

Abstract
Tumour-induced myeloid-derived suppressor cells (MDSC) promote immune suppression and mediate tumour progression. However, the molecular basis for the generation of MDSC, which in mice co-express the CD11b+ and Gr-1+ cell surface markers remains unclear. Because CD11b+Gr-1+ cells expand during progressive tumour growth, this suggests that tumour-induced events alter signalling pathways that affect normal myeloid cell development. Interferon regulatory factor-8 (IRF-8), a member of the IFN-γ regulatory factor family, is essential for normal myelopoiesis. We therefore examined whether IRF-8 modulated tumour-induced CD11b+Gr-1+ cell development or accumulation using both implantable (4T1) and transgenic (MMTV-PyMT) mouse models of mammary tumour growth. In the 4T1 model, both splenic and bone marrow-derived CD11b+Gr-1+ cells of tumour-bearing mice displayed a marked reduction in IRF-8 expression compared to control populations. A causal link between IRF-8 expression and the emergence of tumour-induced CD11b+Gr-1+ cells was explored in vivo using a double transgenic (dTg) mouse model designed to express transgenes for both IRF-8 and mammary carcinoma development. Despite the fact that tumour growth was unaffected, splenomegaly, as well as the frequencies and absolute numbers of CD11b+Gr-1+ cells were significantly lower in dTg mice when compared with single transgenic tumour-bearing mice. Overall, these data reveal that IRF-8 plays an important role in tumour-induced development and/or accumulation of CD11b+Gr-1+ cells, and establishes a molecular basis for the potential manipulation of these myeloid populations for cancer therapy.
Keywords: IRF-8, myeloid-derived suppressor cells, tumour progression, haematopoiesis
Introduction
During host–tumour interactions, a variety of tumour-derived factors (TDF) serve to down-regulate T-cell responses directly or indirectly via recruitment of various immune suppressive cell subpopulations [1–3]. Indeed, a number of distinct immune suppressive cell subsets have been defined that inhibit adaptive T-cell-mediated immunity. For example, in mouse models, tumour growth and progression have been accompanied by the expansion and accumulation of a heterogeneous population of myeloid cells that co-express the CD11b and Gr-1 differentiation markers, and have been termed myeloid-derived suppressor cells (MDSC) [4, 5]. Although more extensively studied in animal models, the proliferation of immunosuppressive myeloid subpopulations have also been observed in human neoplasia, including carcinoma of the head and neck, non-small cell lung carcinoma, renal carcinoma, melanoma and adenocarcinomas of the colon, breast and pancreas [6–10].
In experimental animal models, tumour-induced CD11b+Gr-1+ cells have been implicated in the suppression of T-cell-mediated immunity through diverse pathways, including: (a) the secretion of transforming growth factor-β (TGF-β) which down-regulates CTL induction [11]; (b) the production of nitric oxide, degradation of arginine via arginase-1 activity or the generation of reactive oxygen species that alters signalling, activation and eventually survival of T cells [4, 5, 8, 12] and (c) the promotion of tumour angiogenesis through matrix metalloproteinase-9 (MMP-9) production [13]. Although these MDSC are readily apparent in the bone marrow and peripheral lymphoid organs, they can also infiltrate tumour tissue and constitute approximately 5% of the total cells within the mass [13].
Multiple mechanisms have been reported to account for the immune suppressive behaviour of MDSC, including those outlined above. However, the molecular pathway(s) that underlie their development or expansion remain to be elucidated. Because CD11b+Gr-1+ MDSC aberrantly expand during the process of tumour growth, this suggests that tumour-induced events alter the signalling pathways that affect normal myeloid cell production, differentiation and/or survival. One principal gene that positively regulates normal myeloid cell development, differentiation and function in monocytes/macrophages and dendritic cells is interferon regulatory factor-8 (IRF-8). IRF-8, also known as interferon consensus sequence binding protein, is a member of the IRF family of transcription factors [14–16].
The importance of IRF-8 in regulating normal myelopoiesis has been clearly illustrated in IRF-8 null mice [17–19]. IRF-8-deficiency alters normal haematopoiesis, and leads to a marked accumulation of macrophages, neutrophils and CD11b+Gr-1+ cells in the spleen, lymph nodes, peripheral blood and bone marrow [17–19]. Additionally, IRF-8-deficient macrophages and neutrophils exhibit heightened levels of apoptotic resistance [20]. Ultimately, these mice develop a chronic myelogenous leukaemia-like syndrome [17], revealing that IRF-8 plays an integral role in the regulation of cell death and the pathogenesis of certain haematological malignancies. These observations also raise the notion that the production or accumulation of CD11b+Gr-1+ MDSC in tumour-bearing hosts arise, at least in part, as a consequence of a novel mechanism of tumour-induced IRF-8 down-regulation.
To test this hypothesis, we made use of both implantable (4T1) and transgenic (MMTV-PyMT) mouse models of mammary carcinoma. The 4T1 tumour cell line is a well characterized model, reflecting a spontaneously arising neoplasm in BALB/c mice [21–23]. When implanted orthotopically into a mammary gland of syngeneic mice, 4T1 exhibits progressive tumour growth that is accompanied by spontaneous metastasis to the lymph nodes, lung, liver, bone marrow and brain, as well as a rapid and robust accumulation of MDSC that are readily detectable in peripheral lymphoid tissues. The second model involved the use of transgenic (Tg) mice expressing the polyomavirus middle T antigen (PyMT) under the transcriptional control of the MMTV promoter [24], henceforth termed MTAG mice [25, 26]. These mice develop tissue-specific mammary gland carcinoma, which eventually metastasizes to the lungs. Tumour progression in MTAG mice has been shown to mimic human breast cancer pathology at morphological (disease stage), molecular and immunological levels [25–28]. Moreover, we recently demonstrated that autochthonous primary tumour growth in MTAG mice was accompanied by a significant accumulation of splenic CD11b+Gr-1+ cells [26]. This model has also been used to study the role of tumour-associated macrophages in tumour progression [29].
In both the 4T1 and MTAG models, we showed that tumour-induced CD11b+Gr-1+ cells displayed a marked reduction in constitutive IRF-8 expression levels compared to non-tumour-bearing control populations. Furthermore, through the use of a double Tg (dTg) mouse model, designed to harbour transgenes for both IRF-8 and mammary carcinoma development (MTAG), we observed a significant reduction in both splenomegaly and the frequencies of CD11b+Gr-1+ cells in vivo compared to that observed in single transgenic (sTg) tumour-bearing mice. Taken together, these data reveal an important role for IRF-8 in the development/expansion of CD11b+Gr-1+ cells during tumour progression.
Materials and methods
Mice
Female BALB/c (H-2d) and C57BL/6 (B6; H-2b) mice, 5–6 weeks of age, were obtained from the National Cancer Institute-Frederick Cancer Research Animal Facility (Frederick, MD, USA). Female CB6F1/J hybrid mice (BALB/cJ F × C57BL/6J M) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). The MTAG (MMTV-PyMT) transgenic mouse expresses the polyomavirus middle T antigen driven by the MMTV-LTR promoter [24]. These transgenic mice were originally derived in FVB mice [24] and were later backcrossed on a B6 background, which was kindly provided by S. Gendler (Mayo Clinic, Scottsdale, AZ, USA) [30]. Polyomavirus middle T oncogene expression results in the generation of multifocal mammary carcinomas in 100% of female mice, followed by progression to pulmonary metastases in the vast majority [24, 26]. Only female MTAG mice were used in these experiments and were obtained by breeding Tg male mice with wild-type B6 female mice. Progeny were monitored for transgene expression by PCR analysis. Non-Tg littermates were used as age- and gender-matched controls. All mice in these studies were housed in a specific pathogen-free environment, and all experiments were conducted under approved protocols in accordance with institutional guidelines for animal care and use (IACUC).
In accordance with these regulations, no single tumour mass was allowed to exceed 2 cm3, although MTAG mice may develop up to 10 discrete tumours. MTAG mice were killed once any single tumour mass reached 2 cm3 in size or tumours interfered with normal activity. Tumour growth was measured weekly in two dimensions, using a digital calliper, and tumour volume was calculated as previously described [26] using the formula (w2× l)/2. In MTAG mice, total tumour volume was determined as the sum of each distinct tumour volume within an individual mouse.
Production of IRF-8 Tg mice
Complementary DNA (cDNA) encoding the mouse IRF-8 coding sequence was cloned into the pcDNA3.1(+) plasmid (Invitrogen, Carlsbad, CA, USA) under the control of the cytomegalovirus (CMV) promoter (plasmid kindly provided by K. Ozato, National Institutes of Health [NIH], Bethesda, MD, USA). A 4.3-kb fragment was then generated from the construct by cleavage at an FspI restriction site, and isolation by sucrose gradient purification. This purified material was then microinjected into the pronuclei of one cell embryos of fertilized eggs from B6 mice to obtain several founder mice (Laboratory Animal Sciences Program, SAIC, Frederick, MD, USA). Transgene integration in progeny was verified by Southern blot analysis using the 4.3-kb fragment as the probe and PCR genotyping of tail snips. The founder line, E5, was selected for this study based on the highest IRF-8 copy number and mRNA levels. Furthermore, no atypical effects of IRF-8 transgene expression were observed on various haematological parameters, as determined by phenotypic analysis of the spleen and complete blood count (CBC) on peripheral whole blood. F1 progeny were produced by mating sTg male mice with either wild-type female B6 or BALB/c mice. Moreover, male MTAG mice were bred with female IRF-8 Tg mice to produce dTg mice (in a B6 background) that possessed the transgenes for both IRF-8 and spontaneous mammary carcinoma development (i.e. MMTV-PyMT). Transgene expression was verified by PCR analysis of tail snip DNA using the conditions mentioned in the PCR/RT-PCR section below.
Tumour cells and inoculations
The 4T1 mammary carcinoma cell line was obtained from the American Type Culture Collection (CRL-2539; ATCC, Manassas, VA, USA). 4T1 cells were maintained in RPMI supplemented with 0.1 mM non-essential amino acids, 1mM sodium pyruvate, 2 mM L-glutamine, 15 mM HEPES, 100 U/ml penicillin-100 μg/ml streptomycin solution, 50 μM 2-mercaptoethanol and 10% heat inactivated foetal bovine serum (Gemini, Woodland, CA, USA). 4T1 tumour growth was achieved in vivo by orthotopic implantation of 4T1 cells (5 × 104 cells) into mammary gland #2. Tumour growthwas measured thrice weekly in two dimensions, using a digital calliper, and tumour volume calculated as noted earlier. Mice were euthanized 20–27 days after injection, and spleens and/or bone marrow cells were collected by flushing the femurs and tibias with culture medium, as previously described [31].
Flow cytometric studies
All antibodies (Ab) utilized in these experiments were purchased from BD Biosciences (San Diego, CA, USA). Unfractionated splenocytes or bone marrow cells were stained by direct immunofluorescence with anti-CD11b-PE, anti-Gr-1-FITC, anti-B220-FITC, anti-CD4-PE, anti-CD8-FITC mAb or the appropriate isotype-matched Ab controls, as described [26] and analysed by flow cytometry using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were analysed by CellQuest software (Becton Dickinson).
Purification and culture of CD11b+Gr-1+ cells
CD11b+Gr-1+ cells were purified from the spleens or bone marrow of control or tumour-bearing mice using similarly established protocols [32]. Briefly, single cell suspensions were suspended in ‘depletion buffer’ (PBS, pH 7.2, containing 2 mM ethylenediaminetetraacetic acid and 0.5% bovine serum albumin) before the addition of anti-CD11b MicroBeads (Miltenyi Biotec, Auburn, CA, USA), as per manufacturer’s instructions. Cells were maintained on ice for 30 min., followed by washing with a 15–20 fold (v/v) excess of the same buffer. Cells were then resuspended to an appropriate volume and passed through a MACS pre-separation filter prior to separation on the AutoMACS separator system (Miltenyi Biotec) using a double column depletion protocol. Purity of the positively selected myeloid population was ascertained using anti-CD11b-PE and anti-Gr-1-FITC mAb, as previously outlined. This technique was very effective for the isolation of CD11b+ cells that also co-expressed Gr-1, resulting in populations of double-positive cells that were routinely >90% from both control and tumour-bearing (4T1, MTAG) mice. (In many cases, the purity was even greater.) CD11b+Gr-1+ cells were stimulated by culturing for 24 hrs in the absence or presence of lipopolysaccharide (LPS/L-6143; Sigma-Aldrich, St. Louis, MO, USA) and recombinant mouse IFN-γ (100 U/ml; R&D Systems, Minneapolis, MN, USA). Cell-free supernatants were then collected for cytokine analysis, and the cells retrieved for RNA isolation.
Proliferation assay
Ninety-six-well flat-bottomed plates were pre-coated with anti-CD3 mAb (1 μg/well) by incubation at 4°C overnight; plates were then washed prior to the addition of the various cell combinations. Splenic CD4+ or CD8+ T cells were purified from CB6F1/J hybrid mice (using T cell subset-specific MicroBeads and the AutoMACS separator, and plated (5 × 104 cells/well). Splenic CD11b+Gr-1+ cells were purified, as described above, from the non-tumour-bearing control or 4T1 tumour-bearing hybrid mice and added to the plate at a CD11b+Gr-1+ cell: T-cell ratio of 1:1. Plates were incubated for 48 hrs at 37°C, after which 3H-thymidine (1 μCi/ well) was added for an additional 24 hrs of culture. The extent of proliferation was determined by measuring 3H-thymidine incorporation by liquid scintillation spectroscopy.
RT-PCR and PCR analyses
For RT-PCR, total RNA was isolated from CD11b+Gr-1+ cells using RNeasy Mini kits from Qiagen (Valencia, CA, USA) according to manufacturer’s instructions. The RNeasy kit includes a silica-membrane technology that efficiently removes genomic DNA without DNase treatment. Total RNA was used for first strand cDNA synthesis using the ThermoScript RT-PCR system (Invitrogen). The cDNA was then used as the template for PCR amplification of the following mouse genes: GAPDH, IRF-8, IL-12p40 and inducible nitric oxide synthase (iNOS). PCR reactions were carried out in a PTC-200 thermal cycler (MJ Research, Waltham, MA, USA) under the following conditions: 94°C for 2 min., 30 cycles (94°C for 30 sec., 60°C for 30 sec. and 72°C for 1 min.) and 72°C for 10 min. The PCR primers for GAPDH were as follows: forward primer: 5′-CATCACCATCTTCCAGGAGCG-3′; reverse primer: 5′-ACGGACACATTGGGGGTAGG-3′. The PCR primers for IRF-8 were as follows: forward primer: 5′-CGTGGAAGACGAGGTTACGCTG-3′; reverse primer: 5′- GCTGAATGGTGTGTGTCATAGGC-3′. The PCR primers for IL-12p40 were as follows: 5′-TCACCTGCCCAACTGCCGAG-3′; reverse primer: 5′-GGAACGCACCTTTCTGGTTACAC-3′. The PCR primers for iNOS were as follows: forward primer: 5′-CCAGAGGACCCAGAGACAAGC-3′; reverse primer: 5′-GGCAGCACATCAAAGCGGC-3′. For PCR analysis, various cycle numbers were previously tested, and 30 cycles was found to be optimal for effectively distinguishing differences in IRF-8 levels between control and tumour-induced CD11b+Gr-1+ cells. For genotyping transgenic offspring, PCR analysis was performed with the same IRF-8 primers (30 cycles), which effectively distinguished transgenic IRF-8 levels from endogenous genomic IRF-8 levels (the latter of which were undetectable under these conditions), while the PCR primers (40 cycles) for PyMT were as follows: forward primer: 5′-AGTCACTGCTACTGCACCCAG-3′; reverse primer: 5′-CTCTCCTCAGTTCCTCGCTCC-3′. PCR products were separated on a 1% or 2% agarose gel, and images captured with the BioDocIt imaging system (UVP, Upland, CA, USA).
To quantify PCR band intensities, gel images were first captured as noted above. The PCR band intensity was then quantified using the NIH Image J program (NIH). The IRF-8 band intensity of each sample was normalized over the band intensity of GAPDH corresponding to that same sample. The relative IRF-8 level was expressed as the IRF-8 band intensity/GAPDH band intensity.
Statistical analysis
Two pairwise comparisons were performed to compare distributions of the wild-type (double-negative) versus IRF-8 Tg genotypes, or the sTg (MTAG) versus dTg genotypes for each parameter. For comparisons of the wild-type versus IRF-8 Tg genotypes, or tumour volumes between the sTg (MTAG) and dTg genotypes, anova was used to perform the analysis, with the litter specified as the experimental unit. For other comparisons of sTg (MTAG) versus dTg genotypic parameters, an analysis of covariance was performed, with the litter specified as the experimental unit and tumour volume as the covariate, since the association varied with volume. Specifically, comparisons between genotypes were made at the quartiles of tumour volume if the volume was related to the given parameter. This covariate was dropped from the model only if both slope estimates were equal to zero, in which case the model was reduced to an anova analysis such as for the comparison of the wild-type versus IRF-8 Tg genotypes. Tumour-induced CD11b+Gr-1+ data were analysed with a mixed model anova; treatment (control versus 4T1-bearing mice) was the fixed effect and mouse and sample (‘triplicate’) were the random effects. Two-sample comparisons were performed with a t-test (after verifying normality of distributions). Given the number of analyses performed and the likely relatedness of parameters, only a P-value < 0.01 was considered statistically significant, while P-values between 0.01 and 0.05 would be associated with strong trends.
Results
Accumulation of CD11b+Gr-1+ cells in the spleen and bone marrow of tumour-bearing mice
CD11b+Gr-1+ cells are found at low frequencies in the spleens of wild-type mice. However, these cells have been shown to expand extensively with progressive tumour growth [4, 5, 8, 12, 22]. To understand the relationship between IRF-8 expression and the expansion of CD11b+Gr-1+ cells in tumour-bearing hosts, we examined two models, reflecting implantable (4T1 mammary carcinoma) or autochthonous (MTAG) tumour growth. In the implantable tumour setting, 4T1 cells were injected orthotopically into the mammary gland of female BALB/c or CB6F1/J (H-2b/d) hybrid mice, and tumour measurements recorded. Age-matched control mice did not receive a tumour injection. In the MTAG model, female sTg mice with varying tumour loads (from approximately 1.3–2.5 cm3 total tumour volume) were compared with age-matched, littermate wild-type mice (e.g. see Fig. 6A).
Figure 6.
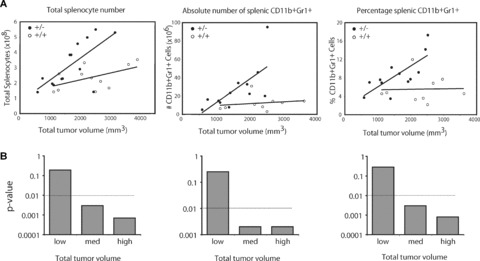
Transgenic expression of IRF-8 reduces splenic CD11b+Gr-1+ cells in a spontaneous tumour model of mammary carcinoma. (A): The role of IRF-8 in the production of tumour-induced CD11b+Gr-1+ cells was analysed in a dTg mouse model as described in Fig. 5. Here, tumour-bearing sTg MTAG mice (denoted as ‘+/–’) were compared to dTg mice (denoted as ‘+/+’) for changes in the percentages and absolute numbers of splenic CD11b+Gr-1+ cells, as well as total splenocytes as a function of overall tumour load. As noted in Fig. 5, since transgenic expression of IRF-8 had no significant impact on tumour growth in dTg mice, both sTg and dTg groups were compared in an age- and littermate-matched fashion. It is also important to note that alterations in these haematopoietic parameters were not observed in IRF-8 sTg mice as compared to wild-type mice (not shown). Each data point represents data from an individual mouse. (B) Statistical summary of the data illustrated in the (A), whereby ‘low, med (medium) and high’ overall tumour loads reflected tumour volumes of approximately 1300, 2000 and 2500 mm3, respectively, in sTg and dTg mice. Horizontal dashed line represents the statistical cut-off.
A robust accumulation of CD11b+Gr-1+ cells was observed in the spleens of 4T1 tumour-bearing mice (Fig. 1A). The results from the five mice illustrated are representative of several independent experiments in two different strains of H-2d-expressing mice. Based on analysis of the five separate mice represented, the percentages of CD11b+Gr-1+ cells in the spleen ranged from 29–44% (Fig. 1A, left panel), which translated to approximately 150–300 × 106 total CD11b+Gr-1+ cells/spleen (Fig. 1B, right panel). Values for splenic CD11b+Gr-1+ cells in non-tumour-bearing mice ranged from 1% to 4%. Moreover, analysis of bone marrow cells in a separate group of 4T1 tumour-bearing mice revealed increases in both the percentages (P= 0.011) and absolute numbers (P= 0.037) of CD11b+Gr-1+ cells as compared to non-tumour-bearing control preparations (Fig. 1B). Representative dot plot analyses of CD11b+Gr-1+ cells from unfractionated spleen and bone marrow compartments of both control and 4T1 tumour-bearing mice are also illustrated (Fig. 2). Functional studies revealed that tumour-derived CD11b+Gr-1+ cells inhibited TCR-induced proliferation of CD4+ or CD8+ T cells compared to CD11b+Gr-1+ cells of non-tumour-bearing control mice (Fig. 1C), an important characteristic of MDSC. These data also support our previous findings in the MTAG model [26], where we observed analogous results in terms of splenomegaly and alterations in the frequencies and numbers of CD11b+Gr-1+ cells as a consequence of autochthonous tumour growth (see also Figs. 5 and 6). In addition, despite the considerable expansion of splenic CD11b+Gr-1+ cell populations, no significant changes were observed in the absolute numbers of various other lymphocyte subsets between MTAG mice with extensive tumour burdens versus non-tumour-bearing littermate control mice [26].
Figure 1.
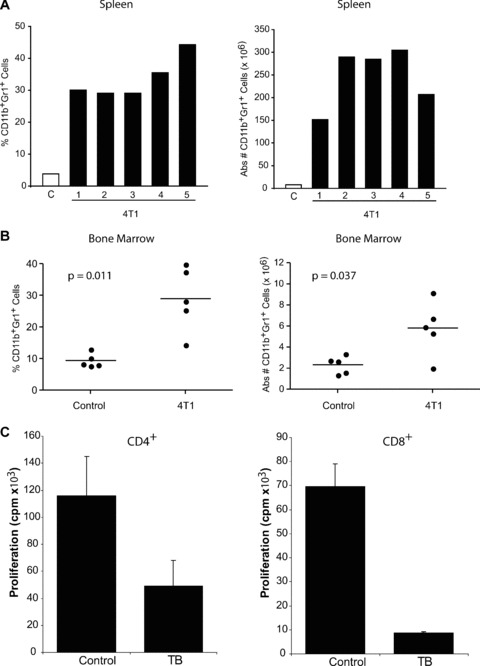
Accumulation of CD11b+Gr-1+ cells in 4T1 tumour-bearing mice. 4T1 mammary tumour cells (5 × 104/mouse) were injected orthotopically in five individual female CB6F1/J (H-2b/d) mice. Control (denoted as ‘C’) mice did not receive a tumour injection. Upon reaching the ethical limit of 2 cm3 tumour volume (at day 26), all mice were euthanized, and splenocytes recovered for enumeration. (A, left panel) Unfractionated splenocytes were evaluated for the percentage of CD11b+Gr-1+ cells using two-colour flow cytometry. (A, right panel) Absolute numbers of CD11b+Gr-1+ cells were calculated by multiplying the percentages determined in (A) by total splenocyte counts. For both panels, splenocytes from the control group were pooled and an average result determined, whereas splenocytes from the 4T1 tumour-bearing mice were analysed separately. Comparable results were observed when BALB/c mice were similarly challenged with 4T1 cells. In a separate experiment, the percentages (B, left panel) and absolute numbers (B, right panel) of CD11b+Gr-1+ cells were determined, as described above from unfractionated bone marrow cells of control and 4T1 tumour-bearing mice. Each data point represents the results of an individual mouse. (C) Splenic CD4+ or CD8+ T cells were purified from non-tumour-bearing wild-type (WT) CB6F1/J mice. T cells were mixed with CD11b+Gr-1+ cells purified from the spleens of either WT or 4T1 tumour-bearing (TB) CB6F1/J mice (1:1 ratio). In the case of the control group, both T cells and CD11b+Gr-1+ cells were obtained from the same mouse. In the case of tumour-induced CD11b+Gr-1+ cells, T cells from a wild-type mouse were used to avoid any in vivo tumour-induced effect on T-cell function. Cultures were then incubated in the absence or presence of immobilized anti-CD3 mAb for 48 hrs. Proliferation was measured by 3H-thymidine uptake after an additional 24 hrs of incubation. Results represent the mean ± S.E.M. of triplicate wells.
Figure 2.
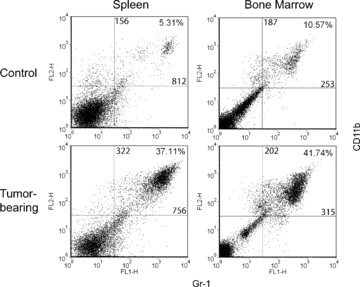
Flow cytometric analysis of CD11b+Gr-1+ cells from both spleen and bone marrow of control and 4T1 tumour-bearing mice. Representative two-colour dot plots for the percentages and MFI values of CD11b+Gr-1+ cells derived from Fig. 1. The percentages and MFI values for two-colour staining are shown in the upper right quadrant. The MFI values for CD11b and Gr-1 staining are shown along the vertical and horizontal lines within the upper right quadrant, respectively. Data are representative of at least five separate mice per group.
Figure 5.
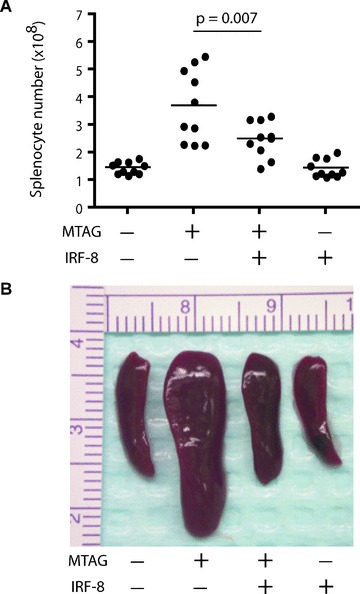
Transgenic expression of IRF-8 reduces splenomegaly in a spontaneous tumour model of mammary carcinoma. (A): The role of IRF-8 on tumour-induced splenomegaly was analysed in a dTg mouse model designed to express the transgenes for both IRF-8 and MMTV-PyMT (denoted as MTAG). Four possible genotypes were produced as a result of mating the two heterozygous sTg mouse colonies. Transgenic expression of IRF-8 did not significantly affect primary tumour growth in dTg mice (2240 ± 268 mm3; mean ± S.E.M. of 10 separate mice), compared with sTg MTAG mice (1950 ± 179 mm3; n= 10). Mice were then killed and splenocyte counts determined alongside age- and (in most cases) littermate-matched double-negative (wild-type) or sTg IRF-8 cohorts. Each data point represents total splenocyte counts from an individual mouse. (B) Photographic illustration of a representative spleen of 10 mice, reflecting the four different genotypes.
IRF-8 is down-regulated in tumour-induced CD11b+Gr-1+ cells
One possibility for the robust expansion or accumulation of tumour-induced CD11b+Gr-1+ cells is altered myelopoiesis. Because IRF-8 is crucial for maintaining normal myelopoiesis [14–19], we examined whether IRF-8 expression was altered in 4T1 tumour-induced CD11b+Gr-1+ cells. CD11b+ cells were purified from the spleens or bone marrow of control and 4T1 tumour-bearing mice. We developed a highly effective technique for the isolation of CD11b+ cells that co-expressed Gr-1 from both control and tumour-bearing (4T1, MTAG) mice. These purified CD11b+Gr-1+ cells were then evaluated for IRF-8 expression levels after treatment with or without IFN-γ/LPS (Fig. 3A and B). IFN-γ/LPS were used to optimally up-regulate IRF-8 expression, as previously described [18, 33]. In some experiments, cell-free supernatants were recovered for cytokine analysis (Fig. 3C and D).
Figure 3.
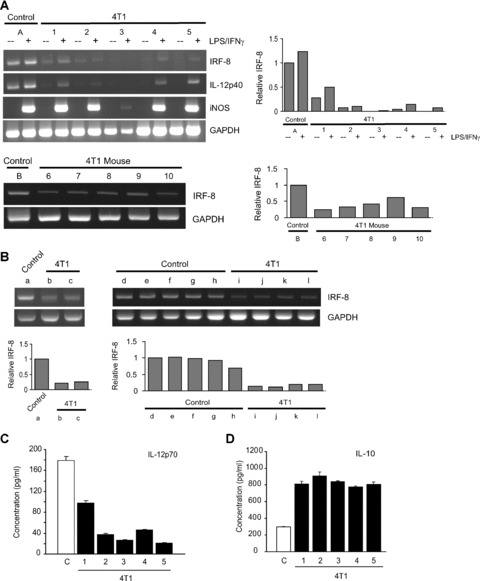
IRF-8 expression is down-regulated in 4T1 tumour-induced CD11b+Gr-1+ cells. CD11b+Gr-1+ cells from the control and 4T1 tumour-bearing groups of mice shown in Fig. 1 were analysed for the expression of the indicated genes by RT-PCR (upper set of genes corresponds to A). CD11b+Gr-1+ cells were first purified by CD11b-magnetic bead cell separation, and incubated overnight in the absence (−) or presence (+) of IFN-γ (100 U/ml) plus LPS (1 μg/ml). (Mouse #3 showed a weak response to iNOS induction, but responded as well as the other 4T1-tumour-bearing mice in terms of IL-10 production; see D). (Lower gel of A) In a separate experiment, IRF-8 expression levels were determined, as described above, from a control group (‘B’) and five individual tumour-bearing mice (‘6–10’). Control CD11b+Gr-1+ cells were first collected from a pool of splenocytes, whereas CD11b+Gr-1+ cells from 4T1 tumour-bearing mice were isolated from individual mice. Experiments were repeated in BALB/c mice with similar results. (B) IRF-8 expression levels of CD11b+Gr-1+ cells purified from bone marrow of control (a; d–h) and 4T1 tumour-bearing CB6F1/J mice (b, c; i–l), as in (A). The results are summarized from two independent experiments shown as, ‘a–c’ and ‘d–l’. Samples shown as ‘d–l’ represent data from individual mice, whereas samples shown as ‘a, b and c’ represent data from pools of five, two and two mice, respectively. The relative levels of IRF-8 shown in (A) and (B) were quantified by analysis of the PCR band intensities, as described in the ‘Materials and methods’. The first lane of each of the four gels was arbitrarily set at a relative intensity of 1. (C and D) Tumour-induced CD11b+Gr-1+ cells also displayed an altered IL-12/IL-10 profile. CD11b+Gr-1+ cells of the same control (‘C’) and 4T1 tumour-bearing groups shown in (A) were also analysed for production of IL-12 (IL-12p70) and IL-10 following stimulation with IFN-γ and LPS overnight. Cytokines were undetectable in the absence of stimulation (not shown). Data represent the mean ± S.E.M. of triplicate determinations for cytokine concentration from each preparation. Experiments were repeated in BALB/c mice with similar results.
In the five separate tumour-derived CD11b+Gr-1+ preparations analysed in Fig. 1, we observed a reproducible reduction of both basal and inducible IRF-8 expression levels as compared to the control preparation (Fig. 3A, upper left and right panels). Due to the low frequencies of CD11b+Gr-1+ cells in non-tumour bearing control mice, RNA was extracted from CD11b+Gr-1+ cells that were isolated from a pool of five control spleens, and was representative of several independent experiments (e.g. see Fig. 3A, lower panel as another independent experiment). The expression of iNOS was measured as a positive control for cellular activation.
In addition, because IL-12p40 subunit expression is IRF-8 dependent [18, 33], we examined control and tumour-induced CD11b+Gr-1+ cells for alterations in IL-12p40 expression levels. As with IRF-8, we observed a reduction in IL-12p40 expression levels in CD11b+Gr-1+ cells from tumour-bearing mice compared with control cells. These latter findings also validated that the reduction in IRF-8 expression levels were significant enough to have downstream functional significance. A reduction in constitutive IRF-8 expression levels in tumour-induced CD11b+Gr-1+ cells, compared to the control cells, was verified in a second independent experiment (Fig. 3A, lower left and right panels). We also observed that CD11b+Gr-1+ cells of tumour-bearing MTAG mice displayed lower IRF-8 levels, particularly basal expression (P= 0.011), with respect to age-matched controls (Fig. 4).
Figure 4.
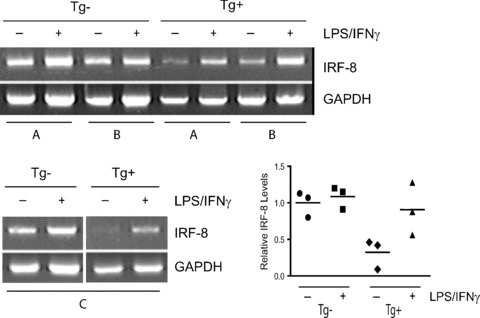
IRF-8 expression is reduced in CD11b+Gr-1+ cells derived from tumour-bearing MTAG mice. CD11b+Gr-1+ cells from control and tumour-bearing MTAG mice were analysed for basal and inducible IRF-8 expression levels, as in Fig. 2. CD11b+Gr-1+ cells were purified and then incubated overnight in the absence (−) or presence (+) of IFN-γ plus LPS. Here, three separate pairs of age-matched control (‘Tg–’) and tumour-bearing MTAG mice (‘Tg+’) (total tumour volume of each mouse, ≥2 cm3), was assessed, indicated as pairs (A), (B) (upper gel) or (C) (lower gel). (Lower right panel) The relative levels of IRF-8 shown for mouse pairs (A)–(C) were then quantified by analysis of the PCR band intensities, as in Fig. 2. We divided (or standardized) each individual ratio (IRF-8/β-actin) by the mean ratio of the Tg- group (without in vitro treatment) to yield a relative ratio. The graph depicts the relative ratio for each of the twelve observations (n= 3 corresponding to groups A, B and C/in vitro treatment). The horizontal lines indicate the means of the standardized values.
The analysis of IRF-8 expression was extended to CD11b+Gr-1+ cells isolated from the bone marrow of individual 4T1 tumour-bearing or control mice. Basal IRF-8 levels of CD11b+Gr-1+ cells purified from the bone marrow of tumour-bearing mice (Fig. 3B, mice b and c; i–l) were found to be greatly reduced compared to controls (Fig. 3B, mice a; d–h). In all cases, IRF-8 expression levels of the controls were similar to each other (Fig. 3B, mice a, d–h) and consistently higher than those observed in samples from tumour-bearing mice (Fig. 3B upper and lower panels, a versus b or c; d–h versus i–l). Therefore, in this 4T1 model, the data indicated that IRF-8 expression within tumour-induced CD11b+Gr-1+ cells were demonstrably lower when compared with control preparations from both the splenic and bone marrow compartments.
After observing that IL-12p40 expression levels were reduced (Fig. 3A, top panel), we examined whether IRF-8 down-regulation correlated with a decrease in the production of IL-12 protein levels. The cell-free supernatants from the same untreated and IFN-γ/ LPS-treated cell preparations were analysed, which revealed that tumour-induced CD11b+Gr-1+ cells produced lower amounts of IL-12 when compared with cells from control mice (P= 0.015; Fig. 3C). Conversely, these tumour-induced CD11b+Gr-1+ cells (including mouse 3) secreted significantly higher levels of IL-10, a MDSC-associated cytokine [34, 35], compared with control cells (P= 0.009; Fig. 2D), which verified that these preparations were functionally intact, but biologically altered.
Transgenic expression of IRF-8 reduces the accumulation of tumour-induced CD11b+Gr-1+ cells
Thus far, these data indicate an inverse correlation between IRF-8 levels and the emergence of tumour-induced CD11b+Gr-1+ cells. To establish a causal link between IRF-8 expression levels, tumour growth and the accumulation of CD11b+Gr-1+ cells, we developed and utilized an IRF-8 Tg mouse. Because of the role that IRF-8 plays in myelopoiesis, we postulated that transgene expression of IRF-8 in the CD11b+Gr-1+ cellular compartment would render these cells more refractory or resistant to tumour-induced alterations. Control experiments verified that IRF-8 expression levels were at least two-fold higher in CD11b+Gr-1+ cells of IRF-8 transgenic mice compared with age- and gender-matched wild-type mice (data not shown). Furthermore, a CBC with differential was performed on peripheral blood from both IRF-8 transgenic and age/gender-matched wild-type mice as readouts for potential haematopoietic changes caused by IRF-8 transgene expression (Table 1). Overall, we found that all haematological parameters measured within the leucocyte compartment of both groups of mice were within normal range. Although some fluctuations were noted in red blood cell numbers, this occurred in both groups of mice. Furthermore, no red blood cell (e.g. red cell mass) or platelet abnormalities were observed in both groups of mice by blood smear analysis. These data support earlier phenotypic studies, which revealed no overt differences in the frequencies and absolute numbers of major splenic leucocyte subsets (e.g. T-cell subsets, B cells and myeloid populations) from IRF-8 transgenic versus wild-type mice (data not shown).
Table 1.
Haematological parameters in peripheral blood of wild-type versus IRF-8 transgenic micea
| Cell type | Wild-type mouse # | IRF-8 transgenic mouse # | Normal range | ||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | ||
| Absolute numbersb | |||||||
| Total leucocytes | 4.86 | 6.10 | 3.74 | 5.96 | 6.60 | 6.04 | 1.8–10.7 |
| Neutrophils | 0.80 | 1.29 | 0.77 | 1.23 | 1.65 | 1.09 | 0.1–2.4 |
| Lymphocytes | 3.80 | 4.46 | 2.82 | 4.59 | 4.74 | 4.70 | 0.9–9.3 |
| Monocytes | 0.26 | 0.34 | 0.15 | 0.13 | 0.20 | 0.24 | 0.0–0.4 |
| Eosinophils | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.0–0.2 |
| Basophils | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.0–0.2 |
| Erythrocytes | 9.58 c | 9.30 | 6.32 | 8.15 | 9.45 | 9.69 | 6.36–9.42 |
| Platelets | 2036.00 | 1049.00 | 1087.00 | 989.00 | 1250.00 | 1076.00 | 592–2972 |
| Percentages | |||||||
| Neutrophils | 16.38 | 21.13 | 20.66 | 20.68 | 24.98 | 18.08 | 6.6–38.9 |
| Lymphocytes | 78.20 | 73.16 | 75.30 | 77.02 | 71.88 | 77.86 | 55.8–91.6 |
| Monocytes | 5.32 | 5.63 | 3.92 | 2.22 | 3.04 | 3.96 | 0.0–7.5 |
| Eosinophils | 0.06 | 0.00 | 0.08 | 0.07 | 0.10 | 0.08 | 0.0–3.9 |
| Basophils | 0.03 | 0.08 | 0.04 | 0.00 | 0.00 | 0.02 | 0.0–2.0 |
CBC was performed on both wild-type and IRF-8 transgenic mice, which were gender- (male), age- (16–20 weeks) and littermate matched. Three mice per group were analysed.
All values are enumerated as (×10−3/μl) except for erythrocytes, which are quantified as (×10−6/μl).
Underlined values are mildly out of normal range, although no abnormalities were observed in red cells by blood smear analysis.
To evaluate the in vivo effects of IRF-8 transgene expression on tumour growth and CD11b+Gr-1+ development/accumulation, dTg mice were produced that expressed the transgenes for both IRF-8 and mammary carcinoma development (MTAG). To do so, we mated sTg IRF-8 and MTAG mice, which shared the same genetic background. Since sTg IRF-8 and MTAG mice are maintained as heterozygous colonies, this also allowed us to examine the resultant progeny from four possible genotypes in an age-, gender-, and in most instances, a littermate-matched manner, including those that were: dTg (i.e. double-positive), wild-type (double-negative) or sTg for either IRF-8 or MTAG (i.e. single-positive). Within these various genotypes, we focused on potential differences in the myeloid compartment, including total splenocyte counts and the percentages and absolute numbers of splenic CD11b+Gr-1+ cells.
Transgenic expression of IRF-8 was found to have a significant impact on constraining splenomegaly and CD11b+Gr-1+ myelopoiesis (Figs. 5 and 6) during autochthonous tumour growth in the dTg mice. It was observed that spleen size and total splenocyte numbers (P= 0.007) were significantly lower in dTg mice, compared to sTg MTAG mice (Fig. 5). The number of splenocytes in dTg mice was also not significantly different from that of wild-type (P= 0.076) or IRF-8 Tg (P= 0.10) mice (Fig. 5). This contrasts sharply with the highly significant differences noted in spleen size and splenocyte number (P < 0.0001) of sTg MTAG mice compared with the wild-type controls. Also, compared to the wild-type controls, IRF-8 expression alone did not significantly affect spleen size, spleen number (P= 0.70; Fig. 5) or the composition of various leucocyte subsets (data not shown). However, no significant (P= 0.37) alteration in the growth of primary mammary gland carcinoma was observed between dTg mice (2240 ± 268 mm3) and sTg MTAG mice (1950 ± 179 mm3), based on the tumour size data illustrated in Fig. 6A. Collectively, these observations suggested that transgenic expression of IRF-8 in dTg tumour-bearing mice resulted in a splenic phenotype similar to that of the non-tumour-bearing control mice. Furthermore, the finding that tumour size was not different between these two genotypes indicated that IRF-8 in dTg mice played an important role in providing resistance to tumour-induced splenomegaly and increased splenic cell number.
Lastly, we evaluated changes in splenocyte number in further detail, as well as the frequencies and absolute numbers of splenic CD11b+Gr-1+ cells as a function of tumour load in sTg MTAG versus dTg mice (Fig. 6). As demonstrated by the slope of the lines, increasing tumour volume was accompanied by a significant increase in the overall number of splenocytes (P= 0.0007), as well as in the number (P= 0.013) and percentage (P= 0.002) of CD11b+Gr-1+ cells in sTg MTAG mice. In contrast, increasing tumour volume had no significant effect on the total number of splenocytes (P= 0.18) or the number (P= 0.62) and percentage (P= 0.75) of splenic CD11b+Gr-1+ in dTg mice (Fig. 6). Thus, in dTg mice, the slope of the line remained essentially unchanged despite increases in overall tumour burden. Therefore, using these particular parameters, as tumour volume increased, so did the differences between the two tumour-bearing genotypes (Fig. 6B). For instance, when tumour volume remained low, no significant differences were observed between these two genotypes in terms of the number of splenocytes or the percentages and numbers of splenic CD11b+Gr-1+ cells (Fig. 6B). However, as tumour volume increased to intermediate and high levels, so did the significance of the differences between these two genotypes in those same haematopoietic parameters (Fig. 6B). Therefore, splenomegaly in sTg MTAG mice was the result of CD11b+Gr-1+ myeloid cell accumulation influenced by the presence and size (volume) of autochthonous primary mammary tumour growth (Fig. 6). In contrast, in dTg mice, splenomegaly and CD11b+Gr-1+ myeloid cell production were significantly dampened regardless of tumour volume, which strengthens the hypothesis that IRF-8 plays an important role in constraining such haematopoietic changes.
Also, in contrast to what was observed with the CD11b+Gr-1+ population, the absolute numbers of CD4+ T cells, CD8+ T cells and NK cells did not significantly vary with tumour volume in both sTg MTAG and dTg mice. The absolute number of B220+ B cells, however, was significantly higher (P= 0.006) in sTg MTAG versus dTg mice at just one level of tumour load (‘med’). Furthermore, there were no significant differences in the total number of splenocytes, as well as the percentages and absolute numbers of these various lymphoid and myeloid subsets between wild-type and IRF-8 sTg mice. This suggested no adverse impact of the IRF-8 transgene on these haematopoietic compartments. Therefore, compared with sTg MTAG mice, transgenic expression of IRF-8 in MTAG mice seemed to constrain the tendency toward tumour-induced splenomegaly and CD11b+Gr-1+ myelopoiesis.
Discussion
CD11b+Gr-1+ MDSC are potentially important negative regulators of host immunosurveillance and antitumour activity. Therefore, efforts to understand their development or function may aid in the design of therapies that impede their involvement, thereby enhancing antitumour immunity. We tested the hypothesis that IRF-8 controls the development/expansion of tumour-induced CD11b+Gr-1+ cells. This notion stemmed from the original discovery that IRF-8 is essential for normal myeloid cell development, differentiation and function [14–19]. Consequently, if IRF-8 expression becomes altered during tumour growth or progression, this may have profound consequences on normal myelopoiesis and the accumulation of CD11b+Gr-1+ cells. In fact, IRF-8-deficiency in null mice causes a myeloproliferative disorder, including the emergence and expansion of CD11b+Gr-1+ myeloid populations. The loss of IRF-8 in tumour-induced CD11b+Gr-1+ cells may also have important implications for the production of IRF-8-dependent cytokines, such as IL-12 [18, 33], which if reduced or inhibited could further compromise host–tumour immunosurveillance.
In this study, we examined whether: (i) IRF-8 expression in the CD11b+Gr-1+ population was down-regulated as a consequence of tumour growth and (ii) IRF-8 expression played a causal role in the emergence of tumour-induced CD11b+Gr-1+ cells in vivo. The approach taken to explore a mechanistic role of IRF-8 in the regulation of this myeloid population in vivo was to produce an IRF-8 Tg mouse. In this model, IRF-8 was transcriptionally regulated via the CMV promoter, and we focused our efforts on the consequences of tumour-induced alterations in the spleen, which included the induction of splenomegaly and an increase in the frequency/absolute numbers of splenic CD11b+Gr-1+ cells. The prediction was if tumour growth altered IRF-8 expression, and IRF-8 levels were important for the development or accumulation of the CD11b+Gr-1+ cell population, then tumour-bearing IRF-8 (dTg) mice would be more refractory to changes in the frequencies of these cells.
In addition to the well-characterized 4T1 mammary tumour model [21–23], we made use of the MTAG mouse model of autochthonous mammary carcinoma [24–28] to address specific aspects of the relationship between IRF-8 and CD11b+Gr-1+ myelopoiesis. As expected, 4T1 tumour growth led to profound splenomegaly, which was predominately attributed to the accumulation of CD11b+Gr-1+ cells. Similar findings have also been recently reported in the MTAG model [25, 26]. Functional studies verified that these cells were immunosuppressive, as judged by their ability to inhibit TCR-mediated lymphoproliferation in vitro. Moreover, in both the 4T1 and MTAG models, we observed an inverse relationship between IRF-8 expression and the emergence of these tumour-induced CD11b+Gr-1+ cells. The exact mechanisms that alter IRF-8 expression, however, remain unclear but may reflect the types and/or quantities of TDF. For instance, several studies have reported that different tumours secrete various factors that potentially modulate the generation, differentiation and/or function of CD11b+Gr-1+ cells, namely VEGF, GM-CSF, CSF-1, IL-10, PGE2 and/or IL-1 [4, 5, 8, 22, 32, 36, 37]. Thus, future work is needed to evaluate the relative contribution of these and/or other TDF that potentially affect IRF-8 expression in these myeloid populations.
Nonetheless, it was clear that transgenic expression of IRF-8 in MTAG mice (through the use of the dTg model) significantly reduced the propensity for splenomegaly and CD11b+Gr-1+ myelopoiesis. This indicates that IRF-8 plays a causal role in the tumour-directed process of CD11b+Gr-1+ cell induction and/or expansion. It is important to emphasize that in this study we focused on IRF-8-induced changes in splenic and bone marrow compartments of tumour-bearing mice, where counterpart control cell populations could be similarly isolated from non-tumour-bearing mice for comparison. Future studies are warranted to evaluate IRF-8 expression levels in tumour-infiltrating myeloid populations, as well as the functional impact of IRF-8 overexpression on MDSC activity and apoptotic phenotype. Additionally, although earlier studies have implicated a role for Stat3 activation in the development of these cells [1, 38], follow-up investigations are needed to reveal the precise molecular or biochemical relationship between these two transcription factors, IRF-8 and Stat3.
Despite the fact that IRF-8 expression in autochthonous tumour-bearing dTg mice resulted in a reversal to a near normal frequency of CD11b+Gr-1+ cells, there was no significant effect on primary tumour growth. It is possible, however, that the reduction in CD11b+Gr-1+ cells per se may be insufficient to affect tumour growth or progression and that additional host factors or mechanisms acting in concert with alterations in these cells may be required to maximally inhibit tumour growth. This notion is consistent with other tumour studies, revealing that biologic- or immune-based therapies mediate stronger antitumour effects when combined with strategies that eliminate MDSC or inhibit their suppressive function [39–43]. It is also possible that the lack of effect on tumour growth in dTg mice may be due to inadequate numbers of immune cells that infiltrate these tumours. This contention is supported by preliminary histopathological findings of tumours derived from sTg MTAG and dTg mice, which revealed low numbers of inflammatory cell infiltrates, as well as apoptosis in both groups of tumours.
Although further studies are necessary to explore the mechanism(s) underlying the lack of immune cell infiltration, our results in dTg mice demonstrated that transgenic IRF-8 expression affected the haematopoietic system and functioned as a suppressor of tumour-induced CD11b+Gr-1+ cell development/accumulation. Interestingly, the cytokine/chemokine profile of CD11b+Gr-1+ cells of tumour-bearing IRF-8 transgenic mice returned to a more ‘normal’, non-tumour-bearing phenotype. In terms of secreted IL-12 levels, there was a tendency toward higher amounts produced by CD11b+Gr-1+ myeloid cells of the IRF-8 transgenic group compared with the non-transgenic group (manuscript submitted). Future studies are also necessary to investigate additional functional aspects of IRF-8 overexpression, such as the effect of IRF-8 on MDSC activity, as well as their apoptotic phenotype. Taken collectively, this study demonstrates for the first time that differential IRF-8 expression plays an important role in the development and accumulation of tumour-induced CD11b+Gr-1+ cells, which may have important implications for the design of improved anti-cancer combination therapies.
Acknowledgments
We thank Drs. Keiko Ozato (NICHD, NIH) for providing the IRF-8 plasmid, Sandra Gendler (Mayo Clinic) for originally providing the B6-MTAG mice and Lionel Feigenbaum (SAIC/NCI) for assistance with the production of the IRF-8 transgenic mouse. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This work was also supported in part by RCPI Institutional Funding (to S.I.A.). K Liu is supported by Grant ROI CA 133085 from the National Cancer Institute/NIH.
References
- 1.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nature Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart TJ, Greeneltch KM, Lutsiak ME, et al. Immunological responses can have both pro- and antitumour effects: implications for immunotherapy. Expert Rev Mol Med. 2007;9:1–20. doi: 10.1017/S1462399407000233. [DOI] [PubMed] [Google Scholar]
- 4.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–23. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 5.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–45. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–89. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 7.Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–53. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 8.Ochoa AC, Zea AH, Hernandez C, et al. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13:721s–6s. doi: 10.1158/1078-0432.CCR-06-2197. [DOI] [PubMed] [Google Scholar]
- 9.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001;61:4756–60. [PubMed] [Google Scholar]
- 10.Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 11.Terabe M, Matsui S, Park JM, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198:1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature Rev. 2005;5:641–54. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 14.Nagamura-Inoue T, Tamura T, Ozato K. Transcription factors that regulate growth and differentiation of myeloid cells. Int Rev Immunol. 2001;20:83–105. doi: 10.3109/08830180109056724. [DOI] [PubMed] [Google Scholar]
- 15.Schiavoni G, Mattei F, Borghi P, et al. ICSBP is critically involved in the normal development and trafficking of Langerhans cells and dermal dendritic cells. Blood. 2004;103:2221–8. doi: 10.1182/blood-2003-09-3007. [DOI] [PubMed] [Google Scholar]
- 16.Tsujimura H, Nagamura-Inoue T, Tamura T, et al. IFN consensus sequence binding protein/IFN regulatory factor-8 guides bone marrow progenitor cells toward the macrophage lineage. J Immunol. 2002;169:1261–9. doi: 10.4049/jimmunol.169.3.1261. [DOI] [PubMed] [Google Scholar]
- 17.Holtschke T, Lohler J, Kanno Y, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–17. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 18.Masumi A, Tamaoki S, Wang IM, et al. IRF-8/ICSBP and IRF-1 cooperatively stimulate mouse IL-12 promoter activity in macrophages. FEBS Lett. 2002;531:348–53. doi: 10.1016/s0014-5793(02)03556-1. [DOI] [PubMed] [Google Scholar]
- 19.Tamura T, Ozato K. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res. 2002;22:145–52. doi: 10.1089/107999002753452755. [DOI] [PubMed] [Google Scholar]
- 20.Gabriele L, Phung J, Fukumoto J, et al. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J Exp Med. 1999;190:411–21. doi: 10.1084/jem.190.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–405. [PubMed] [Google Scholar]
- 22.Bunt SK, Yang L, Sinha P, et al. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–26. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinha P, Okoro C, Foell D, et al. Proinflammatory s100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–61. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–9. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 26.Stewart TJ, Lutsiak ME, Abrams SI. Immune consequences of protracted host-tumor interactions in a transgenic mouse model of mammary carcinoma. Cancer Invest. 2008;26:237–49. doi: 10.1080/07357900701708419. [DOI] [PubMed] [Google Scholar]
- 27.Maglione JE, Moghanaki D, Young LJ, et al. Transgenic polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–305. [PubMed] [Google Scholar]
- 28.Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67:5064–6. doi: 10.1158/0008-5472.CAN-07-0912. [DOI] [PubMed] [Google Scholar]
- 30.Chen D, Xia J, Tanaka Y, et al. Immunotherapy of spontaneous mammary carcinoma with fusions of dendritic cells and mucin 1-positive carcinoma cells. Immunology. 2003;109:300–7. doi: 10.1046/j.1365-2567.2003.01656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greeneltch KM, Haudenschild CC, Keegan AD, et al. The opioid antagonist naltrexone blocks acute endotoxic shock by inhibiting tumor necrosis factor-alpha production. Brain Behav Immun. 2004;18:476–84. doi: 10.1016/j.bbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Serafini P, Carbley R, Noonan KA, et al. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–43. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 33.Wang IM, Contursi C, Masumi A, et al. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol. 2000;165:271–9. doi: 10.4049/jimmunol.165.1.271. [DOI] [PubMed] [Google Scholar]
- 34.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 35.Sinha P, Clements VK, Bunt SK, et al. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–83. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez PC, Hernandez CP, Quiceno D, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–9. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinha P, Clements VK, Fulton AM, et al. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–13. doi: 10.1158/0008-5472.CAN-06-4174. [DOI] [PubMed] [Google Scholar]
- 38.Nefedova Y, Huang M, Kusmartsev S, et al. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol. 2004;172:464–74. doi: 10.4049/jimmunol.172.1.464. [DOI] [PubMed] [Google Scholar]
- 39.Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9. [PubMed] [Google Scholar]
- 40.Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Santo C, Serafini P, Marigo I, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci USA. 2005;102:4185–90. doi: 10.1073/pnas.0409783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki E, Kapoor V, Jassar AS, et al. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 43.Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]