

Abstract
Single-molecule approaches permit an unrivalled view of how complex systems operate and have recently been used to understand DNA–protein interactions. These tools have enabled advances in a particularly challenging problem, the search for damaged sites on DNA. DNA repair proteins are present at the level of just a few hundred copies in bacterial cells to just a few thousand in human cells, and they scan the entire genome in search of their specific substrates. How do these proteins achieve this herculean task when their targets may differ from undamaged DNA by only a single hydrogen bond? Here we examine, using single-molecule approaches, how the prokaryotic nucleotide excision repair system balances the necessity for speed against specificity. We discuss issues at a theoretical, biological, and technical level and finally pose questions for future research.
I. Structural Insights of Bacterial Nucleotide Excision Repair
A. Overview of the Process
Bacterial nucleotide excision repair (NER) is mediated by six proteins1 (Fig. 1). Initial damage recognition is performed by UvrA, which acts as a dimer. The cocrystal structure of a UvrA dimer bound to a DNA fragment containing a fluorescein-modified thymine indicates that the damaged DNA fits snugly into a channel formed by the UvrA dimer.2 It is believed that positively charged amino acids along this channel help to stabilize the binding of DNA to UvrA.2,3 Damage recognition is apparently mediated by the C-terminal zinc fingers.4 Most recently, it has been shown that these zinc fingers help to communicate with the ATPase sites within the protein.5 ATP binding promotes UvrA dimerization, whereas ATP hydrolysis promotes monomerization. However, ATP hydrolysis is required for damage discrimination, as ATP-γ-S abrogates damage recognition by increasing the nonspecific binding of UvrA.6,7 These ATP-binding sites are also important for allowing damage verification by UvrB,5 as hydrolysis by UvrA is believed to promote UvrA release during loading of UvrB at the site of damage.8,9 UvrB has been determined to melt 4–6 bases by inserting a beta-hairpin through the two DNA strands containing the DNA lesion.10 Tyr96 at the base of the beta-hairpin is essential for damage verification.11,12 UvrB resembles monomeric helicases, and it is believed that ATP hydrolysis of UvrB’s helicase-like fold is essential for damage verification and proper lesion positioning for binding and incision by UvrC.8
Fig. 1.
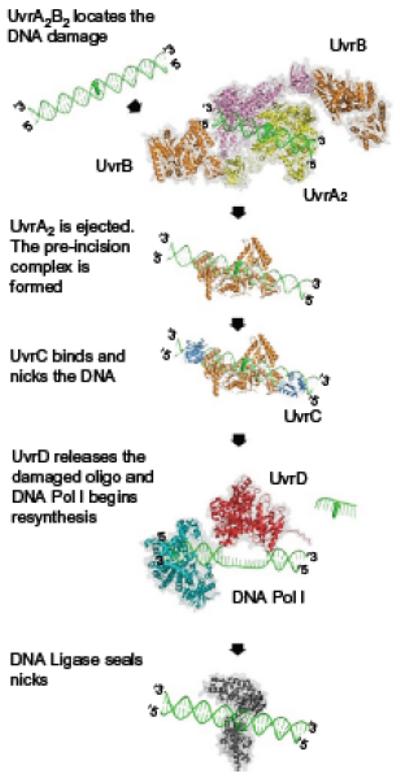
Structural model of bacterial nucleotide excision repair mediated by six proteins. The process of NER is a complex multiprotein cascade of events. Each step requires the recruitment of another protein to the lesion, with UvrB remaining at the lesion site as it interacts with each component of the reaction. Remarkably, despite this central role, UvrB is incapable of binding to the lesion site directly, requiring loading by UvrA2.
UvrC contains two nuclease domains in the N-terminal and C-terminal regions, required for dual incisions 3′ and 5′ on the damaged nucleotide, respectively.13,14 Once UvrC performs both endonucleolytic cuts, two additional proteins, UvrD and DNA polymerase I (Pol I), are required to displace the postincision complex, fill in the repair patch, and allow UvrB and UvrC to turn over.15 The final step in prokaryotic NER is the sealing of the remaining nick by DNA ligase I.
B. Dynamics of the UvrA2B2–DNA Complex
Determination of the stoichiometry of the UvrAB complex has been controversial, but two definitive experiments indicate that the working complex consists of two subunits each of UvrA and UvrB.16,17 There are three important problems regarding the dynamics of the interaction of UvrA2B2 with damaged DNA: (1) the way the damage is handed off from the UvrA dimer to UvrB; (2) the nature of the search mechanism; and (3) the rate-limiting step in damage processing by these two proteins. Regarding the first question of damage handoff, one alternative is that the damage site slides from the central location of the two zinc fingers at the UvrA dimer interface to the beta-hairpin of UvrB.2,18,19 Another more likely alternative is that the UvrA2UvrB2–DNA complex and interaction interface between the proteins is flexible enough to allow UvrB to clamp down onto the DNA face opposite UvrA2 in order to be positioned for a direct handoff of the lesion.2,18,19 In this model, UvrA2 may bend the DNA to facilitate localized unwinding of the helix to allow easy insertion of UvrB’s beta-hairpin.1 Still unresolved is how the UvrA/UvrB interface is weakened to allow dissociation of the UvrA dimer during loading of UvrB. One plausible explanation is that the conformation of this interface is altered by damage-induced hydrolysis of ATP by both UvrA and UvrB.20 This review describes how single-molecule techniques offer a new approach toward understanding these problems. In particular, we focus on novel imaging platforms that have recently been developed to allow the process of NER to be examined one molecule at a time without the influence of flow or surfaces and to highlight the use of current protein-tagging technologies, particularly quantum dots.
C. Kinetic Proofreading as Part of a Dynamic DNA Damage Recognition Process: Role of ATP
As explained in the previous section, DNA damage recognition is a dynamic, ATP-requiring process. Damage recognition, damage verification, and finally DNA damage processing leading to incision, all require a high degree of specificity, which is probably achieved by what has been termed “kinetic proofreading”.21–23 It is postulated that to cope with the frequent small differences in binding energy between target and nontarget sites during NER, high-energy protein–substrate intermediate complexes are created using ATP; however, if the target is not confirmed, the complex can dissociate. This process utilizes energy to generate a time gap between initial protein binding and subsequent catalysis during which the complex can dissociate and the catalysis (DNA incision) is aborted. By including multiple proofreading steps prior to catalysis, damage specificity is improved. For NER, we envision that once UvrA2 senses damage, UvrA2 utilizes ATP hydrolysis to position UvrB for damage verification, representing the first kinetic proofreading step. Following this, in the second kinetic proofreading step, UvrB hydrolyses ATP and releases UvrA2, making reversal of this process improbable; this also helps to impose a specific conformation on the damaged DNA, promoting efficient incision11 (see Fig. 1). In this way, UvrC is probably “capturing” this preincision UvrB–DNA complex during an ATPase cycle on UvrB.11This occurs prior to the enzymatic commitment step, which in this case is the dual incision of the DNA that alters the integrity of the genome.21–23
II. So Few DNA Repair Proteins, So Much DNA: Defining the Big Problem
A. Challenge of Repair Inside a Bacterial Cell
The average size of an Escherichia coli cell is about 1 μmm in diameter. Packed inside this volume of ~1 × 10−15 L is 4.6 × 106 bp of genomic DNA, making the base pair concentration in the millimolar range. An undamaged E. coli cell contains about 20, 50, and 20 copies of UvrA, UvrB, and UvrC respectively, and their concentrations are approximately equivalent to 20, 50, and 20 nM, respectively. During DNA damage-induced SOS induction, which takes ~20–40 min, the levels of UvrA and UvrB proteins may rise about 10-fold, whereas the uvrC gene is not under SOS control. Assuming UvrC employs the same search mechanism as UvrA and UvrB, it is likely that UvrC’s search process for the UvrB–DNA preincision complex would be rate-limiting for NER. In addition to global NER, transcription-coupled repair occurs when damage blocks the progression of transcribing RNA polymerase (RNAP).24 The MFD or transcription-coupling protein, which contains a fold similar to UvrB, helps recruit the repair machinery to the site of the damage-induced stalled RNAP.25E. coli K12 WT strain showed about 10% survival after 22 J/m2 of 254 nm UV light (UVC),26 and quantitative polymerase chain reaction (PCR) analysis of the damaged E. coli indicated that UVC induced cyclobutane pyrimdine dimers (CPD) and 6–4 photoproducts at a ratio of 2.1:1, respectively.27 A fluence of 100 J/m2 of UVC produced about 0.4 photoproducts/kb, of which about 16.5% and 36.7% of these lesions were repaired in 15 min on the nontranscribed and transcribed strands of the lacI gene, respectively.27,28 Thus, assuming an initial noninduced state, some 10 UvrA2B2 complexes were capable of processing about 1000 photoproducts in 15 min or about 6.6 lesions/complex/min. If, however, there was ample induction of UvrA and UvrB proteins, this rate of repair could be up to 10 times slower. Using a complementary approach of monitoring removal of ~4 CPD/pBR322 in E. coli, it was found that the UvrABC system could remove about 0.045 dimers/plasmid (4.4 kb)/ min.29 As pBR322 exists at about 10 copies per cell and E. coli has 4.6 × 106 bp genome, also irradiated in this experiment, that is equivalent to a total of about 45 CPD/min being processed by roughly 10 UvrA2B2 complexes or about 4.5 CPD/complex/min. Together these conditions give a repair rate of 1 photoproduct/1–2 kbp in roughly 10–100 s/UvrA2B2 complex. How can such a small number of proteins search through a vast sea of undamaged DNA to quickly repair that quantity of DNA damage? This fundamental problem in NER is discussed in more detail in the following section.
B. Potential Modes of Damage Site Location
The question of how proteins find their cognate binding sites in the presence of excess DNA has been of great interest for over 50 years and several search mechanisms have been proposed (reviewed in Ref. 30). Despite the large number of proteins packed into the small volume of a bacterial cell, proteins diffuse rapidly. The green fluorescence protein (GFP), MW of 30 kDa, was found to have a three-dimensional (3D) diffusion constant of about 7.7 μm2/s.31 This rapid Brownian motion is caused by collisions with water molecules such that a single GFP molecule can diffuse the width of a bacterium in ~22 ms.32 However, the likelihood of a UvrA2B2 complex landing directly on a lesion in DNA has a low probability, and a large number of binding and dissociation steps would be required to adequately find DNA damage during a period of cellular division using a 3D searching strategy.
This problem is exacerbated by the presence of a vast excess of undamaged DNA for which the UvrA2B2 complex has affinity. In reality, however, the presence of excess nontarget sequences has been shown to accelerate target acquisition33, suggesting that such a 3D searching strategy is an inadequate explanation.
Alternative mechanisms of target search include DNA sliding or hopping, which reduces the search space from three dimensions to one. Using techniques described below, researchers have found several proteins to slide various distances on DNA (see Table I). It should be noted, however, that the one-dimensional (1D) diffusion constants laid out in Table I are all slower than those expected considering the hydrodynamics of motion. The helical structure of DNA offers a unique challenge to a one-dimensionally diffusing molecule. Consider by analogy a modern rollercoaster ride, in which there are corkscrew sections analogous to the helical groove of DNA. The rollercoaster car spins as it traverses these sections whereas over the linear sections the car slides rapidly unimpeded by any spiraling. In direct analogy, the drag experienced by a protein that slides along DNA without being confined to the helical structure of the DNA is much less than that by a protein corkscrewing around the DNA along the grooves. Let us first consider the case of a sphere the size of a protein (r) sliding in one dimension. Ignoring the DNA helix, we calculate the maximum possible diffusion constant (= κBT/6πηr) and compare this to the measured value from Table I. This difference in diffusion constant provides information on the magnitude of the energy barrier to diffusion. In all cases, the value is extraordinarily large. Instead, if we calculate the maximum diffusion constant based on the protein spiraling around the DNA, we find that the drag due to rotation (effective friction factor = 1 + 4/3[2π]2 + [r/3.4 × 10−9]2) has a major effect on the diffusion constant,53 thus lowering the theoretical maximum value. In turn, this lowers the difference between the expected and measured diffusion constants and thus the associated energy barrier to reasonable values ranging from near zero to just a few κBT.
TABLE I.
Single-Molecule Analysis of Protein Movement on DNA
| Protein | Process | MSD (μm2/s) | Mode | References |
|---|---|---|---|---|
| EcoRI | Host restriction | 3.5×10−3 | Sliding | 34 |
| EcoRV | Host restriction | 1.1×10−2 | Sliding and jumping |
35 |
| EcoRV | Host restriction | 3.15×10−3 | Sliding | 36 |
| Fpg | BER | 2.3×10−2 | Sliding | 37 |
| Lac I repressor | Transcription | 2.3×10−4–1.3×10−1 | Sliding | 38 |
| Lac I repressor | Transcription | 4.6×10−2 | Sliding | 39 |
| Lac I repressor | Transcription | 0.4 | Sliding | 39 |
| Lac I repressor | Transcription | 3a | 3D search | 39 |
| Mlh1 | MMR | 1.37×10−1 | Hopping (?) | 40 |
| Mlh1-Pms1 | MMR | 0.2–9.9×10−1 | Hoppingb | 40 |
| Msh2-Msh6 | MMR | 1.2×10−2 | Sliding | 41 |
| MutM | BER | 4.0×10−2 | Sliding | 42 |
| MutS | MMR | 3.6×10−2 | Sliding/paused1c | 43 |
| Nei | BER | 3.4×10−2 | Sliding | 37 |
| Nth | BER | 5.8×10−2 | Sliding | 37 |
| Ogg1 | BER | 5.8×10−1 | Sliding | 42 |
| PCNA | Replication | 1.16 | Sliding (direct and rotational) |
44 |
| p53 | DDR | 1.62×10−1 | Complex | 45 |
| p53 (core) | DDR | 2.39×10−2 | Hopping | 45 |
| p53 (C-ter) | DDR | 7.76×10−1 | Sliding | 45 |
| Rad51 | Recombination | 4.2×10−2 | 46 | |
| RecBCD | Recombination | 1250 bp/sd | Directed motion | 47,48 |
| RNAP (T7) | Transcription | 6.1×10−3–4.3×10−1 | Sliding | 49 |
| RNAP (Escherichia coli) |
Transcription | 1.0×10−2 | Sliding | 50 |
| T7 DNA pol | Replication | 1.86 (34 mM)e | Hopping | 51 |
| T7 DNA pol/ thioredoxin |
Replication | 4×10−1 | Sliding | 51 |
| Uracil glycosylase |
BER | 1.15×10−3 | Sliding | 52 |
| UvrA | NER | None | Jumping | 16 |
| UvrAB | NER | 4.4×10−4 | Sliding | 16 |
| UvrAB | NER | 1.3×10−3 (μm/s)f | Paused, directed | 16 |
Conversions:
1×10−15 m2/s = 1×10−11 cm2 = 1×10−3 μm2/s =8.6×103 bp2/s
BER, base excision repair; DDR, DNA damage response; Fpg, formamidopyrimidine DNA glycosylase; MMR, mismatch repair; Nei, endonuclease VIII; Nth, endonuclease III; NER, nucleotide excision repair.
3D diffusion constant.
D increased with increasing salt, evidence for hopping.
short sliding events, followed by long ATP-induced pauses.
rate for translocating through lambda DNA.
at this ionic strength.
directed motion, therefore units = μm/s.
Early studies looking at the processivity of UvrABC-mediated repair of DNA damage in plasmids suggested that the UvrA2B2 complex is capable of limited 1D DNA scanning. These studies showed that following a lag period after UV-induced damage, during which plasmids were bound, UvrA2B2 utilized some type of scanning mechanism to search for and repair lesions.29 This study also implied that incision and gap filling were highly coupled, preserving the topological state of the plasmid with no accumulation of nicked plasmids.
It is valid to assume that the distance a protein could slide before encountering another protein bound to DNA is very short given the large numbers of DNA-binding proteins. Thus, an alternative to DNA sliding has been proposed, DNA hopping, in which the protein performs microdissociation and reassociation without undergoing macro-dissociation away from the DNA helix (see Fig. 2). This interaction is thought to be mediated by an ionic interaction between the protein and DNA; most DNA-binding proteins possess a positively charged DNA-binding cleft such as that found at the dimer interface of UvrA.2 In order to discriminate between sliding and hopping, researchers use buffers containing different ionic strengths.54 The rate of sliding is believed to be independent of ionic strength in a purely ionic-mediated system because there is no change in the number of ions bound as the proteins move. However, hopping is greatly affected by the ionic strength as the ionic-mediated association of the protein to the DNA will be shielded with increasing salt concentrations, leading to shorter attached lifetimes and potentially more rapid diffusion.55 One extreme form of hopping is jumping, in which the UvrA2B2 complex could rebind to a second DNA helix at a great sequence distance from the initial DNA-binding site. This could occur inside a living cell where the local DNA concentration is very high. However, regardless of the two types (sliding or hopping) of movement on DNA, with no input of external energy, the probability of stepping in one direction versus the other is equal and therefore there is no net movement in any one direction. Thus, directed motion in which the protein migrates in only one direction requires an input of energy. It is possible that the ATPase sites in UvrA and UvrB could provide directed motion.
Fig. 2.
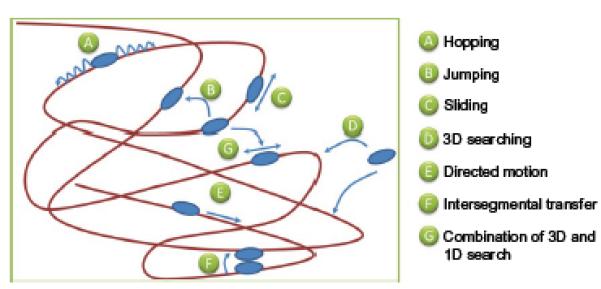
Potential modes of DNA damage searching by UvrA and UvrAB repair proteins. A number of mechanisms by which proteins can search for lesions are depicted. Hopping is distinguished from jumping by the distance over which the translocation occurs; however, in both cases, the protein remains within close proximity of the DNA. Sliding suggests that the protein remains in constant contact with the DNA, making it difficult to separate from hopping. If the protein dissociates from DNA into bulk solution, then a 3D search is employed to find the target site. Directed motion requires the input of energy in the form of nucleotide.
In another example of how UvrA2B2 could search the genome, it has been noted that this complex can, in principle, bind multiple DNA helices56 and, therefore, it offers a mechanism for transfer from one DNA region to another without ever dissociating in what is called intersegmental transfer (see Fig. 2). In this mode of searching, two DNA molecules would have to make close contact. Such mechanisms of protein movement on DNA have been proposed for restriction endonucleases and for lac repressor.57,58
Clearly, there are a number of proposed mechanisms by which proteins can negotiate DNA to find their target sites; however, definitive evidence for one mechanism versus another is extremely difficult to obtain using bulk methods. Therefore, more recently, there has been an explosion of studies using a variety of single-molecule techniques that permit each individual protein complex to be followed with good time and spatial resolution.40,43,45,59–69 These studies are likely to provide a clear understanding of which method(s) of protein transfer occurs. Understanding the precise mechanism of how proteins locate their target sites is of crucial importance and is discussed in more detail below.
C. Necessary Experimental Components to Observe Single Molecules in Action
The original presentation of the lac repressor finding its target site faster than the Smoluchowski-defined diffusional limit70,71 was the first experimental demonstration that the search mechanism may involve a 1D component instead of being purely 3D. This subsequently prompted a number of papers exploring the theoretical aspects of this potential mode of target location (see Ref. 55). In addition, a number of experimental studies have provided evidence for and against these models which are summarized below. Single-molecule techniques offer a direct method of determining how proteins find their targets and have more recently been deployed in a number of experimental formats. A few of these are summarized here:
Optical tweezers: By using a focused (normally infrared) beam, it is possible to exert forces on dielectric spheres, thus “trapping” them at the waist of the incident laser. This approach allows for the position of the trapped bead to be manipulated in three dimensions, and by attaching a single strand of DNA to the bead, the DNA is likewise manipulated. In an ingenious experiment where one end of DNA was tethered to a bead and the other to RNAP attached to a surface, it was shown that RNAP scans the groove of DNA in search of its promoter72 because movement of the surface led to rotation of the trapped bead. These data have provided strong evidence that sliding is used by RNAP. In another study, Yanagida and coworkers50 suspended a single extended DNA filament between two trapped beads and found that RNAP performed a random walk along the DNA. Upon release of the DNA tension, the DNA reformed a random coil conformation and it was noted that the proteins bound to the DNA targets with greater efficiency. This could imply either that DNA tension is preventing efficient target location or that the proteins bind and release rapidly and that their redistribution to the promoter sites is favored by the formation of a compact DNA structure that allows the proteins to three-dimensionally hop between sites on the DNA, as suggested by a later study using restriction endonucleases.73 The use of laser tweezers has permitted a number of elegant studies on the single-molecule properties of DNA–protein interactions,74–78 including obtaining base pair resolution of RNAP stepping.79 Despite these advances, optical trapping is not a trivial technique to apply as it requires specialist equipment and expertise. In addition, data collection is not multiplexed and is therefore slow as each investigation typically provides information on a single DNA strand. However, the recent use of multichannel fluidics could overcome some of the latter shortcomings.47
Magnetic tweezers offer an approach that harnesses much of the power of laser tweezers with the potential for more multiplexing. This approach has been used to investigate the function of a number of enzymes bound to DNA that alter its end-to-end length. Such length variations occur through alterations in single-stranded and double-stranded character80 or topoisomerase-induced relaxation of DNA,81 among others. For magnetic tweezers to provide detailed information on the physical basis of target site location, however, further development to combine this with fluorescence detection is required.
Direct imaging is currently in a position to provide direct information on the mechanism of target site acquisition for DNA-binding proteins. To date, a number of studies have provided diffusion constants and hopping distances (many summarized in Table I). These studies have invariably required the extension of the DNA substrate from its random coil to facilitate imaging of protein motions. To achieve this, DNA can be laid directly down on a surface by combing.82,83 This approach allows the highest signal to noise ratio because imaging can be performed in the evanescent field of a total internal reflection microscope (TIRF-equipped microscope). Typically, the surface is activated with a hydrophobic moiety such as polystyrene or polymethylmethacrylate.82 At low pH, DNA binds to these surfaces at two or more points; indeed, the number of binding points can be visualized by snapping the DNA under intense illumination, at which time the DNA recoils to its bound points on the surface.83 This approach is very simple and requires no further flow during data acquisition. Other methods of confining DNA close to the surface have been achieved by tagging one end of the DNA with biotin and conjugating to surface-immobilized streptavidin.42 Alternatively, lipid-immobilized DNA molecules can be spatially aligned on the coverslip such that under flow they elongate into “curtains”.84 The disadvantages of these methods are, first, that they all require flow (with the exception of combing), which will tend to bias the dataset toward sliders as proteins that dissociate from the DNA are likely to be carried away by the flow. In addition, the effects of the surface are unknown: Are moving complexes stalling due to the binding of the target site or because of interaction with the surface? Finally, as these structures are often located within the same focal plane as the surface, it becomes difficult to distinguish between surface-immobilized beads and those that are immobile but attached to the DNA. Stopping and starting flow using DNA curtains helps to circumvent this latter problem.30
Elevated DNA platforms or “tightropes”16 offer perhaps the best compromise between many of the methods described here as they are microns away from the surface and they elongate the DNA, and anything that appears stationary for long enough to see within the visual field must be bound to DNA (acquisitions are performed using high numerical aperture objectives, which shorten the depth of field) (see Fig. 3). These structures can be simply constructed by sequentially flowing the components into a surface-passivated observation flowcell.16 The simple flow sequence for setting up tightropes is as follows: (i) poly-l-lysine-coated 5 μm sized beads, (ii) DNA, (iii) wash, and (iv) Qdot-bound proteins. TIRF is adapted to illuminate the proteins through the lens by steering the excitation laser to a subcritical angle; in practice, this simply requires moving the excitation laser toward the center of the objective until the DNA tightropes can be seen. Using this “oblique angle fluorescence” (OAF) configuration provides considerable improvement in the signal to noise ratio over epi-fluorescence. Visualization of specific proteins is mediated through tagging with quantum dots (Qdots) (discussed later). Qdots are suitable for long acquisitions because of their resistance to photobleaching while their extreme brightness permits short integration times. Qdots also have very broad excitation spectra permitting multicolor emissions to be recorded using just a single excitation source. This latter point is extremely important as most DNA enzymes work with partners to perform their job; such is the case for all systems that exploit kinetic proofreading mechanisms. Moreover, as the proteins are elevated far from any surface-bound Qdots, they do not interfere with fluorescent spot discrimination. Finally, the density of DNA can be tuned to high or low values. This permits a single protein on a single DNA strand to be examined in the context of other closely juxtaposed DNA molecules. This is close, but not a perfect equivalent, to random coil DNA, where sequence-distant strands come within close spatial proximity of the DNA-bound proteins, thereby making it possible to study intersegmental transfer or even observe hopping directly16.
To image proteins working on DNA using any of the approaches above first requires the conjugation of a fluorophore. Here we focus on strategies for Qdot conjugation to allow single-molecule imaging during NER. In order to watch UvrA and UvrB work together to search DNA, two different strategies for conjugating Qdots were necessary16,85 (see Fig. 4). Qdots are commercially available with a number of different ligands attached to the surface and a wide variety of approaches have been used to visualize single molecules both in vitro and in vivo.86,87 The two most common are streptavidin and antibodies. In order to conjugate UvrA to Qdots, a biotin ligase site was engineered into the C terminus of UvrA. This 15-amino acid-sequence contains a specific lysine that is covalently attached to biotin by biotin ligase. This modification can be done during protein production within E. coli or after purification using purified biotin ligase to levels approaching 95% of the total protein. Mixing the biotinylated UvrA with excess Qdots ensures that only one UvrA monomer is attached per Qdot. As multiple streptavidin moieties are bound to the Qdot surface, atomic force microscopy is routinely used to assure a 1:1 stoichiometry of attachment of protein to DNA.16,85
Fig. 3.

Oblique angle fluorescence permits a high signal to noise view of DNA tightropes. To generate OAF (top right), a standard TIRF (bottom right) optical path is steered to a subcritical angle, resulting in a far-field illumination beam emerging at a steep angle. Although this was achieved using a custom-built system (left), this is possible using off-the-shelf systems with little difficulty.
Fig. 4.
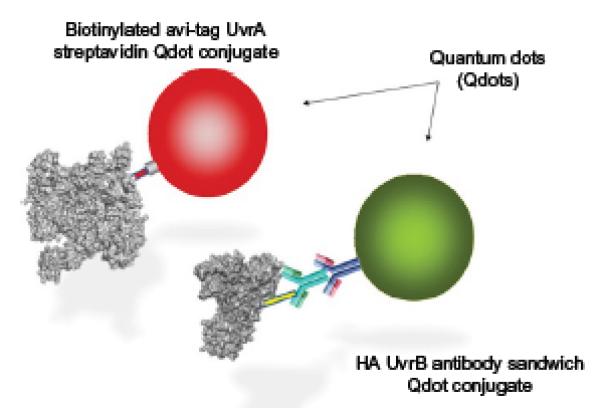
Quantum dot conjugation strategies. To ensure that there is no cross talk between different quantum dots, a differential conjugation strategy is important. Two approaches that were used for UvrA and UvrB conjugations are highlighted. UvrA (left) was bound to quantum dots using a short peptide sequence extension, which is biotinylated either endogenously or more efficiently using biotin ligase. UvrB could be differentially labeled by using an HA tag, which can be labeled using an antibody sandwich strategy.
Importantly, it is essential to ensure that the attachment of a Qdot does not affect the function of the protein. Therefore, electrophoretic mobility shift assays in agarose gels are used to ensure that the attachment of the Qdot to the protein does not interfere with its DNA binding or interaction with other proteins.16 UvrB was conjugated to Qdots by placing antibody recognition sequence of hemagglutinin, HA tag, on its N terminus. Specific high-affinity mouse monoclonal antibodies bind to the HA tag. Qdots coated with goat F (ab′)2 antimouse IgG conjugates are then used to make an “antibody sandwich”.85 Using this dual labeling approach, both proteins could be observed simultaneously as UvrA loads UvrB onto damaged sites.16 As many researchers use polyhistidine tags and nickel chromatography to purify and tag mammalian proteins, Piehler and coworkers have developed a chemical tag that binds to six or more histidines.88 They have used this approach to effectively label single molecules both in vitro and in vivo.89–91
Application of these methods allows the physical biochemistry of the DNA search to be examined. This is a problem that is pertinent not only to the mechanism of NER, but also to all protein systems that interact with DNA, and which can be addressed only now with the development of single-molecule methods. Specifically, using direct imaging (see approach 4 above), the first glimpses of how the bacterial NER proteins find lesions have been obtained after over four decades since the discovery of genes.
III. Damage Searching by UvrA2 and UvrA2B2
The first aspect of any DNA protein interaction is specificity, which often refers to the affinity of a protein for its cognate binding site. However, there is a vast amount of very similar DNA, which leads to the question: How are targets found? This is the central theme of this chapter and by using the single-molecule techniques outlined earlier, we are able to begin to address this question. It is likely that the NER system uses a combination of the many possible mechanisms such as sliding, hopping, and intersegmental transfer. Investigations at the single-molecule level have permitted direct observation of these processes and also allow quantitative characterization of their relative abundance. With more details emerging from such analyses, it may also become clear why one mechanism is favored over another (Fig. 5).
Fig. 5.
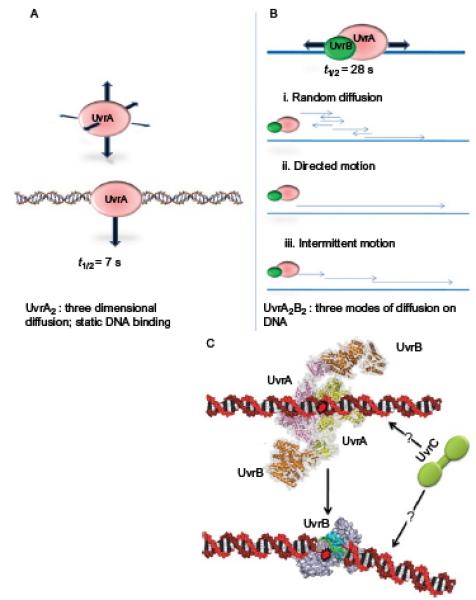
Summary of motion of UvrA2 and UvrA2B2 on DNA. (A) UvrA2 has been shown to perform a 3D search with an average bound life time of 7 s on DNA. UvrA2 was also observed jumping from DNA helix to another helix over distances of 1–2 μm (not shown). (B) UvrB collapses UvrA’s search mode from 3D to 1D sliding and increases its average lifetime on DNA to 40 s. Three different sliding modes were observed for UvrA2B2. (C) The nature of how UvrC finds its way to the UvrB–DNA preincision complex is not known. The low concentrations of UvrC in the cell would make a 3D search very inefficient if UvrC remained statically bound for longer than 1 s.
Observation of UvrA2 bound to DNA found it to remain statically bound for ~7 s before detaching and binding elsewhere. This is exactly the equivalent of a 3D search mechanism, where the DNA is searched through random encounters. However, it was noted that UvrA2 appeared to jump to nearby strands, with an average jump distance of 1.2 μm.16 The significance of this distance has yet to be determined, but it may be a result of the architecture of the assay. A high density of DNA tightropes resembles a sheet of binding surface; as a molecule is released from the DNA, what is the probability that it will pass back onto the sheet versus diffusing into solution? Does the relatively high incidence of rebinding suggest that DNA keeps the protein close to itself until it finds the right orientation to bind, possibly through electrostatics? If it is the latter, then further studies using salt to shield these charges will provide valuable insight. An approximation of the search time for a lesion suggested that the 3D mode of search was very inefficient16; therefore, it is unlikely that UvrA2 alone can search the DNA effectively. Surprisingly, however, it was found that the addition of UvrB to form a UvrA2B2 complex led to a change in the way UvrA2B2 searched the DNA relative to UvrA2 alone: UvrB collapsed the 3D search into 1D diffusion along the DNA.16 Recalculation of the 1D search time resulted in a huge increase in the rate at which DNA could be examined, bringing the search time for the UvrA2B2 molecules within the cells down from hours to minutes. It is interesting to note that not all of the molecules participated in this process; why is this so? One possibility is that the complex composition (UvrA2B2 vs. UvrA2B) may affect motility. It was found that the UvrAB complexes used three different modes of motion: 1D diffusion, directed, and paused. Directed motion was observed only in the presence of ATP; however, it is possible that these represented under-sampled populations of diffusers. Paused movers appeared to diffuse and then pause, often pausing at the same site many times, and although the DNA was not intentionally damaged, some nicks were detected. Perhaps these “pausers” were sensing damage but not forming a preincision complex16. Although the dominant UvrAB species was determined to comprise two each of UvrA and UvrB, there remains the possibility that other complex combinations18,92,93 may underlie the one-dimensionally diffusion-competent complex.
A comparison of the diffusion constants in Table I shows that the NER proteins are the slowest of all the complexes. This is not a consequence of the DNA tightrope assay as studies of base excision repair have shown much faster diffusion.37Therefore, the NER proteins engage in a slow search; is this because the proteins are hunting for lesions as they move? Or has this to do with the multienzyme processing that is occurring? It is tempting to speculate that there are components of both of these scenarios present. Lesions are thought to be found by detecting deformation of the DNA,9 which makes the proteins sensitive to the conformational state of the DNA; furthermore, as UvrB wraps the DNA around itself,94 this may lead to substantial local energy barriers if the DNA is held at each end as in the tightrope assay. UvrA2 is thought to pass the lesion to UvrB for checking, one of the kinetic proofreading steps; this process will not be instantaneous and therefore may also slow the search process. Wallace and coworkers37 showed that there is a correlation between the diffusion constant of protein motion and the “alpha” value. The alpha value is a description of how the molecule moves: if α = 1, then the protein is diffusing freely; α = 2 is directed motion; and α < 1 is subdiffusion or anomalous diffusion. It was found that slower movers have α < 1. Furthermore, they showed that a critical phenylalanine residue in the tip of an interrogation loop was responsible for this subdiffusive behavior of a DNA glycosylase as it moves along the DNA.37 Models of subdiffusion95,96 propose a number of explanations including the possibility that the molecule pauses and then continues. Our own simulations suggest the same, and therefore suggest that perhaps the slow diffusion of the UvrA2B2 is due to interrogation of the DNA. Interestingly, we have found that pause distributions occur over very long and very short timescales, offering a potential explanation for the observed paused movers. It is interesting to note that UvrA also contains a Phe residue at the tip of the zinc finger, which has been shown to be essential for damage recognition.4
Target acquisition is not only the domain of UvrA2B2 but also the problem that faces the other components of NER, which include UvrC, UvrD, Pol I, and DNA ligase. UvrC needs to efficiently locate a preincision complex with UvrB bound to the DNA. As the very slow release of UvrB is on the order of hours,92,97 it means that as damage accumulates, UvrB becomes trapped on the DNA. Therefore, efficient location by UvrC frees UvrB to search for more lesions. However, UvrC is present only at 20 molecules/cell, making this search process the likely rate-limiting step of the whole repair reaction.98 As UvrC is an endonuclease, its cellular concentration reflects a balance between its molecular imperative to cut DNA99,100 and its preincision complex search efficiency. In addition, as it has an affinity for UvrB and shares a binding site with UvrA on UvrB, it seems that a useful switch has evolved, preventing interaction with a searching complex until a lesion is found. However, UvrB is in excess of UvrA and therefore it is possible that UvrB interacts with UvrC,101 sequestering it from interacting with DNA until a preincision complex is found. Therefore, perhaps, UvrB forms a complex with UvrC and accompanies it to the preincision complex and at the same time protects DNA from spurious incisions.102
Single-molecule approaches offer another advantage beyond the physical description of lesion search: direct observation of repair in action being made possible, revealing both the sequence of events and their kinetics. The canonical mechanism of NER involves a complex of UvrA and UvrB searching for lesions, followed by UvrA’s ejection upon encountering damage. By observing this process, it will be possible to confirm this sequence of events and also determine the kinetics of UvrA release once the lesion is encountered. Furthermore, abortive complexes formed stochastically99,102 that may contribute to kinetic proofreading will also be detected. Together these data will provide a comprehensive understanding of how the preincision complex is formed. The next process of UvrC recruitment can also be studied similarly and, as DNA helicase working together with DNA polymerase I has been proposed to remove UvrC and the damaged oligonucleotide,103 we will also be able to discover whether UvrB is associated with the damaged strand or bound to the opposite strand, and whether UvrB is present as a dimer or a monomer. This latter question links back to our earlier discussion of hopping versus sliding; one of the key arguments against sliding is that it probes only one strand. Therefore, is the role of the UvrA2B2 complex to permit UvrB loading onto either strand? All these questions can be studied using multicolor direct imaging of repair proteins labeled with differently colored Qdots.
The physical principles that underlie NER are critical to our understanding of this process. Therefore, it is important that bulk and single-molecule studies go hand in hand. Furthermore, investigations in simple bacterial systems are an ideal platform for understanding these new concepts; diving into more complex mammalian counterpart systems will preclude a thorough understanding. Therefore, it is imperative that we revisit the bacterial system to achieve a robust grounding in the principles that likely underlie mammalian NER.
IV. Future Directions
A. Observing Protein Nanomachines at Work
The end goal of the investigations of protein machines working on DNA is to provide a kinetically correct, ordered mechanism. NER offers an ideal platform for creating such a view of DNA repair; it is a relatively simple multiprotein system that has been studied for many years, providing a body of knowledge from which we can now apply single-molecule methods to address the physical principles that underlie the mechanism. It has only recently become possible to image multiple proteins at the single-molecule level albeit currently there are many limitations, such as protein labeling, excitation of multiple dyes, and cross talk between multiple-emission color channels. However, with the introduction of Qdots that are all excited by a single wavelength, the possibilities for multiplexing are rapidly becoming reinforced. This still leaves key problems such as multiple specific protein labeling, and although imaging multiple colors is challenging, the use of image splitting and recombining technologies has enabled multiple differentially filtered images to be projected onto a single camera chip, permitting real-time multicolor imaging. Furthermore, with the very recent commercialization of complementary metaloxide semiconductor (CMOS) cameras, the available field of view has increased enormously without the cost of readout speed. Although issues of sensitivity and quantum efficiency still loom before CMOS, the rapid advances in EMCCD tell us that a new generation of highly sensitive rapid-imaging cameras is around the corner. Much larger image chips enable more colors to be imaged at the same time; three and four colors are currently possible with even more being conceivable in the near future. These new cameras combined with multicolor protein tagging and single-molecule tracking may allow real-time imaging of all the steps of DNA repair processes in living cells in the future.
B. Overcoming the Brownian Motion Barrier
What happens as a diffusing molecule moves? We return to the problem that it is not trivial to distinguish hopping from sliding. Evidence for both scenarios exists but the definitive test is to directly observe motion along DNA (see Table I). Currently, the time resolution for such motions is limited by photon capture, that is, an image is formed by the photons collected by the detector; more photons give a more precise estimation of the position of the protein. However, more photons take longer to collect, in which time a diffuser can move, reducing the accuracy of its position determination. This paradox can be addressed by collecting photons more quickly; one way to do this is to use dark-field microscopy. A gold nanoparticle attached to the protein of interest will scatter light in proportion to the rate (intensity) of photons striking it. If the incident light enters from an oblique angle, then it will not be directly detected, thus creating a “dark” background. This approach has been successfully used to measure the motion of the molecular motor myosin V104 with submillisecond time resolution. By observing slowly diffusing DNA–protein complexes moving with high spatial and temporal resolution, it will be possible to resolve whether proteins slide or hop. Additionally, gold nanorods offer the ability to follow the polarization of the molecule as it negotiates DNA; this is an important measure of whether or not the molecule follows the helical pitch of the DNA.
Recent technical advances have allowed for the development of new single-molecule approaches to study how proteins interact with DNA. A physical description of these interactions is vital as simply knowing the sequence of events only describes half of the picture. Furthermore, bulk methods average behavior across the entire population, whereas single-molecule approaches allow for a complete description of the various heterogeneous pathways and processes that molecules undertake in reality. This is clearly evident from the number of modes of motion that UvrAB employs to find its target site. The next challenge that faces the field is to construct more complex systems, and watch the entire process of NER occur in real time. First, this will be achieved in vitro and, ultimately, in vivo.39 Though this is not an easy challenge, as our approaches become more interdisciplinary, it is both a worthy and attainable one.
References
- 1.Peng Y, Wang H, Santana dos Santos L, Kisker C, Van Houten B. Chapter 13. Nucleotide excision repair from bacteria to humans: Structure-function studies. In: Penning TM, editor. Chemical carcinogenesis, current cancer research. Springer Science + Business Media, LLC; New York, NY: 2011. [Google Scholar]
- 2.Jaciuk M, Nowak E, Skowronek K, Tanska A, Nowotny M. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat Struct Mol Biol. 2011;18:191–7. doi: 10.1038/nsmb.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Croteau DL, DellaVecchia MJ, Perera L, Van Houten B. Cooperative damage recognition by UvrA and UvrB: identification of UvrA residues that mediate DNA binding. DNA Repair (Amst) 2008;7:392–404. doi: 10.1016/j.dnarep.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Croteau DL, DellaVecchia MJ, Wang H, Bienstock RJ, Melton MA, Van Houten B. The C-terminal zinc finger of UvrA does not bind DNA directly but regulates damage-specific DNA binding. J Biol Chem. 2006;281:26370–81. doi: 10.1074/jbc.M603093200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner K, Moolenaar GF, Goosen N. Role of the insertion domain and the zinc-finger motif of Escherichia coli UvrA in damage recognition and ATP hydrolysis. DNA Repair (Amst) 2011;10:483–96. doi: 10.1016/j.dnarep.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Wagner K, Moolenaar GF, Goosen N. Role of the two ATPase domains of Escherichia coli UvrA in binding non-bulky DNA lesions and interaction with UvrB. DNA Repair (Amst) 2010;9:1176–86. doi: 10.1016/j.dnarep.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Van Houten B, Gamper H, Hearst JE, Sancar A. Analysis of sequential steps of nucleotide excision repair in Escherichia coli using synthetic substrates containing single psoralen adducts. J Biol Chem. 1988;263:16553–60. [PubMed] [Google Scholar]
- 8.Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev. 2006;106:233–52. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- 9.Van Houten B, Croteau DL, DellaVecchia MJ, Wang H, Kisker C. “Close-fitting sleeves”: DNA damage recognition by the UvrABC nuclease system. Mutat Res. 2005;577:92–117. doi: 10.1016/j.mrfmmm.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Truglio JJ, Karakas E, Rhau B, Wang H, DellaVecchia MJ, Van Houten B, et al. Structural basis for DNA recognition and processing by UvrB. Nat Struct Mol Biol. 2006;13:360–4. doi: 10.1038/nsmb1072. [DOI] [PubMed] [Google Scholar]
- 11.Skorvaga M, DellaVecchia MJ, Croteau DL, Theis K, Truglio JJ, Mandavilli BS, et al. Identification of residues within UvrB that are important for efficient DNA binding and damage processing. J Biol Chem. 2004;279:51574–80. doi: 10.1074/jbc.M409266200. [DOI] [PubMed] [Google Scholar]
- 12.Skorvaga M, Theis K, Mandavilli BS, Kisker C, Van Houten B. The beta -hairpin motif of UvrB is essential for DNA binding, damage processing, and UvrC-mediated incisions. J Biol Chem. 2002;277:1553–9. doi: 10.1074/jbc.M108847200. [DOI] [PubMed] [Google Scholar]
- 13.Karakas E, Truglio JJ, Croteau D, Rhau B, Wang L, Van Houten B, et al. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007;26:613–22. doi: 10.1038/sj.emboj.7601497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truglio JJ, Rhau B, Croteau DL, Wang L, Skorvaga M, Karakas E, et al. Structural insights into the first incision reaction during nucleotide excision repair. EMBO J. 2005;24:885–94. doi: 10.1038/sj.emboj.7600568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Husain I, Van Houten B, Thomas DC, Abdel-Monem M, Sancar A. Effect of DNA polymerase I and DNA helicase II on the turnover rate of UvrABC excision nuclease. Proc Natl Acad Sci USA. 1985;82:6774–8. doi: 10.1073/pnas.82.20.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol Cell. 2010;37:702–13. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malta E, Moolenaar GF, Goosen N. Dynamics of the UvrABC nucleotide excision repair proteins analyzed by fluorescence resonance energy transfer. Biochemistry. 2007;46:9080–8. doi: 10.1021/bi7002235. [DOI] [PubMed] [Google Scholar]
- 18.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, et al. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–33. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pakotiprapha D, Liu Y, Verdine GL, Jeruzalmi D. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J Biol Chem. 2009;284:12837–44. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DellaVecchia MJ, Croteau DL, Skorvaga M, Dezhurov SV, Lavrik OI, Van Houten B. Analyzing the handoff of DNA from UvrA to UvrB utilizing DNA-protein photoaffinity labeling. J Biol Chem. 2004;279:45245–56. doi: 10.1074/jbc.M408659200. [DOI] [PubMed] [Google Scholar]
- 21.Reardon JT, Sancar A. Thermodynamic cooperativity and kinetic proofreading in DNA damage recognition and repair. Cell cycle (Georgetown, Tex.) 2004;3:141–4. [PubMed] [Google Scholar]
- 22.Luijsterburg MS, von Bornstaedt G, Gourdin AM, Politi AZ, Mone MJ, Warmerdam DO, et al. Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J Cell Biol. 2010;189:445–63. doi: 10.1083/jcb.200909175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA. 1974;71:4135–9. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellon I, Hanawalt PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342:95–8. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 25.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–8. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 26.Gascon J, Oubina A, Perez-Lezaun A, Urmeneta J. Sensitivity of selected bacterial species to UV radiation. Curr Microbiol. 1995;30:177–82. doi: 10.1007/BF00296205. [DOI] [PubMed] [Google Scholar]
- 27.Chandrasekhar D, Van Houten B. In vivo formation and repair of cyclobutane pyrimidine dimers and 6-4 photoproducts measured at the gene and nucleotide level in Escherichia coli. Mutat Res. 2000;450:19–40. doi: 10.1016/s0027-5107(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 28.Chandrasekhar D, Van Houten B. High resolution mapping of UV-induced photoproducts in the Escherichia coli lacI gene. Inefficient repair of the non-transcribed strand correlates with high mutation frequency. J Mol Biol. 1994;238:319–32. doi: 10.1006/jmbi.1994.1295. [DOI] [PubMed] [Google Scholar]
- 29.Gruskin EA, Lloyd RS. Molecular analysis of plasmid DNA repair within ultraviolet-irradiated Escherichia coli.II. UvrABC-initiated excision repair and photolyase-catalyzed dimer monomerization. J Biol Chem. 1988;263:12738–43. [PubMed] [Google Scholar]
- 30.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–74. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 31.Elowitz MB, Surette MG, Wolf PE, Stock JB, Leibler S. Protein mobility in the cytoplasm of Escherichia coli. J Bacteriol. 1999;181:197–203. doi: 10.1128/jb.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berg HC. Random walks in biology. Princeton University Press; Princeton, N.J: 1983. [Google Scholar]
- 33.Gowers DM, Halford SE. Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. EMBO J. 2003;22:1410–8. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rau DC, Sidorova NY. Diffusion of the restriction nuclease EcoRI along DNA. J Mol Biol. 2010;395:408–16. doi: 10.1016/j.jmb.2009.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnet I, Biebricher A, Porte PL, Loverdo C, Benichou O, Voituriez R, et al. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008;36:4118–27. doi: 10.1093/nar/gkn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biebricher A, Wende W, Escude C, Pingoud A, Desbiolles P. Tracking of single quantum dot labeled EcoRV sliding along DNA manipulated by double optical tweezers. Biophys J. 2009;96:L50–2. doi: 10.1016/j.bpj.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunn AR, Kad NM, Nelson SR, Warshaw DM, Wallace SS. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011;39:7487–98. doi: 10.1093/nar/gkr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, DellaVecchia MJ, Skorvaga M, Croteau DL, Erie DA, Van Houten B. UvrB domain 4, an autoinhibitory gate for regulation of DNA binding and ATPase activity. J Biol Chem. 2006;281:15227–37. doi: 10.1074/jbc.M601476200. [DOI] [PubMed] [Google Scholar]
- 39.Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science. 2007;316:1191–4. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorman J, Plys AJ, Visnapuu ML, Alani E, Greene EC. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol. 2010;17:932–8. doi: 10.1038/nsmb.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorman J, Chowdhury A, Surtees JA, Shimada J, Reichman DR, Alani E, et al. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28:359–70. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci USA. 2006;103:5752–7. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong C, Cho WK, Song KM, Cook C, Yoon TY, Ban C, et al. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat Struct Mol Biol. 2011;18:379–85. doi: 10.1038/nsmb.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochaniak AB, Habuchi S, Loparo JJ, Chang DJ, Cimprich KA, Walter JC, et al. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem. 2009;284:17700–10. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci USA. 2011;108:563–8. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graneli A, Yeykal CC, Robertson RB, Greene EC. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc Natl Acad Sci USA. 2006;103:1221–6. doi: 10.1073/pnas.0508366103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amitani I, Liu B, Dombrowski CC, Baskin RJ, Kowalczykowski SC. Watching individual proteins acting on single molecules of DNA. Methods Enzymol. 2010;472:261–91. doi: 10.1016/S0076-6879(10)72007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finkelstein IJ, Visnapuu ML, Greene EC. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature. 2010;468:983–7. doi: 10.1038/nature09561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JH, Larson RG. Single-molecule analysis of 1D diffusion and transcription elongation of T7 RNA polymerase along individual stretched DNA molecules. Nucleic Acids Res. 2007;35:3848–58. doi: 10.1093/nar/gkm332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harada Y, Funatsu T, Murakami K, Nonoyama Y, Ishihama A, Yanagida T. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophys J. 1999;76:709–15. doi: 10.1016/S0006-3495(99)77237-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Etson CM, Hamdan SM, Richardson CC, van Oijen AM. Thioredoxin suppresses microscopic hopping of T7 DNA polymerase on duplex DNA. Proc Natl Acad Sci USA. 2010;107:1900–5. doi: 10.1073/pnas.0912664107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc Natl Acad Sci USA. 2008;105:10791–6. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schurr JM. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys Chem. 1979;9:413–4. [PubMed] [Google Scholar]
- 54.von Hippel PH, Berg OG. Facilitated target location in biological systems. J Biol Chem. 1989;264:675–8. [PubMed] [Google Scholar]
- 55.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–48. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 56.Wagner K, Moolenaar G, van Noort J, Goosen N. Single-molecule analysis reveals two separate DNA-binding domains in the Escherichia coli UvrA dimer. Nucleic Acids Res. 2009;37:1962–72. doi: 10.1093/nar/gkp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halford SE, Welsh AJ, Szczelkun MD. Enzyme-mediated DNA looping. Annu Rev Biophys Biomol Struct. 2004;33:1–24. doi: 10.1146/annurev.biophys.33.110502.132711. [DOI] [PubMed] [Google Scholar]
- 58.Fickert R, Muller-Hill B. How Lac repressor finds lac operator in vitro. J Mol Biol. 1992;226:59–68. doi: 10.1016/0022-2836(92)90124-3. [DOI] [PubMed] [Google Scholar]
- 59.Wang F, Greene EC. Single-molecule studies of transcription: from one RNA polymerase at a time to the gene expression profile of a cell. J Mol Biol. 2011;412:814–31. doi: 10.1016/j.jmb.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Oijen AM, Loparo JJ. Single-molecule studies of the replisome. Annu Rev Biophys. 2010;39:429–48. doi: 10.1146/annurev.biophys.093008.131327. [DOI] [PubMed] [Google Scholar]
- 61.Rigler R. Fluorescence and single molecule analysis in cell biology. Biochem Biophys Res Commun. 2010;396:170–5. doi: 10.1016/j.bbrc.2010.04.070. [DOI] [PubMed] [Google Scholar]
- 62.Rajagopalan S, Huang F, Fersht AR. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2010;39:2294–303. doi: 10.1093/nar/gkq800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manley S, Gillette JM, Lippincott-Schwartz J. Single-particle tracking photoactivated localization microscopy for mapping single-molecule dynamics. Methods Enzymol. 2010;475:109–20. doi: 10.1016/S0076-6879(10)75005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lymperopoulos K, Kiel A, Seefeld A, Stohr K, Herten DP. Fluorescent probes and delivery methods for single-molecule experiments. Chemphyschem. 2010;11:43–53. doi: 10.1002/cphc.200900359. [DOI] [PubMed] [Google Scholar]
- 65.Pierobon P, Cappello G. Quantum dots to tail single bio-molecules inside living cells. Adv Drug Deliv Rev. 2012;64:167–78. doi: 10.1016/j.addr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 66.Lavelle C, Praly E, Bensimon D, Le Cam E, Croquette V. Nucleosome remodeling machines and other molecular motors observed at the single molecule level. FEBS J. 2011;278:3596–607. doi: 10.1111/j.1742-4658.2011.08280.x. [DOI] [PubMed] [Google Scholar]
- 67.Gill JP, Wang J, Millar DP. DNA polymerase activity at the single-molecule level. Biochem Soc Trans. 2011;39:595–9. doi: 10.1042/BST0390595. [DOI] [PubMed] [Google Scholar]
- 68.English BP, Hauryliuk V, Sanamrad A, Tankov S, Dekker NH, Elf J. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci USA. 2011;108:E365–73. doi: 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Friedman JI, Stivers JT. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010;49:4957–67. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Riggs AD, Bourgeois S, Cohn M. The lac repressor-operator interaction. 3. Kinetic studies. J Mol Biol. 1970;53:401–17. doi: 10.1016/0022-2836(70)90074-4. [DOI] [PubMed] [Google Scholar]
- 71.Riggs AD, Suzuki H, Bourgeois S. Lac repressor-operator interaction I. Equilibrium studies. J Mol Biol. 1970;48:67–83. doi: 10.1016/0022-2836(70)90219-6. [DOI] [PubMed] [Google Scholar]
- 72.Sakata-Sogawa K, Shimamoto N. RNA polymerase can track a DNA groove during promoter search. Proc Natl Acad Sci USA. 2004;101:14731–5. doi: 10.1073/pnas.0406441101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van den Broek B, Lomholt MA, Kalisch SM, Metzler R, Wuite GJ. How DNA coiling enhances target localization by proteins. Proc Natl Acad Sci USA. 2008;105:15738–42. doi: 10.1073/pnas.0804248105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bianco PR, Kowalczykowski SC. Translocation step size and mechanism of the RecBC DNA helicase. Nature. 2000;405:368–72. doi: 10.1038/35012652. [DOI] [PubMed] [Google Scholar]
- 75.Brower-Toland BD, Smith CL, Yeh RC, Lis JT, Peterson CL, Wang MD. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc Natl Acad Sci USA. 2002;99:1960–5. doi: 10.1073/pnas.022638399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perkins TT, Li HW, Dalal RV, Gelles J, Block SM. Forward and reverse motion of single RecBCD molecules on DNA. Biophys J. 2004;86:1640–8. doi: 10.1016/S0006-3495(04)74232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skinner GM, Baumann CG, Quinn DM, Molloy JE, Hoggett JG. Promoter binding, initiation, and elongation by bacteriophage T7 RNA polymerase. A single-molecule view of the transcription cycle. J Biol Chem. 2004;279:3239–44. doi: 10.1074/jbc.M310471200. [DOI] [PubMed] [Google Scholar]
- 78.van Oijen AM, Blainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS. Single-molecule kinetics of lambda exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–8. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]
- 79.Abbondanzieri EA, Greenleaf WJ, Shaevitz JW, Landick R, Block SM. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005;438:460–5. doi: 10.1038/nature04268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee JB, Hite RK, Hamdan SM, Xie XS, Richardson CC, van Oijen AM. DNA primase acts as a molecular brake in DNA replication. Nature. 2006;439:621–4. doi: 10.1038/nature04317. [DOI] [PubMed] [Google Scholar]
- 81.Strick TR, Croquette V, Bensimon D. Single-molecule analysis of DNA uncoiling by a type II topoisomerase. Nature. 2000;404:901–4. doi: 10.1038/35009144. [DOI] [PubMed] [Google Scholar]
- 82.Allemand JF, Bensimon D, Jullien L, Bensimon A, Croquette V. pH-dependent specific binding and combing of DNA. Biophys J. 1997;73:2064–70. doi: 10.1016/S0006-3495(97)78236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lyon WA, Fang MM, Haskins WE, Nie S. A dual-beam optical microscope for observation and cleavage of single DNA molecules. Anal Chem. 1998;70:1743–8. doi: 10.1021/ac980040+. [DOI] [PubMed] [Google Scholar]
- 84.Graneli A, Yeykal CC, Prasad TK, Greene EC. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir. 2006;22:292–9. doi: 10.1021/la051944a. [DOI] [PubMed] [Google Scholar]
- 85.Wang H, Tessmer I, Croteau DL, Erie DA, Van Houten B. Functional characterization and atomic force microscopy of a DNA repair protein conjugated to a quantum dot. Nano Lett. 2008;8:1631–7. doi: 10.1021/nl080316l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pinaud F, Clarke S, Sittner A, Dahan M. Probing cellular events, one quantum dot at a time. Nat Methods. 2010;7:275–85. doi: 10.1038/nmeth.1444. [DOI] [PubMed] [Google Scholar]
- 87.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat Mater. 2005;4:435–46. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 88.Clarke S, Pinaud F, Beutel O, You C, Piehler J, Dahan M. Covalent monofunctionalization of peptide-coated quantum dots for single-molecule assays. Nano Lett. 2010;10:2147–54. doi: 10.1021/nl100825n. [DOI] [PubMed] [Google Scholar]
- 89.DeRocco V, Anderson T, Piehler J, Erie DA, Weninger K. Four-color single-molecule fluorescence with noncovalent dye labeling to monitor dynamic multimolecular complexes. Biotechniques. 2010;49:807–16. doi: 10.2144/000113551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roullier V, Clarke S, You C, Pinaud F, Gouzer GG, Schaible D, et al. High-affinity labeling and tracking of individual histidine-tagged proteins in live cells using Ni2+ tris-nitrilotriacetic acid quantum dot conjugates. Nano Lett. 2009;9:1228–34. doi: 10.1021/nl9001298. [DOI] [PubMed] [Google Scholar]
- 91.You C, Wilmes S, Beutel O, Lochte S, Podoplelowa Y, Roder F, et al. Self-controlled monofunctionalization of quantum dots for multiplexed protein tracking in live cells. Angewandte Chemie. 2010;49:4108–12. doi: 10.1002/anie.200907032. (International ed. in English) [DOI] [PubMed] [Google Scholar]
- 92.Orren DK, Sancar A. The (A)BC excinuclease of Escherichia coli has only the UvrB and UvrC subunits in the incision complex. Proc Natl Acad Sci USA. 1989;86:5237–41. doi: 10.1073/pnas.86.14.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Orren DK, Sancar A. Formation and enzymatic properties of the UvrB.DNA complex. J Biol Chem. 1990;265:15796–803. [PubMed] [Google Scholar]
- 94.Wang H, Lu M, Tang MS, Van Houten B, Ross JB, Weinfeld M, et al. DNA wrapping is required for DNA damage recognition in the Escherichia coli DNA nucleotide excision repair pathway. Proc Natl Acad Sci USA. 2009;106:12849–54. doi: 10.1073/pnas.0902281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saxton MJ. A biological interpretation of transient anomalous subdiffusion I. Qualitative model. Biophys J. 2007;92:1178–91. doi: 10.1529/biophysj.106.092619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guigas G, Weiss M. Sampling the cell with anomalous diffusion - the discovery of slowness. Biophys J. 2008;94:90–4. doi: 10.1529/biophysj.107.117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yeung AT, Mattes WB, Grossman L. Protein complexes formed during the incision reaction catalyzed by the Escherichia coli UvrABC endonuclease. Nucleic Acids Res. 1986;14:2567–82. doi: 10.1093/nar/14.6.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Van Houten B. Nucleotide excision repair in Escherichia coli. Microbiol Rev. 1990;54:18–51. doi: 10.1128/mr.54.1.18-51.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Caron PR, Grossman L. Incision of damaged versus nondamaged DNA by the Escherichia coli UvrABC proteins. Nucleic Acids Res. 1988;16:7855–65. doi: 10.1093/nar/16.16.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Verhoeven EE, van Kesteren M, Moolenaar GF, Visse R, Goosen N. Catalytic sites for 3′and 5′incision of Escherichia coli nucleotide excision repair are both located in UvrC. J Biol Chem. 2000;275:5120–3. doi: 10.1074/jbc.275.7.5120. [DOI] [PubMed] [Google Scholar]
- 101.Moolenaar GF, Franken KL, van de Putte P, Goosen N. Function of the homologous regions of the Escherichia coli DNA excision repair proteins UvrB and UvrC in stabilization of the UvrBC-DNA complex and in 3′-incision. Mutat Res. 1997;385:195–203. doi: 10.1016/s0921-8777(97)00042-6. [DOI] [PubMed] [Google Scholar]
- 102.Moolenaar GF, Hoglund L, Goosen N. Clue to damage recognition by UvrB: residues in the beta-hairpin structure prevent binding to non-damaged DNA. EMBO J. 2001;20:6140–9. doi: 10.1093/emboj/20.21.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Orren DK, Selby CP, Hearst JE, Sancar A. Post-incision steps of nucleotide excision repair in Escherichia coli. Disassembly of the UvrBC-DNA complex by helicase II and DNA polymerase I. J Biol Chem. 1992;267:780–8. [PubMed] [Google Scholar]
- 104.Dunn AR, Spudich JA. Dynamics of the unbound head during myosin V processive translocation. Nat Struct Mol Biol. 2007;14:246–8. doi: 10.1038/nsmb1206. [DOI] [PubMed] [Google Scholar]