Wilson, 2007).
The mechanisms underlying the generation of EEG slow waves, however, remain controversial. Because (1) lesions of thalamic nuclei do not suppress slow waves in anesthetized cats (Steriade et al., 1993b) and (2) UP and DOWN states are recorded in neocortical slices (Sanchez-Vives and McCormick, 2000; Cossart et al., 2003) and in an isolated cortical gyrus in vivo during anesthesia (Timofeev et al., 2000), these EEG slow waves are exclusively and consistently viewed as a cortically generated rhythm (Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000; Chauvette et al., 2011; Brown et al., 2012). However, (1) increasing thalamic inhibition alters EEG slow waves in anesthetized rats (Doi et al., 2007) and suppresses whisking-induced cortical UP states in head-restrained mice (Poulet et al., 2012); (2) UP and DOWN states, and associated slow waves, can be recorded in thalamic slices (Hughes et al., 2002, 2004; Blethyn et al., 2006); and (3) selective thalamic degeneration modifies slow waves of non-REM sleep in humans (Gemignani et al., 2012). These findings, together with other mechanistic in vitro studies and investigations in anesthetized animals (for review, see Crunelli and Hughes, 2010), question the current corticocentric view of slow wave generation and led us to suggest that the full expression of these EEG waves of natural sleep requires a dynamic interplay of cortical and thalamic oscillators (Crunelli and Hughes, 2010). Unfortunately, the resolution of this controversy is still hampered by the lack of any study that has directly and systematically addressed this issue in unrestrained, naturally waking-sleeping animals. Moreover, our current mechanistic knowledge of slow waves of natural sleep continues to be clouded by the speculative extrapolations of findings obtained in anesthetized conditions.
Using a combination of neuronal ensemble recordings, thalamus-selective pharmacological inactivation, and optogenetic activation of thalamocortical (TC) neurons in naturally sleeping or anesthetized rats, here we show, for the first time, that the thalamus is required for finely tuning the frequency of slow waves during non-REM sleep and anesthesia. Moreover, we demonstrate that the entrainment of EEG slow waves by selective thalamic activation is dependent on T-type calcium channels. Together, these results provide the first conclusive evidence that cortical and thalamic oscillators are necessary for the full expression of slow waves of non-REM sleep.
Materials and Methods
All experimental procedures were performed in accordance with the United Kingdom Animals (Scientific Procedure) Act 1986 and local ethics committee guidelines. All efforts were made to minimize animal suffering and the number of animals used. Experiments were performed on male adult Wistar rats (260–400 g, Harlan Laboratories), maintained on a normal diet and under a 8:00 A.M. to 8:00 P.M. light-on regimen.
Experiments in anesthetized rats
Surgery.
After anesthesia induction with 5% isoflurane, rats received an intraperitoneal injection of ketamine (120 mg/kg) and xylazine (20 mg/kg). Anesthesia was then maintained with a constant flow of ketamine (42 mg/kg/h) and xylazine (7 mg/kg/h) delivered via an intraperitoneal catheter connected to a pump (NewEra NE-300 syringe pump). Body temperature was maintained at 37°C with a heating pad and rectal probe. Rats were implanted with gold-plated skull screws (diameter 1 mm, length 3 mm) for EEG recordings in S1: anteroposterior (AP) = −2.2 mm, mediolateral (ML) = ±5.5 mm from bregma (Paxinos and Watson, 2007). An additional screw (ground electrode) was placed anterior to the bregma, and two other screws above the cerebellum were used as reference electrodes for EEG and thalamic recordings.
For measuring the spatial extent of the action of 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2), a potent and selective T-type channel antagonist (Uebele et al., 2009; Dreyfus et al., 2010), or of the sodium channel blocker TTX in the thalamus (i.e., see experiments illustrated in Fig. 3), a 1 mm hole was drilled unilaterally above the ventrobasal thalamic nucleus (VB), the dura was carefully removed, and a 32-channel silicone probe (10 mm length, 0.6 mm width, 1–4 mΩ) with four shanks (200 μm recording point distance, 15 μm thickness) (NeuroNexus Technologies) was slowly lowered in the VB (dorsoventral [DV] = −4.5 to −5.5 mm). A second hole in the same hemisphere was used to slowly lower (200 μm every 5 min) a microdialysis probe (CMA 12 Elite, 2 mm dialysis membrane length, 20 kDa cutoff, with a 16° angle with respect to the vertical axis; see Fig. 3A) to a final position that was between 0.05 and 1 mm away from the silicone probe. The 16° angle positioning of the dialysis probe was dictated by the space constraints of the silicone and dialysis probes connecting devices on the animal skull.
Figure 3.

Block of TC neuron firing by TTA-P2 directly applied by reverse microdialysis in the thalamus under anesthesia. A, Coronal brain section showing the position of a microdialysis (DP) (inserted with a 16° angle with respect to the vertical axis, see Materials and Methods) and a silicone probe (SP) in the VB, both stained with a red fluorescent dye. B1, Distance–response curve of TTA-P2-elicited block of high-frequency bursts in VB TC neurons (n = 533 neurons from 37 rats). Burst rate was measured between 50 and 60 min from the start of TTA-P2 or TTX dialysis. Data are normalized to the burst rate measured during the last 10 min of the preceding 1 h of aCSF dialysis (see Materials and Methods). Different TTA-P2 concentrations are color-coded as illustrated and refer to the drug concentration in the inlet dialysis tube. There is similarity in the action of 1 and 3 mm TTA-P2. The effect of TTX is also depicted (n = 33 neurons from 11 rats). B2, Same as B1, but for total TC neuron firing (i.e., high-frequency bursts plus single action potentials). C, Distance dependence of time of half-block of high-frequency bursts by VB microdialysis of 300 μm TTA-P2 (top) and of time of half-block of total firing by VB microdialysis of 50 μm TTX (bottom). Black lines indicate the best fit of a fourth-order parabolic function. D, Schematic brain drawing (from Paxinos and Watson, 2007) showing that the area of burst firing block achieved with the dialysis of 300 μm TTA-P2 (green) (calculated from the data shown in B2) covers almost the entire VB. Only a small increase in the area of block (which now covers a small portion of the NRT) is achieved with 3 mm TTA-P2 (red). This drawing assumes a fully vertical position of the dialysis probe as it was used for all the experiments described in Figures 4, 5, 6, and 7. E, In vivo recovery of TTA-P2 applied by dialysis (n = 6 rats for both concentrations), which was estimated using the formula ([X]in − [X]out)/[X]in, where [X]in and [X]out are the TTA-P2 concentration in the inlet and outlet dialysis tubes, respectively (Chan and Chan, 1999).
For measuring the effect of TTX and TTA-P2 (applied by microdialysis in the VB) on slow and spindle waves during anesthesia (i.e., see experiments illustrated in Fig. 4), EEG electrodes were implanted as above, and two microdialysis probes (one in each VB) were slowly lowered fully vertically until their tips rested 6.5 mm below the skull (i.e., in the most ventral part of the VB). In some rats, a silicone probe was also inserted unilaterally in the VB with a 16° angle with respect to the vertical axis.
Figure 4.
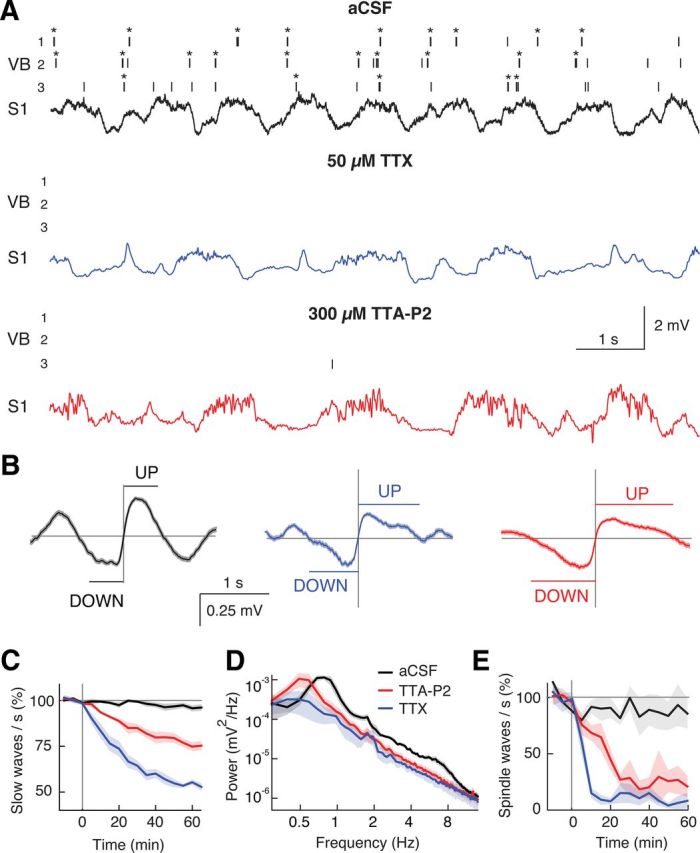
Block of thalamic firing decelerates EEG slow waves during anesthesia. A, Spike raster plots (top three traces; *bursts) from 3 VB TC neurons and EEG from S1 (bottom trace) show the effects of 50 μm TTX and 300 μm TTA-P2 dialysis in the VB. The predominant burst firing during aCSF is virtually abolished by TTX and TTA-P2, an effect accompanied by slowing of the EEG rhythm. B, Event-triggered averages of raw EEG traces centered on the middle point of DOWN to UP state transitions were calculated after 1 h of aCSF, TTX, and TTA-P2 dialysis (n = 438, 243, and 222 transitions, respectively). C, Normalized (to predrug period), time-dependent decrease of slow waves by TTX (n = 5) and TTA-P2 (n = 5) (drug dialysis starts at 0). D, EEG power spectra 60 min after start of drug dialysis. E, Normalized (to predrug period), time-dependent reduction of spindle waves by TTX and TTA-P2. C–E, Solid lines indicate the mean; color shadings indicate SEM. In this and the following figures, illustrated drug concentrations during microdialysis are those of the inlet dialysis tube (for brain concentration delivered by dialysis probes, see Fig. 3E). In this experiment and those depicted in Figures 5, 6, and 7, the dialysis probes were inserted in a fully vertical position.
For measuring the effect of systemic TTA-P2 injection (i.e., see experiments illustrated in Fig. 5), rats were implanted with EEG electrodes and a unilateral silicone probe in the VB (as described above).
Figure 5.
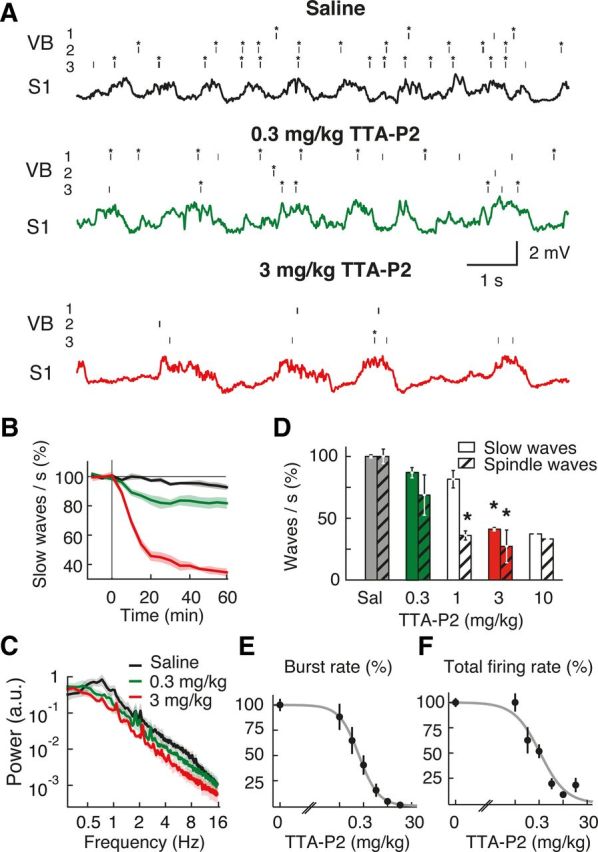
Systemic injection of TTA-P2 markedly decreases the frequency of slow and spindle waves during anesthesia. A, Spike raster plots (top three traces; *bursts) from 3 different TC neurons in the VB and EEG (bottom trace) from S1 show the effect of two doses of intraperitoneally injected TTA-P2 on neuronal firing and slow waves. B, Time-dependent block of slow waves after 0.3 and 3 mg/kg intraperitoneally of TTA-P2 injected at time 0. C, Power spectra calculated 1 h after TTA-P2 injection. D, Summary data showing the percentage reduction in slow and spindle waves produced by different doses of TTA-P2 (measured 1 h after intraperitoneal injection). Number of animals for saline (Sal) and TTA-P2 0.3, 1, 3, and 10 mg/kg injections are 4, 3, 3, 3, and 1, respectively. Error bars indicate SEM. *p < 0.01 compared with saline injection (Mann–Whitney U test). E, F, Dose–response curve of burst and total spike rate measured 40 min after systemic intraperitoneal injection of TTA-P2 (logistic regression fits, p < 0.05) (ED50 for bursts: 0.18 ± 0.05 mg/kg; ED50 for total spikes: 0.26 ± 0.06 mg/kg). The 3 and 10 mg/kg TTA-P2 abolish bursts (p < 10−6 compared with saline injection, Mann–Whitney U test, n = 40 TC neurons). B, C, Solid lines indicate the mean; color shadings indicate SEM. Color code in C also applies to B and to the traces in A.
Systemic and microdialysis solutions.
For intraperitoneal injection, TTA-P2 was dissolved in saline containing 4% DMSO and the pH adjusted with potassium hydroxide (1 mm). Control intraperitoneal injections contained 4% DMSO in saline. For reverse microdialysis injection, TTA-P2 was dissolved in aCSF with 4% DMSO. Tetrodotoxin citrate (TTX) was dissolved in aCSF. Flow rate of the microdialysis injection was set at 1 μl/min.
Injections.
For the systemic injections, once stable EEG slow waves were recorded for at least 30 min and high amplitude well isolated units were present in some of the silicone probe channels, a control period of at least 40 min was recorded before injecting intraperitoneally either saline/DMSO or TTA-P2 while continuing recording for at least another 2 h. For intrathalamic drug application, reverse microdialysis injection of aCSF was initiated as soon as the microdialysis probes were in position, and continued for at least 1 h before electrical recordings commenced. Once stable, high-amplitude, well-isolated units could be recorded from some of the silicone probe channels, recording of a control period of at least 40 min was initiated. This was followed either by continuation of aCSF application or the inlet tubing was switched to a solution containing either TTX or TTA-P2. Recordings and dialysis application then continued for at least 1 h. At the end of the recording session, rats were transcardially perfused.
Recordings.
Thalamic signals were amplified with an Omnetics preamplifier (gain 20, bandwidth 0.8 Hz to 54 kHz) and a Plexon recorder/64 channel amplifier (gain 7500–12,500, bandwidth 1–6000 Hz, Plexon).
The EEG signal was amplified with a combination of SuperTech BioAMP (Pecs) pre- (bandwidth 0.1–500 Hz) and main-amplifiers (bandwidth DC to 500 Hz). When combined unit and EEG recordings were made, signals were digitized with a Plexon recorder/64 system at 20 kHz with 16-bit resolution. EEG recordings were digitized using the Plexon recorder input via the IP16 event input breakout panel. The digitized data were converted to Spike2 format (version 5.13, CED). For all further analyses, data were converted to a raw binary format using tools of the freely available Klusters, Neuroscope, and NDManager software suite (Hazan et al., 2006). EEG data were low-pass filtered with a windowed sinc filter at 100 Hz and downsampled to 200 Hz.
Experiments in freely moving rats
Surgery.
Rats were anesthetized and implanted with EEG electrodes, as described above, and an EMG electrode was positioned in the neck muscle. Two or four microdialysis guide cannulae (one or two in each VB, respectively) (i.e., see experiments illustrated in Fig. 6 for two probes in each VB) were slowly lowered fully vertically so their tip was just above the VB (DV = −4.4 mm) (Paxinos and Watson, 2007). When two guide cannulae were inserted in each VB, their AP coordinates were as follows: −2.6 and −3.8 mm (Paxinos and Watson, 2007). Both EEG electrodes and guide cannulae were fixed to the skull with dental acrylic cement. Rats were allowed at least 7 d to recover from surgery (single housed) and to habituate to the recording cage (4 h/d). At the end of the recording sessions, rats were transcardially perfused.
Figure 6.
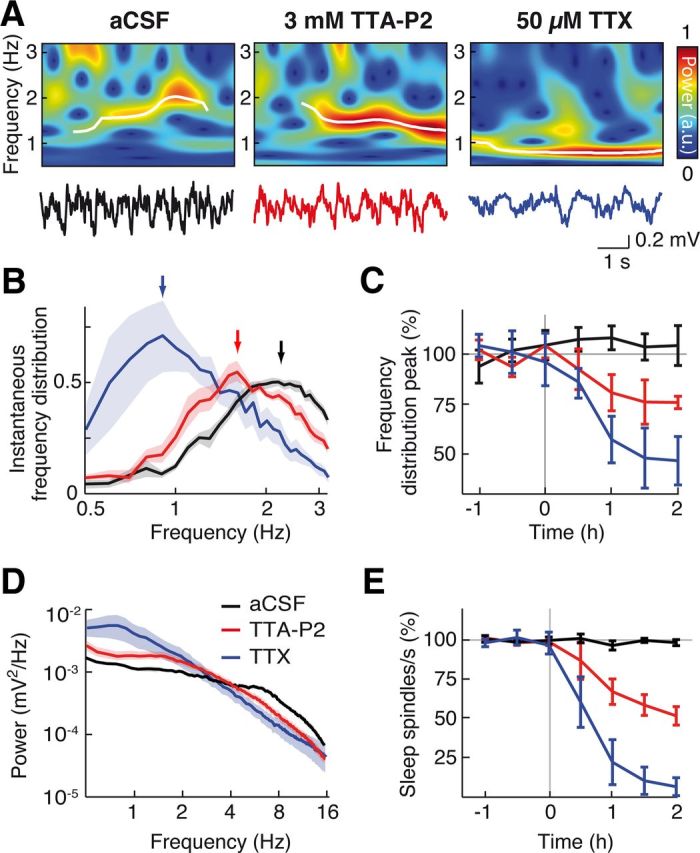
Block of thalamic firing decreases slow wave frequency during natural sleep. A, Non-REM sleep EEG (bottom) and corresponding wavelet spectra (top) during VB microdialysis of aCSF, 3 mm TTA-P2, and 50 μm TTX. Transient slow waves (white lines) were detected as ridges in the wavelet spectra (see Materials and Methods). B, Slow wave frequency density distribution during aCSF, TTA-P2, and TTX dialysis, 1 h after the start of drug dialysis (arrows indicate the measured peaks). C, Time dependence of TTA-P2 (n = 7 rats) and TTX (n = 5 rats) effects on the normalized peak of the slow wave frequency distribution. D, Raw non-REM EEG power spectra show TTA-P2- and TTX-elicited decrease of power in sleep spindle frequency range and increase of power in slow wave frequency range. E, Time dependence of TTA-P2 and TTX effects on sleep spindles normalized count. B, D, Solid lines indicate the mean; color shading indicates SEM. C, E, Error bars indicate SEM.
Injections.
For the systemic injections, on the day of the experiment rats were transferred to the recording cage, connected to the electrical recording and microdialysis apparatus (as appropriate), and allowed to move freely in their cage for at least 1 h before any recording commenced. A control period of 1.5 h was then recorded before an intraperitoneal injection of either saline or TTA-P2 was made while continuing recording for at least another 2 h. Four days were allowed between two consecutive recording days in each rat. For intrathalamic drug application, 24 h before recording, microdialysis probes were slowly inserted into the brain to replace the dummy probe in the guide cannulae. On the day of the experiments, rats were transferred to the recording cage and connected to the dialysis probes and electrical recording apparatus. aCSF dialysis was initiated immediately while the rats were allowed to habituate for 1 h. A control period of 2.5 h was then recorded while administering aCSF (same conditions as described for the anesthetized condition). Animals were then recorded for an additional 2 h while receiving either aCSF or drug-containing aCSF (in a random order on consecutive recording days, each separated by at least 4 d). Video recording was performed simultaneously with electrical recordings in all experiments in freely moving rats.
TTA-P2 levels in the VB.
The concentration of TTA-P2 in samples taken from the inlet and outlet dialysis tubes was measured, following protein precipitation with acetonitrile, by liquid chromatography-mass spectrometry under a validated analytical protocol (Shipe et al., 2008; Uebele et al., 2009). TTA-P2 concentration in the brain tissue outside the dialysis membrane was estimated according to the equilibrium equation described previously (Chan and Chan, 1999).
Optogenetics
Viral injection.
pAAV-CaMKIIα-hChR2(H134R)-mCherry plasmids (K. Deisseroth laboratory, Addgene plasmid 26975) were packaged into recombinant AAV2 vectors and serotyped with AAV1 coat proteins. Viral suspensions were titered to 1.14 × 1013 genome copies/ml (GC) (University of Pennsylvania Vector Core). Concentrated stock virus was diluted with 0.1 m PBS tinted with Fast Green FCF (Sigma), giving a final viral concentration of 5.70 × 108 to 2.28 × 109 GC/μl for injection. The dorsal surface of the skull was exposed, a small craniotomy made over one VB (AP = −3.14 mm, ML = 2.80 mm from bregma) (Paxinos and Watson, 2007), and the dura reflected. A 10 μl Gastight Hamilton syringe and 34 Ga needle were front filled with mineral oil and diluted virus. Needles were then lowered slowly into the thalamus (DV = −5.75 mm from the pia) and left in place for 10 min. A 1 μl virus (5.70 × 108 to 2.28 × 109 infectious units) was injected at a rate of 100 nl/min using a programmable micro-pump (UMP3–1, WPI) and allowed to disperse for a further 10 min before the needle was slowly retracted. Rats were then individually housed and allowed to recover with ad libitum access to food and water for a minimum of 3 weeks to allow viral gene expression.
Optical stimulation and recording.
Rats previously injected with rAAV-CaMKIIα-hChR2 (H134R)-mCherry were anesthetized and EEG screws and two microdialysis probes (one in each VB) were implanted (as above) with a 16° angle with respect to the vertical axis. A 200 μm multimode (0.39 NA) optic fiber (CFM12L20; Thorlabs) was custom-glued to a 32-channel silicone probe, with the fiber tip ∼400 μm above the top recording sites. The fiber ferrule was connected to a compatible patch-cord and 473 nm laser diode (70 mW Stradus; Vortran Laser Technology), and the silicon probe and EEG wires connected to a Digital Lynx 10SX recording system (with Hybrid Input Boards; Neuralynx) via HS-36 unity gain preamplifiers. One optrode was slowly lowered to just above one VB, 300 μm posterior and medial to the virus injection site (AP = −3.44 mm, ML = 2.50 mm from bregma) (Paxinos and Watson, 2007), and DV = −4.60 mm from the pia, and then moved in small steps to locate light-responsive ChR2+ cells exhibiting characteristic TC neuron bursts (see experiments illustrated in Figs. 9 and 10). Extracellular action potentials (sampling frequency: 32 kHz per channel, filtered from 600 Hz to 9 kHz), continuous extracellular signal (sampling frequency: 32 kHz per channel and broadband filtered from 0.1 Hz to 9 kHz), EEG signals (sampling frequency: 4 kHz, filtered from 0.1 Hz to 1 kHz), and light stimulation events were simultaneously recorded using Cheetah 5 Data Acquisition software (Neuralynx). Digital laser modulation was controlled with pClamp software and a 1322A Digidata (Molecular Devices), synchronized with the Digital Lynx 10SX. Laser output power for 5, 20, and 100 ms pulses was 40 mW, which equated to <10 mW at the fiber tip (dependent on stimulation frequency), quantified with a digital power meter and photodiode sensor (PM120D; Thorlabs).
Figure 9.
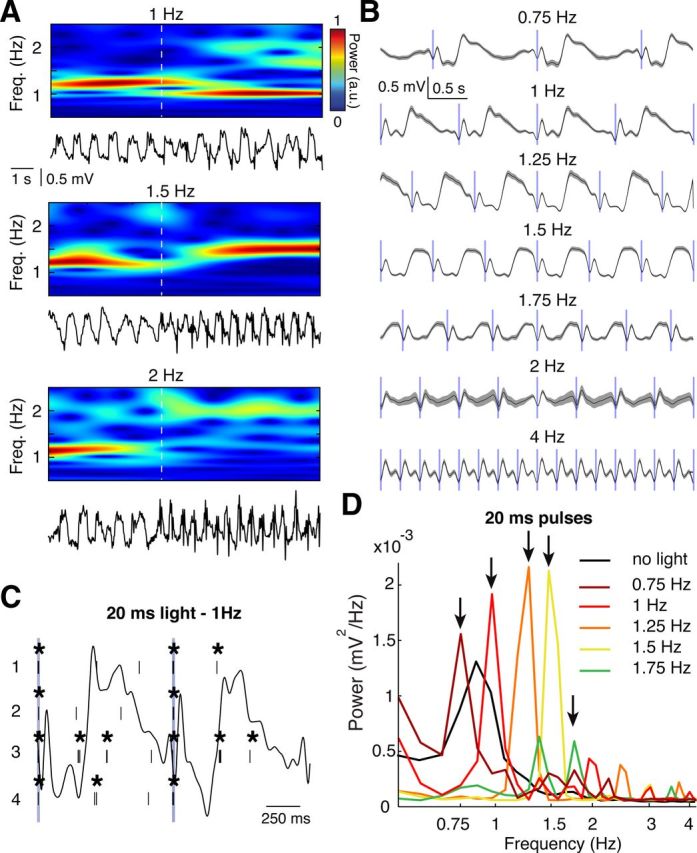
Thalamic entrainment of EEG slow waves during anesthesia. A, EEG trace (bottom) and wavelet transform (top) showing the effect of 20 ms, 473 nm light pulses at 1, 1.5, and 2 Hz. Dashed white line indicates the start of the first pulse. B, Event-triggered EEG averages centered on the 20 ms light pulses (blue vertical bars) for the illustrated stimulation frequencies. Gray areas indicate SEM. A total of 10 s of EEG was used for each average. C, Raster plots of firing of 4 VB TC neurons in responses to two consecutive 20 ms pulses (at 1 Hz) (blue vertical lines). *Bursts. The corresponding EEG trace is superimposed as a black line. D, EEG power spectra in response to 20 ms light pulses at the illustrated frequencies (indicated by arrows). There are frequency-dependent amplification and the shift of the peak of the power spectra compared with control (i.e., without light stimulation, black line).
Figure 10.
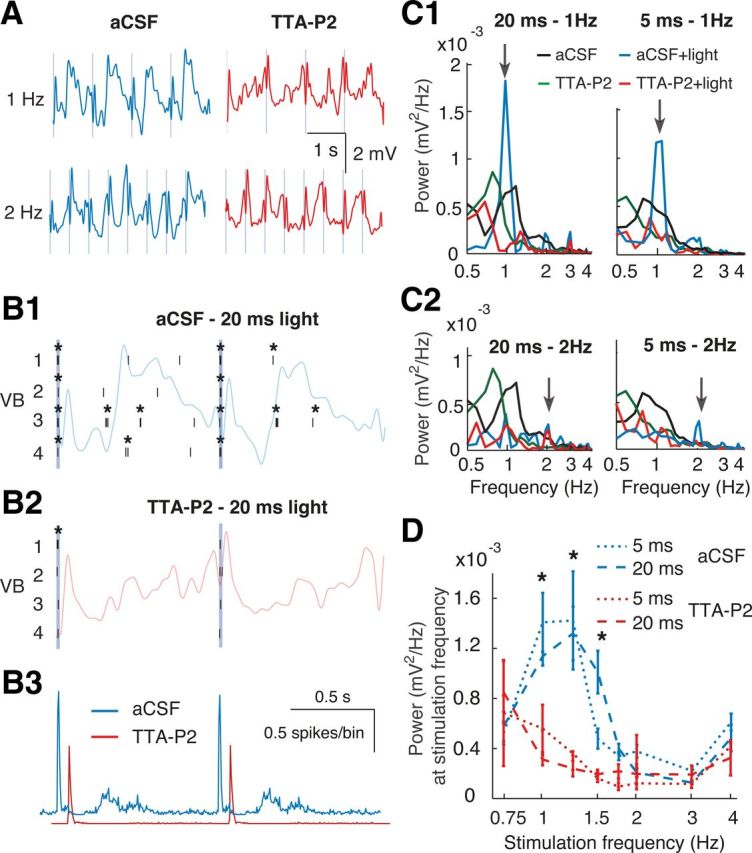
Thalamic entrainment of EEG slow waves requires thalamic T-type calcium channels during anesthesia. A, EEG traces showing the effect of 20 ms, 473 nm light pulses (vertical blue lines) at 1 and 2 Hz during aCSF and TTA-P2 dialysis in VB. B, Raster plots of firing of 4 VB TC neurons in responses to 20 ms pulses (at 1 Hz) during aCSF (B1) and TTA-P2 dialysis (B2). *Bursts. The corresponding EEG trace is superimposed. B3, Light pulse-triggered spike rates (red trace is right-shifted for clarity) from 41 stimulation epochs. C, EEG power spectra in response to 20 ms light stimulation at 1 (C1) and 2 (C2) Hz (arrows) during thalamic aCSF (blue, n = 7 rats) and TTA-P2 (red, n = 3 rats) dialysis compared with power spectra without light stimulation. Black represents aCSF; green represents TTA-P2. D, The power values at a given stimulation frequency (obtained from spectra as in C) for different light stimulation frequencies are plotted during aCSF and TTA-P2 dialysis. Error bars indicate SEM. *p < 0.05 for 5 and 20 ms pulses (Mann–Whitney U test).
Data analysis
Spike sorting and data preprocessing were performed with the Klusters, Neuroscope, NDManager software suite (Hazan et al., 2006). All other analyses were performed with routines based on the free toolboxes SciPy 0.8 (Jones et al., 2001), OpenElectrophy 0.2 (Garcia and Fourcaud-Trocmé, 2009), running under Python 2.6.6 and MATLAB (R2010b, MathWorks) on a 64-bit Linux computer.
Data preprocessing and spike sorting.
To extract spikes from the extracellular field potential, the signal was high-pass filtered with median filter (0.5 ms window half-length). Spikes were detected by thresholding at 1.2 SD and clustered by an expectation maximization algorithm (Klustakwik; Harris et al., 2000) on the basis of their first three principal components. All results of the automatic clustering were verified post hoc by visual inspection. Units were excluded from further analyses if more than one high amplitude cell was present on a single channel, if their autocorrelogram did not show a refractory period of at least 2 ms, and if their spike amplitude markedly changed during the experimental session.
Burst analysis.
Bursts were defined as two or more spikes that were preceded by at least 100 ms of silence and had interspike intervals (ISIs) <10 ms, and were visually examined post hoc. For each burst, the following parameters were calculated (Fig. 1): (1) ISI, (2) interburst interval, (3) number of spikes per second in 5 min windows (spike rate), (4) number of bursts per second in 5 min windows (burst rate), (5) ratio of all spikes taking part in a burst, (6) number of spikes in each burst, and (7) ISI as a function of the ISI number within a burst (i.e., burst signature). Units with a decelerando burst signature (Fig. 1C,D) were classified as TC neurons, whereas those with a burst signature and spike autocorrelogram typical of thalamic reticular neurons (Huguenard and Prince, 1992) were discarded.
Figure 1.

Properties of high-frequency bursts in VB TC neurons during ketamine-xylazine anesthesia and natural sleep. A, B, Local field potential in VB during anesthesia (A) and natural sleep (B). *Bursts. C, Burst properties of a representative TC neuron in the VB during anesthesia: ISIs for different burst lengths (left), and distribution of interburst intervals (IBI) (right) (n = 5102 bursts). D, Same as C for a different representative TC neuron in the VB during natural sleep (n = 825 bursts). C, D Insets, Representative bursts. E, F, Population data, as in C and D, for n = 85 and n = 9 TC neurons from n = 18 anesthetized and n = 3 naturally sleeping rats, respectively.
Detection of slow waves and spindles under anesthesia.
To quantify EEG slow waves beyond power spectral analysis, a slow wave detection algorithm similar to those described previously (Mölle et al., 2009; Nir et al., 2011) was implemented (Fig. 2A–D). On the 0.2 to 4.5 Hz bandpass-filtered signal, all negative to positive zero-crossings were detected as slow waves. To discard spurious slow waves, the local minimum and maximum around a crossing were determined. If the difference between these was <60% of the mean maximum-to-minimum distance, the slow wave was discarded. For the analysis of UP and DOWN state duration, the periods above and below the midpoint of the DOWN to UP transitions in the bandpass-filtered signal were labeled as UP and DOWN states, respectively. States with a magnitude of <25% of the average magnitude during the control period were discarded and the neighboring states of equal type (UP or DOWN) merged. Spindle waves were detected in the frequency range 5–12.5 Hz (Fig. 2E,F).
Figure 2.
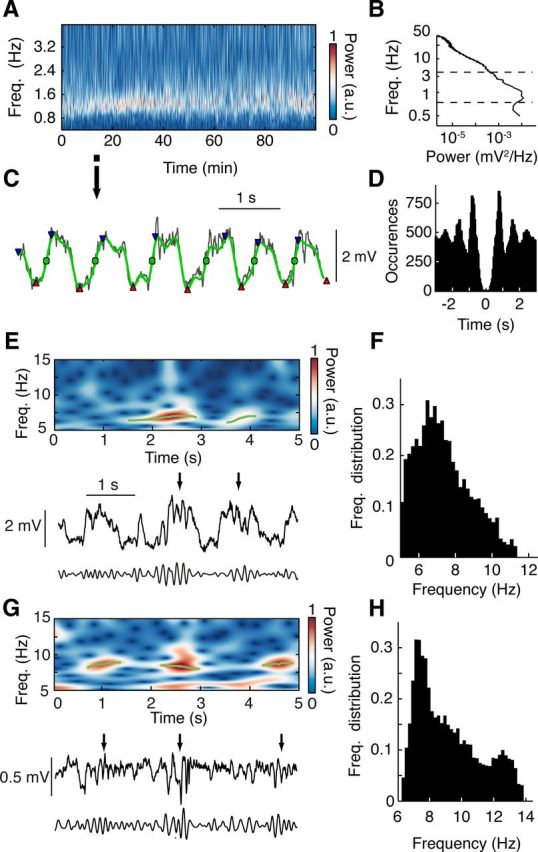
Detection of slow and spindle waves. A, Representative EEG wavelet power spectrum of slow wave frequency band (0.2–4.5 Hz) during ketamine-xylazine anesthesia. B, Representative EEG broadband power spectrum. Dashed lines indicate the frequency band shown in A. C, Expanded EEG trace (black) (from time period marked in A with arrow) illustrating the identification of slow waves (green circles) by means of negative to positive zero-crossing detection on the 0.2–4.5 Hz bandpass-filtered signal (green trace). Waves with peak-to-peak amplitude (red triangles to blue triangles) of <60% of the mean peak-to-peak amplitude were discarded. D, Autocorrelogram of EEG slow waves detected as shown in C from the first 20 min of data shown in A. E, Representative raw (middle), 5–12.5 Hz bandpass-filtered (bottom) EEG traces showing spindle waves (black arrows) and wavelet power spectrum (top) with detected spindle wave episodes (green lines) during anesthesia. F, Average frequency distribution of spindles during anesthesia (n = 7 rats). G, H, Data for spindles during natural sleep are illustrated in the same format as E and F, respectively (n = 6 rats).
Spectral analysis.
Power spectral densities were estimated with Welch's average periodogram method using a Hanning window with length of 4096 data points. Total power in a frequency band was determined as the integral over that frequency band.
Detection of non-REM sleep epochs.
The power of the EEG signal between 0.5 and 4 Hz was computed for every 10 s of recording. If the calculated power was higher than twice the power calculated during a period of active wakefulness, the 10 s epoch was classified as non-REM sleep. To ensure detection reliability and behavioral state stability over the selected periods, only consecutive epochs of sleep lasting at least 60 s were included in further analysis.
Detection of slow waves and spindles during natural sleep.
The EEG signal was convolved with complex Morlet wavelets of 2.5 cycles for sleep spindles and 1.0 cycle for slow waves at a frequency resolution of 0.1 Hz for spindles and 0.01 Hz for slow waves (Kronland-Martinet et al., 1987). Using a wavelet ridge extraction method, each oscillatory epoch of the EEG was extracted with an energy threshold to detect its beginning and end (see Figs. 2G, 6A, and 7A) (Roux et al., 2007; Garcia and Fourcaud-Trocmé, 2009). The boundary frequencies of wave detection were chosen as from 0.5 to 4 Hz for slow waves (see Fig. 6B) and from 6 to 14 Hz for spindle oscillations (see Fig. 2G,H). The threshold was defined as 3 times the average energy during a non-REM sleep period during the control session. Slow and spindle waves with <2 and 3 cycles, respectively, were discarded. When overlapping oscillations were detected, the wave with the highest energy was selected.
Figure 7.
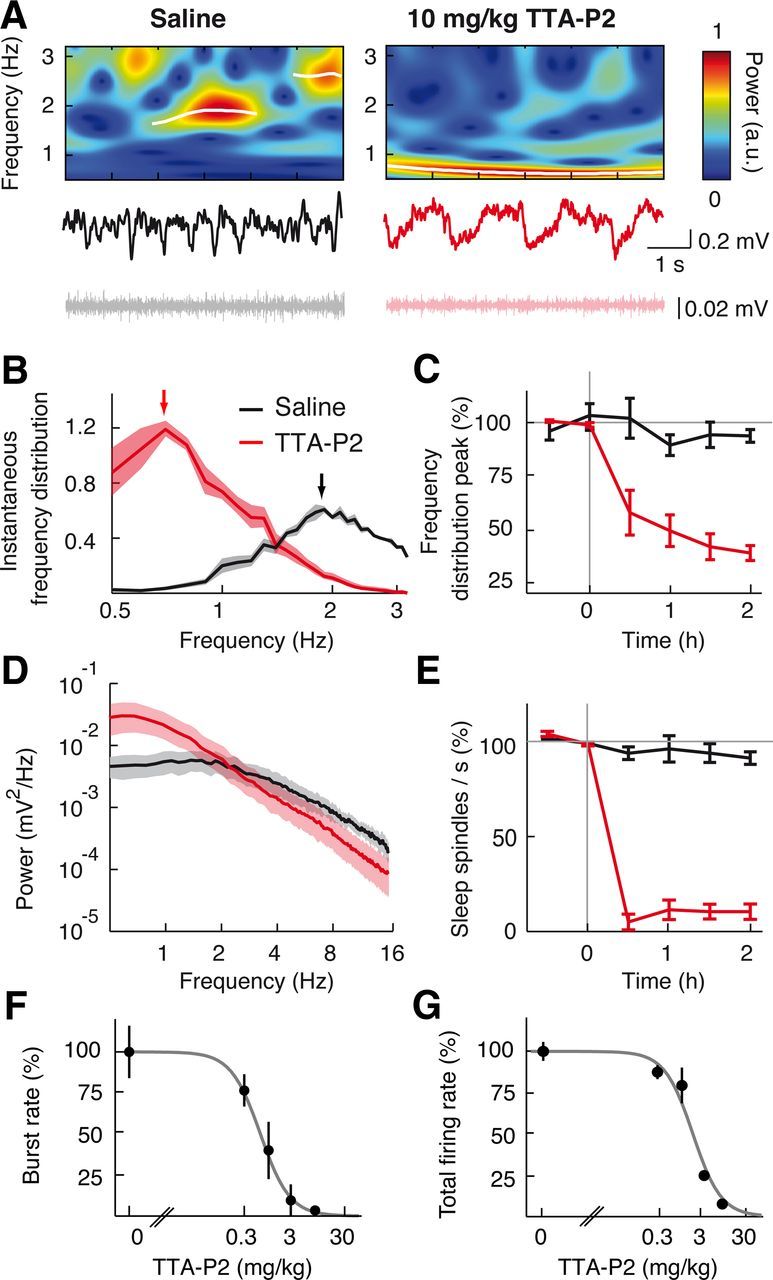
Systemic injection of TTA-P2 markedly decreases the frequency of slow waves and abolishes spindles during natural sleep. A, Non-REM sleep EEG (middle), corresponding wavelet spectra (top), and EMG (bottom) after intraperitoneal injection of saline (left) and 10 mg/kg TTA-P2 (right). Transient slow waves (white lines) were detected as ridges in the wavelet spectra as in Figure 2. B, Slow wave frequency distribution after saline and TTA-P2 injection. There is a shift of the peak (arrows) from ∼2 Hz to ∼0.7 Hz. C, Time dependence of TTA-P2 (n = 4 rats) and saline (n = 6 rats) effects on the normalized peak of the slow wave frequency distribution. D, Raw non-REM sleep EEG power spectra show TTA-P2-elicited decrease of power in sleep spindle frequency range and increase of power in slow wave frequency range compared with saline injection. E, Time dependence of TTA-P2 effects on sleep spindles normalized count. F, G, Dose–response curve of burst (F) and total spike (G) rate measured 40 min after systemic intraperitoneal injection of TTA-P2 (logistic regression fits, p < 0.05) (ED50 for bursts: 0.55 ± 0.03 mg/kg; ED50 for total spikes: 1.71 ± 0.11 mg/kg). The 3 and 10 mg/kg TTA-P2 abolish bursts recorded during natural sleep (p < 10−6 compared with saline injection, Mann–Whitney U test, n = 42 TC neurons). B, D, Solid lines indicate the mean; color shading indicates SEM. C, E–G, Error bars indicate SEM.
Statistical analysis.
Group comparisons were performed using the Mann–Whitney U test. Paired data were tested with Wilcoxon's signed ranks test. All quantitative data in figures and text are given as mean ± SEM.
Histology
Electrode and microdialysis probe tracking.
Before insertion, silicone and microdialysis probes were immersed for 1/2 h and 5 min, respectively, in a 1% Vybrant Dil (Invitrogen) dye solution. At the end of the experiment, rats were injected with a lethal dose of urethane (40%), and the brains were removed and placed in a 4% PFA solution for 48 h. The brains were then transferred and stored in a 0.1 m PBS. Sections (100 μm thick) containing the VB were cut with a vibratome (Leica VT1000S) and mounted on coverslips to measure the relative position of microdialysis and silicone probes, which were visualized using a fluorescent microscope (Leica).
Immunofluorescence.
Rats were given an overdose of ketamine-xylazine and transcardially perfused with 4% PFA. Brains were fixed in 4% PFA and then cryoprotected in 20% (w/v) sucrose in 0.1 m PBS. Each brain was blocked to give coronal sections, mounted onto a freezing microtome (Leica), and cut into 50 μm sections. Free-floating sections were processed for NeuN and RFP immunofluorescence. Briefly, sections were washed 3 × 10 min with fresh 0.1 m PBS and blocked for 1.5 h in 1 × PGT [0.1 m PBS, 3% NGS, and 0.2% Triton X-100 (Sigma-Aldrich)]. Sections were gently shaken at room temperature for 2 h, then overnight at 4°C with primary antibodies against both NeuN (1:500 mouse monoclonal, Millipore), and RFP (1:1000 Living Colors DsRed rabbit polyclonal, Clontech) in 1 × PGT. Sections were rinsed 3 × 10 min with fresh 0.1 m PBS and incubated for 2 to 3 h at room temperature with 1:200 goat anti-mouse AlexaFluor-488 (Invitrogen) and 1:200 goat anti-rabbit AlexaFluor-594 (Invitrogen) secondary antibodies in 1 × PGT. Sections were washed 3 × 10 min in fresh 0.1 m PBS and mounted onto gelatin-subbed Superfrost Plus microscope slides (Thermo Scientific). Slides were coverslipped with VectaShield fluorescent mounting medium (Vector Laboratories) and visualized using a fluorescence microscope (Leica).
Results
Effect of thalamic inactivation on slow waves during anesthesia
We first abolished the somatosensory thalamic output to the cortex by bilateral reverse microdialysis of TTX in the ventrobasal thalamus (VB) of ketamine-xylazine anesthetized rats, while simultaneously recording slow waves from primary somatosensory cortex (S1) and the firing of different single TC neurons in the VB. TTX abolished action potentials in TC neurons around the VB injection site (predrug: 2.51 ± 0.11 spikes/s, TTX: 0.001 ± 0.002, n = 11 rats, n = 33 neurons, p < 107, Wilcoxon signed-rank test) (Fig. 3B1,B2, blue lines, C, bottom plot; see also Fig. 4A, unit recordings). This effect led to a concomitant marked reduction (45 ± 1.4%) in the number of EEG slow waves after 1 h of drug dialysis (Fig. 4A,B) and thus to a frequency decrease from 1.19 ± 0.02 to 0.63 ± 0.03 slow waves/s (n = 5, p = 0.0017, Mann–Whitney U test compared with aCSF, n = 6) (Fig. 4A–D). The duration of both UP and DOWN states was increased by TTX from 0.40 ± 0.01 (UP) and 0.42 ± 0.03 s (DOWN) to 0.68 ± 0.07 and 0.58 ± 0.06 s, respectively (p < 0.006, Mann–Whitney U test compared with aCSF) (Fig. 4B). In the same rats, the simultaneously recorded spindle waves, which are a thalamically generated rhythm (Morison and Bassett, 1945; Steriade et al., 1985; De Gennaro and Ferrara, 2003; Astori et al., 2011), were abolished by TTX (predrug: 0.13 ± 0.02 spindles/s, TTX: 0.009 ± 0.008, p = 0.0016) (Fig. 4E), indicating the effectiveness of our thalamic inactivation by microdialysis administration of TTX.
Because T-type calcium channels play a key role in the thalamic output to cortex (Llinás and Jahnsen, 1982; Crunelli et al., 1989; Deleuze et al., 2012) and underlie TC neuron UP states (Hughes et al., 2002), we next investigated the effect on slow waves of the potent and selective T-type calcium channel antagonist, TTA-P2 (Uebele et al., 2009; Dreyfus et al., 2010), directly applied in the VB by reverse microdialysis. This drug produced a block of high-frequency bursts of TC neurons, which was dependent on its concentration in the dialysis inlet tube and on the distance between the recorded neuron and the dialysis probe (Fig. 3B1; see also unit recordings in Fig. 4A). Interestingly, single action potential firing (which accounted for ∼18.4 ± 1.2% of the total firing) was also markedly decreased by TTA-P2 (Fig. 3B2) (see Fig. 4A, unit recordings) so that, at a distance of 500 μm from the dialysis probe, only 3.3 ± 2.1% of total spikes (i.e., in bursts and as single action potentials) remained after 1 h of TTA-P2 application. We chose a microdialysis inlet tube concentration of 300 μm TTA-P2 because: (1) it virtually abolished the total TC neuron firing in a region that almost fully covered the mediolateral extent of the VB (Fig. 3C,D), and (2) it resulted in a tissue concentration around the probe of ∼42 μm (based on a 14% recovery rate) (for explanation, see Fig. 3E), which is well in the range of concentrations that we previously showed to be required to abolish intrinsic and synaptically driven TC neuron firing in the VB in vitro (Dreyfus et al., 2010, their Fig. 1C). Thus, TTA-P2 applied in the VB at 300 μm elicited a clear reduction (25 ± 3%, n = 5, p = 0.02 compared with aCSF, n = 6) of slow waves (Fig. 4A,B), leading to a frequency decrease from 1.00 ± 0.02 to 0.69 ± 0.03 slow waves/s (p = 0.020) (Fig. 4A,C,D). The durations of UP and DOWN states were both prolonged from 0.59 ± 0.04 (UP) and 0.58 ± 0.02 (DOWN) to 0.77 ± 0.06 and 0.70 ± 0.03 s, respectively (p < 0.05, Mann–Whitney U test compared with aCSF) (Fig. 4B). Moreover, in the same experiments, spindle waves were markedly suppressed (88 ± 5%) by microdialysis injection of TTA-P2 in the VB (aCSF: 0.15 ± 0.01 spindles/s; TTA-P2: 0.019 ± 0.008; p = 0.02) (Fig. 4E), indicating the effectiveness of our thalamic inactivation by microdialysis administration of TTA-P2.
TTA-P2 also elicited a dose-dependent decrease in slow waves when applied systemically (Fig. 5A–D), with a similar ED50 on burst (0.18 ± 0.05 mg/kg) (Fig. 5E) and total firing (0.26 ± 0.06 mg/kg) (Fig. 5F). In particular, at a dose (3 mg/kg) that abolished burst firing in the VB (Fig. 5E), TTA-P2 produced a stronger decrease (63 ± 1%, n = 3, p = 0.018) of slow waves (Fig. 5B) than an equipotent (on burst firing) intra-VB application (compare with Fig. 3B1), indicating that T-type calcium channels in other thalamic nuclei and brain regions effectively contribute to the full expression of EEG slow waves in S1 during anesthesia.
Effect of thalamic inactivation on slow waves during natural sleep
Because anesthesia does not fully reproduce the spatiotemporal dynamics of slow waves during non-REM sleep (Chauvette et al., 2011; Nir et al., 2011; Vyazovskiy et al., 2011), we then applied TTX and TTA-P2 by reverse microdialysis in the thalamus of naturally sleeping-waking rats. The same inlet dialysis concentration of TTA-P2 (300 μm) and one-probe-per-VB configuration that was effective during anesthesia had no effect on slow waves of natural sleep, nor did 1 and 3 mm TTA-P2, most likely because the volume of tissue where a firing block was achieved with these higher TTAP-2 concentrations was not much bigger than the region affected by 300 μm (Fig. 3B1,B2,D). We therefore enlarged the thalamic area affected by microdialysis application by implanting a group of rats with four dialysis probes, so that in each VB one probe was close to its rostral and one to its caudal end (see Materials and Methods). In these animals, TTA-P2 (3 mm) induced a significant decrease (23 ± 7.8%, n = 7, p = 0.0095, Mann–Whitney U test compared with aCSF, n = 12) in the peak of the instantaneous frequency distribution of slow waves during non-REM sleep (predrug: 2.19 ± 0.09 Hz; TTA-P2: 1.72 ± 0.13 Hz), as did TTX (50 μm) (TTX: 0.92 ± 0.13 Hz, 50 ± 13% reduction, n = 5, p = 0.0061) (Fig. 6A–D). As expected, in the same animals, sleep spindles were more potently decreased by TTX and TTA-P2 (94 ± 7% and 48 ± 6%, respectively) than slow waves (predrug: 0.36 ± 0.02 spindles/s; TTX: 0.05 ± 0.03, TTA-P2: 0.19 ± 0.02; p = 0.001 for both compared with aCSF) (Fig. 6D,E).
As it was observed during anesthesia, also in naturally sleeping rats systemic injections of TTA-P2 dose-dependently blocked burst and total firing in VB TC neurons during non-REM sleep (with an ED50 of 0.55 ± 0.03 and 1.71 ± 0.11 mg/kg, respectively) (Fig. 7F,G), as well as slow waves and sleep spindles (Fig. 7A–E). In particular, a dose of 10 mg/kg of TTA-P2 markedly reduced slow wave (saline: 1.94 ± 0.13 Hz, n = 6; TTA-P2: 0.8 ± 0.01 Hz, n = 4 rats) and sleep spindle frequencies (saline: 0.32 ± 0.13 spindles/s, TTA-P2: 0.05 ± 0.01) by 73 ± 5% (p = 0.0095) and 86 ± 4% (p = 0.00031), respectively (Fig. 7E).
Entrainment of EEG slow waves by optogenetic stimulation of TC neurons during anesthesia
Because thalamic inactivation by either TTX or by selective block of thalamic T-type calcium channels with TTA-P2 decreased the frequency of slow waves during anesthesia and natural sleep, the thalamic input to the neocortex should be able to entrain these EEG waves. Because electrical stimulation of the thalamus leads to antidromic excitation of the somatotopic cortical region, which may in turn affect the cortical slow wave oscillator, we addressed this question using selective optogenetic activation of channelrhodopsin2-expressing TC neurons (Fig. 8) with short (5, 20, or 100 ms) 473 nm light pulses at stimulation frequencies that ranged from 0.75 to 4 Hz (n = 7 rats) (Fig. 9). At the cellular level, each light pulse elicited a single high-frequency burst of action potentials, which was invariably followed by a 100–250 ms period of electrical silence before firing resumed (Fig. 9C). For stimulation frequencies from 0.75 to 1.5 Hz, this pattern of TC neuron activation elicited a clear peak in the EEG wavelet (Fig. 9A) and FFT power spectrum (Fig. 9D) at the respective stimulation frequency, which had higher amplitude than the peak of the control EEG (i.e., with no light stimulation) (Fig. 9D, black line). As seen from the light-pulse triggered averages (Fig. 9B), light stimulation for frequencies >1.5 Hz not only failed to entrain EEG slow waves but also markedly flattened the power spectra eliminating the peak present at ∼1 Hz during the control condition (i.e., without light stimulation; Fig. 9D, black line). When T-type calcium channels in VB were blocked by microdialysis of 300 μm TTA-P2 (n = 3 rats) (Fig. 10A), the cellular burst response during light stimulation was strongly reduced (burst reduction: 89 ± 4%, n = 1056 pulses, n = 11 neurons, p = 0.0020, Wilcoxon signed-rank test), the TC neuron firing between stimulations was virtually abolished (Fig. 10B), and no entrainment of slow waves occurred for stimulation frequencies between 0.75 and 1.5 Hz (Fig. 10C1,D). Moreover, a similar block of EEG slow wave entrainment was observed after systemic injection of TTA-P2 (3 mg/kg) at these stimulation frequencies (data not shown). Finally, no significant effect of TTA-P2 injected locally (Fig. 10C2,D) or systemically (data not shown) was observed at stimulation frequencies ≥ 1.75 Hz.
Figure 8.
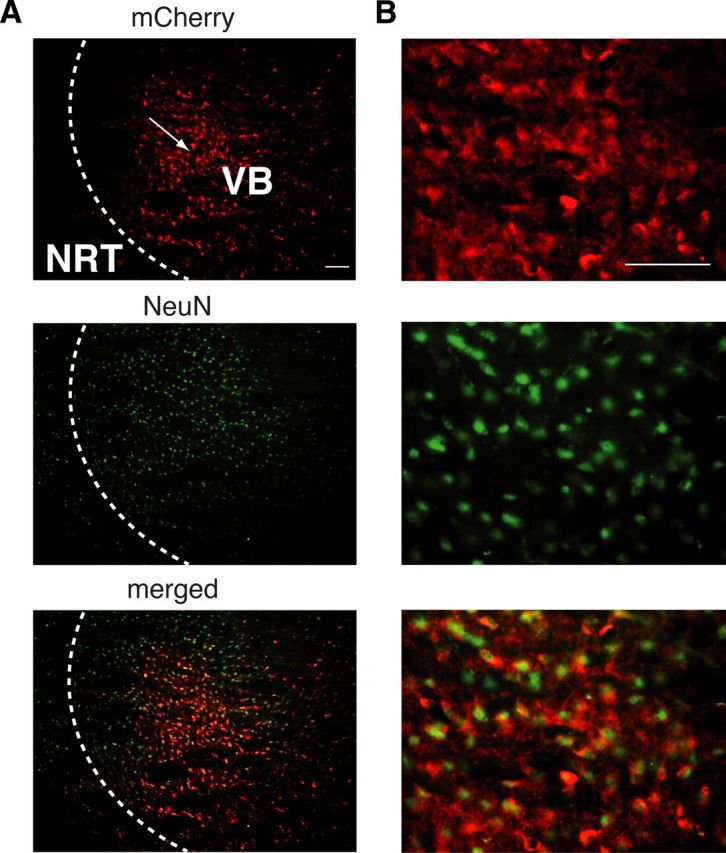
Channelrhodopsin-2 expression in VB TC neurons. A, Immunostaining of channelrhodopsin-2-mCherry (red) showing that the expression of the channelrhodopsin-2 protein is restricted to VB TC neurons and some TC axons passing through the NRT (top). Arrow indicates the putative site of the virus injection. NeuN staining (green) is evident in both VB and NRT somata (middle). Merged images (bottom) demonstrate colocalization of channelorhodopsin-2 and NeuN in TC, but not NRT, neurons. B, Higher magnification of a portion of the respective panels in A. Scale bars, A, B: 300 μm.
Discussion
Our results conclusively demonstrate that the full manifestation of EEG slow waves during non-REM sleep in freely moving, naturally waking-sleeping rats requires a dynamic interplay of cortical and thalamic neuronal ensembles because inactivation of the thalamic output to the neocortex brings about a marked deceleration of slow waves and selective excitation of TC neurons entrains EEG slow waves.
Contribution of thalamic oscillators to sleep rhythms
There is a general consensus that an isolated neocortex can generate and maintain the UP and DOWN state dynamics that underlie EEG slow waves of natural sleep via an intricate balance of excitation and inhibition that is mostly generated by synaptically driven cortical slow wave oscillators (Sanchez-Vives and McCormick, 2000; Timofeev et al., 2000; Bazhenov et al., 2002; Cossart et al., 2003; Shu et al., 2003; Le Bon-Jego and Yuste, 2007; Beltramo et al., 2013). In line with this interpretation, the cortical firing (during UP states) and the electrical silence (during DOWN states) impose similar UP/DOWN state transitions on thalamic neurons, making the thalamus fully subservient to corticofugal activity. Over the last 10 years, however, this view has been challenged by solid experimental evidence demonstrating that an isolated thalamus as well as single TC and nucleus reticularis thalami (NRT) neurons in vitro can sustain slow waves and an intrinsic UP/DOWN states dynamics, respectively, which are similar to those observed during natural sleep (Crunelli and Hughes, 2010). This thalamic activity only occurs when the metabotropic glutamate receptors (which are present postsynaptically to the corticofugal terminals) of TC and NRT neurons (Godwin et al., 1996) are active, making these neurons “conditional oscillators” for slow waves (Crunelli and Hughes, 2010).
The present findings strongly support this hypothesis: removal of the thalamic input to cortex reduces the frequency of slow waves of non-REM sleep, demonstrating, for the first time, a necessary role for the thalamus in this physiological rhythm. The generation of slow waves during natural sleep, therefore, derives from cortical networks as primary oscillators with the thalamic oscillator providing a fine-tuning by imposing its faster frequency and phase properties to the cortical slow wave rhythm. The full physiological rhythm might, indeed, be the result of two competing oscillators (Gutierrez et al., 2013), with the thalamus being able to reset the phase of the slower cortical networks, as indicated by the ability of selective optogenetic excitation of thalamofugal fibers to entrain EEG slow waves (this work) and to induce isolated UP states in awake, head-restrained rats (Poulet et al., 2012). Importantly, the ability of the thalamus to modulate cortical UP states depends on thalamic T-type calcium channels, and in particular on the high-frequency burst of action potentials that invariably marks the start of TC neuron UP states (Contreras and Steriade, 1995, their Fig. 9; Slézia et al., 2011; Ushimaru et al., 2012). The effect of this thalamic modulation will also be important in determining the slight acceleration of slow waves observed from sleep Stage 2 to 4 (Crunelli and Hughes, 2010; Brown et al., 2012) because the intrinsic slow wave thalamic oscillator increases its frequency with the progressive hyperpolarization that accompanies non-REM sleep deepening (Hughes et al., 2002; Crunelli and Hughes, 2010). Support to the thalamic modulation of EEG slow waves also comes from data showing that an acceleration of these waves is accompanied by a phase shift of the TC neuron firing toward the start of an UP state (Slézia et al., 2011).
In the same animals where slow waves of non-REM sleep were decreased in frequency, the simultaneously recorded sleep spindles, which are a thalamically generated rhythm (Morison and Bassett, 1945; Steriade et al., 1985; De Gennaro and Ferrara, 2003; Astori et al., 2011), were abolished by inactivation of the same thalamic region. This demonstrates that, whereas both cortex and thalamus are required for the full expression of slow waves and spindles of natural sleep, the relative contribution of these brain areas to these oscillations is markedly different, reflecting the diverse cellular/network generators that underlie these two EEG rhythms.
By comparing, for the first time, slow waves during natural sleep and anesthesia under the same laboratory conditions, we could identify that a larger area of thalamic inactivation was necessary to obtain a significant effect on slow waves during non-REM sleep compared with anesthesia. This indicates that the thalamic modulation of slow waves during anesthesia is very different from that during natural sleep, stressing the diverse nature of the cortical and thalamic neuronal dynamics underlying these behavioral states, the limitations associated with extrapolating results from one experimental condition to the other, and a potentially different involvement of the “core” and “matrix” thalamic projection systems (Jones, 2001).
Thalamic T-type calcium channels and the slow rhythm
Our investigation is the first to provide direct evidence that the T-type calcium channels of thalamic (i.e., TC and NRT) neurons are required for the full expression of slow waves during natural sleep, although we could not distinguish between TC and NRT neuron contribution because of TTA-P2 spread to the latter neuronal population at the highest concentrations. A previous study reported an increased number of awakenings in mice carrying a supposedly thalamic-selective deletion of CaV3.1 T-type channels expression (Anderson et al., 2005). However, the presence of recombination in piriform cortex, some hypothalamic nuclei, and other brain areas questions the selectivity of this genetic approach and weakens its conclusions. Another study in anesthetized CaV3.1 T-type calcium channel KO mice (Lee et al., 2004) suggested a contribution of these channels to δ waves (defined as power in the 1–4 Hz frequency band) but not to slow waves (defined as power in the <1 Hz frequency band). Our results in anesthetized rats instead indicate that slow waves peaking at ∼1 Hz are decreased in number by TTA-P2, leading to a slowing down in frequency to ∼0.6 Hz. Although these contradictory data may be the result of the use of different species or anesthetics, they may also result from the different wave classifications. Indeed, it is debatable (Luczak and Barthó, 2012) whether EEG waves occurring at the upper end of the δ frequency range (2–4 Hz) in naturally sleeping rats do represent a separate entity from slow (<2 Hz) waves because the underlying UP and DOWN state firing dynamics of slow waves in the 2–4 Hz band is similar to that in 0.5–2 Hz band (Ji and Wilson, 2007, their Fig. 2).
It is not surprising that the vast majority of the total TC neuron firing (i.e., high-frequency bursts plus single action potentials) depends on T-type calcium channels (i.e., it is blocked by TTA-P2). Indeed, although these channels are classically viewed as underlying only high-frequency bursts of TC neurons, recent evidence has indicated that the continuous opening of a very small number of T-type channels that occurs at ∼−60 mV generate a depolarizing window current (Dreyfus et al., 2010, their Fig. 2) that is crucial for the maintenance of the UP state (Hughes et al., 2002; Crunelli and Hughes, 2010). This strong reliance of the thalamic output on T-type channels also indicates that during slow waves synaptic activity, and in particular cortical inputs to the thalamus, are not sufficient to drive a TC neuron output in the absence of these channels.
Resetting and entrainment of EEG slow waves can be achieved by whisker stimulation (Civillico and Contreras, 2012) or by optogenetic activation of cortical neurons in anesthetized rats (Kuki et al., 2013), whereas long (>4 s) optogenetic stimulation of TC neurons in head-restrained mice can induce UP states in neocortical neurons that do not outlast the light stimulus (Poulet et al., 2012). Our results show that short (5 ms) optogenetic stimuli, which evoke high-frequency bursts in TC neurons, strongly entrain EEG slow waves, and that this response is only present when thalamic T-type calcium channels are functionally active. This strengthens the suggestion that thalamic T-type channel-mediated bursts may critically control UP states in related cortical territories during non-REM sleep by finely tuning the frequency of the EEG slow waves that occur during this behavioral state (Crunelli and Hughes, 2010). In contrast, a recent imaging study has reported that thalamic slow waves always follow cortical slow waves during anesthesia (Stroh et al., 2013). However, these results shed little light on the relative contribution of thalamic and cortical neuronal ensembles to slow waves of natural sleep, because, as indicated by Wester and Contreras (2013), they may be confounded by the experimental conditions, in particular the use of an anesthetic agent that is known to markedly block T-type calcium channels (Joksovic and Todorovic, 2010; Eckle et al., 2012).
In conclusion, these findings provide conclusive demonstration of our hypothesis (Crunelli and Hughes, 2010) that both cortical and thalamic population activities are required for the full manifestation of EEG slow waves of natural sleep, and suggest that a decreased thalamic output to the neocortex resulting from thalamic pathologies (Schmahmann, 2003; Kopelman et al., 2009; Parnaudeau et al., 2013) will bring about a slowing down of slow waves during non-REM sleep with deleterious consequences for memory processes.
Footnotes
This work was supported by the Wellcome Trust (91882), ANR-MNMP-2009, and Centre National de la Recherche Scientifique (LEA 528). We thank Dr. K. Deisseroth for the channelrhodopsin-2 viral construct, Drs. W.M. Connelly and S.W. Hughes for critical comments on the manuscript, and Mr. T. Gould for histological processing.
V.N.U. and J.J.R. are employees of Merck and Co., Inc. and potentially own stock and/or stock options in the company. The remaining authors declare no competing financial interests.
References
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 2005;102:1743–1748. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Lüthi A. The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci U S A. 2011;108:13823–13828. doi: 10.1073/pnas.1105115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Model of thalamocortical slow-wave sleep oscillations and transitions to activated States. J Neurosci. 2002;22:8691–8704. doi: 10.1523/JNEUROSCI.22-19-08691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo R, D'Urso G, Dal Maschio M, Farisello P, Bovetti S, Clovis Y, Lassi G, Tucci V, De Pietri Tonelli D, Fellin T. Layer-specific excitatory circuits differentially control recurrent network dynamics in the neocortex. Nat Neurosci. 2013;16:227–234. doi: 10.1038/nn.3306. [DOI] [PubMed] [Google Scholar]
- Blethyn KL, Hughes SW, Tóth TI, Cope DW, Crunelli V. Neuronal basis of the slow (<1 Hz) oscillation in neurons of the nucleus reticularis thalami in vitro. J Neurosci. 2006;26:2474–2486. doi: 10.1523/JNEUROSCI.3607-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SHH, Chan JYH. Application of reverse microdialysis in the evaluation of neural regulation of cardiovascular functions. Anal Chim Acta. 1999;379:275–279. doi: 10.1016/S0003-2670(98)00406-1. [DOI] [Google Scholar]
- Chauvette S, Crochet S, Volgushev M, Timofeev I. Properties of slow oscillation during slow-wave sleep and anesthesia in cats. J Neurosci. 2011;31:14998–15008. doi: 10.1523/JNEUROSCI.2339-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civillico EF, Contreras D. Spatiotemporal properties of sensory responses in vivo are strongly dependent on network context. Front Syst Neurosci. 2012;6:25. doi: 10.3389/fnsys.2012.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Aronov D, Yuste R. Attractor dynamics of network UP states in the neocortex. Nature. 2003;423:283–288. doi: 10.1038/nature01614. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Hughes SW. The slow (1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci. 2010;13:9–17. doi: 10.1038/nn.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Lightowler S, Pollard CE. A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J Physiol. 1989;413:543–561. doi: 10.1113/jphysiol.1989.sp017668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Lörincz ML, Errington AC, Hughes SW. Activity of cortical and thalamic neurons during the slow (<1 Hz) rhythm in the mouse in vivo. Pflugers Arch. 2012;463:73–88. doi: 10.1007/s00424-011-1011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gennaro L, Ferrara M. Sleep spindles: an overview. Sleep Med Rev. 2003;7:423–440. doi: 10.1053/smrv.2002.0252. [DOI] [PubMed] [Google Scholar]
- Deleuze C, David F, Béhuret S, Sadoc G, Shin HS, Uebele VN, Renger JJ, Lambert RC, Leresche N, Bal T. T-type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci. 2012;32:12228–12236. doi: 10.1523/JNEUROSCI.1362-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi A, Mizuno M, Katafuchi T, Furue H, Koga K, Yoshimura M. Slow oscillation of membrane currents mediated by glutamatergic inputs of rat somatosensory cortical neurons: in vivo patch-clamp analysis. Eur J Neurosci. 2007;26:2565–2575. doi: 10.1111/j.1460-9568.2007.05885.x. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. J Neurosci. 2010;30:99–109. doi: 10.1523/JNEUROSCI.4305-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle VS, Digruccio MR, Uebele VN, Renger JJ, Todorovic SM. Inhibition of T-type calcium current in rat thalamocortical neurons by isoflurane. Neuropharmacology. 2012;63:266–273. doi: 10.1016/j.neuropharm.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S, Fourcaud-Trocmé N. OpenElectrophy: an electrophysiological data- and analysis-sharing framework. Front Neuroinform. 2009;3:14. doi: 10.3389/neuro.11.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemignani A, Laurino M, Provini F, Piarulli A, Barletta G, d'Ascanio P, Bedini R, Lodi R, Manners DN, Allegrini P, Menicucci D, Cortelli P. Thalamic contribution to sleep slow oscillation features in humans: a single case cross sectional EEG study in fatal familial insomnia. Sleep Med. 2012;13:946–952. doi: 10.1016/j.sleep.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Godwin DW, Van Horn SC, Eriir A, Sesma M, Romano C, Sherman SM. Ultrastructural localization suggests that retinal and cortical inputs access different metabotropic glutamate receptors in the lateral geniculate nucleus. J Neurosci. 1996;16:8181–8192. doi: 10.1523/JNEUROSCI.16-24-08181.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez GJ, O'Leary T, Marder E. Multiple mechanisms switch an electrically coupled, synaptically inhibited neuron between competing rhythmic oscillators. Neuron. 2013;77:845–858. doi: 10.1016/j.neuron.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Henze DA, Csicsvari J, Hirase H, Buzsáki G. Accuracy of tetrode spike separation as determined by simultaneous intracellular and extracellular measurements. J Neurophysiol. 2000;84:401–414. doi: 10.1152/jn.2000.84.1.401. [DOI] [PubMed] [Google Scholar]
- Hazan L, Zugaro M, Buzsáki G. Klusters, NeuroScope, NDManager: a free software suite for neurophysiological data processing and visualization. J Neurosci Methods. 2006;155:207–216. doi: 10.1016/j.jneumeth.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron. 2002;33:947–958. doi: 10.1016/S0896-6273(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lörincz M, Cope DW, Blethyn KL, Kékesi KA, Parri HR, Juhász G, Crunelli V. Synchronized oscillations at α and θ frequencies in the lateral geniculate nucleus. Neuron. 2004;42:253–268. doi: 10.1016/S0896-6273(04)00191-6. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji D, Wilson MA. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat Neurosci. 2007;10:100–107. doi: 10.1038/nn1825. [DOI] [PubMed] [Google Scholar]
- Joksovic PM, Todorovic SM. Isoflurane modulates neuronal excitability of the nucleus reticularis thalami in vitro. Ann N Y Acad Sci. 2010;1199:36–42. doi: 10.1111/j.1749-6632.2009.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The thalamic matrix and thalamocortical synchrony. Trends Neurosci. 2001;24:595–601. doi: 10.1016/S0166-2236(00)01922-6. [DOI] [PubMed] [Google Scholar]
- Jones E, Oliphant T, Peterson P. SciPy: Open Source Scientific Tools for Python. 2001. Available at: http://www.scipy.org.
- Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The Korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol. 2009;44:148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- Kronland-Martinet R, Morlet J, Grossmann A. Analysis of sound patterns through wavelet transforms. Int J Pattern Recogn. 1987;1:273–302. doi: 10.1142/S0218001487000205. [DOI] [Google Scholar]
- Kuki T, Ohshiro T, Ito S, Ji ZG, Fukazawa Y, Matsuzaka Y, Yawo H, Mushiake H. Frequency-dependent entrainment of neocortical slow oscillation to repeated optogenetic stimulation in the anesthetized rat. Neurosci Res. 2013;75:35–45. doi: 10.1016/j.neures.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Le Bon-Jego M, Yuste R. Persistently active, pacemaker-like neurons in neocortex. Front Neurosci. 2007;1:123–129. doi: 10.3389/neuro.01.1.1.009.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim D, Shin HS. Lack of δ waves and sleep disturbances during non-rapid eye movement sleep in mice lacking α1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A. 2004;101:18195–18199. doi: 10.1073/pnas.0408089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Jahnsen H. Electrophysiology of mammalian thalamic neurones in vitro. Nature. 1982;297:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Luczak A, Barthó P. Consistent sequential activity across diverse forms of UP states under ketamine anesthesia. Eur J Neurosci. 2012;36:2830–2838. doi: 10.1111/j.1460-9568.2012.08201.x. [DOI] [PubMed] [Google Scholar]
- Marshall L, Helgadóttir H, Mölle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- Mölle M, Eschenko O, Gais S, Sara SJ, Born J. The influence of learning on sleep slow oscillations and associated spindles and ripples in humans and rats. Eur J Neurosci. 2009;29:1071–1081. doi: 10.1111/j.1460-9568.2009.06654.x. [DOI] [PubMed] [Google Scholar]
- Morison R, Bassett D. Electrical activity of the thalamus and basal ganglia in decorticate cats. J Neurophysiol. 1945:309–314. [Google Scholar]
- Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, Tononi G. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–169. doi: 10.1016/j.neuron.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnaudeau S, O'Neill PK, Bolkan SS, Ward RD, Abbas AI, Roth BL, Balsam PD, Gordon JA, Kellendonk C. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron. 2013;77:1151–1162. doi: 10.1016/j.neuron.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Petersen CC, Hahn TT, Mehta M, Grinvald A, Sakmann B. Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc Natl Acad Sci U S A. 2003;100:13638–13643. doi: 10.1073/pnas.2235811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulet JF, Fernandez LM, Crochet S, Petersen CC. Thalamic control of cortical states. Nat Neurosci. 2012;15:370–372. doi: 10.1038/nn.3035. [DOI] [PubMed] [Google Scholar]
- Roux SG, Cenier T, Garcia S, Litaudon P, Buonviso N. A wavelet-based method for local phase extraction from a multi-frequency oscillatory signal. J Neurosci Methods. 2007;160:135–143. doi: 10.1016/j.jneumeth.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD. Vascular syndromes of the thalamus. Stroke. 2003;34:2264–2278. doi: 10.1161/01.STR.0000087786.38997.9E. [DOI] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, et al. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem. 2008;51:3692–3695. doi: 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Sirota A, Buzsáki G. Interaction between neocortical and hippocampal networks via slow oscillations. Thalamus Relat Syst. 2005;3:245–259. doi: 10.1017/S1472928807000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slézia A, Hangya B, Ulbert I, Acsády L. Phase advancement and nucleus-specific timing of thalamocortical activity during slow cortical oscillation. J Neurosci. 2011;31:607–617. doi: 10.1523/JNEUROSCI.3375-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M. Synchronized activities of coupled oscillators in the cerebral cortex and thalamus at different levels of vigilance. Cereb Cortex. 1997;7:583–604. doi: 10.1093/cercor/7.6.583. [DOI] [PubMed] [Google Scholar]
- Steriade M, Deschênes M, Domich L, Mulle C. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol. 1985;54:1473–1497. doi: 10.1152/jn.1985.54.6.1473. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993a;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. Intracellular analysis of relations between the slow (< 1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993b;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroh A, Adelsberger H, Groh A, Rühlmann C, Fischer S, Schierloh A, Deisseroth K, Konnerth A. Making waves: initiation and propagation of corticothalamic Ca2+ waves in vivo. Neuron. 2013;77:1136–1150. doi: 10.1016/j.neuron.2013.01.031. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M. Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex. 2000;10:1185–1199. doi: 10.1093/cercor/10.12.1185. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Some considerations on sleep and neural plasticity. Arch Ital Biol. 2001;139:221–241. [PubMed] [Google Scholar]
- Uebele VN, Nuss CE, Fox SV, Garson SL, Cristescu R, Doran SM, Kraus RL, Santarelli VP, Li Y, Barrow JC, Yang ZQ, Schlegel KA, Rittle KE, Reger TS, Bednar RA, Lemaire W, Mullen FA, Ballard JE, Tang C, Dai G, et al. Positive allosteric interaction of structurally diverse T-type calcium channel antagonists. Cell Biochem Biophys. 2009;55:81–93. doi: 10.1007/s12013-009-9057-4. [DOI] [PubMed] [Google Scholar]
- Ushimaru M, Ueta Y, Kawaguchi Y. Differentiated participation of thalamocortical subnetworks in slow/spindle waves and desynchronization. J Neurosci. 2012;32:1730–1746. doi: 10.1523/JNEUROSCI.4883-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Hanlon EC, Nir Y, Cirelli C, Tononi G. Local sleep in awake rats. Nature. 2011;472:443–447. doi: 10.1038/nature10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wester JC, Contreras D. Generating waves in corticothalamocortical networks. Neuron. 2013;77:995–997. doi: 10.1016/j.neuron.2013.03.002. [DOI] [PubMed] [Google Scholar]